

Summary
Bacteriophages, the historic model organisms facilitating the initiation of molecular biology, are still important candidates of numerous useful or promising biotechnological applications. Development of generally applicable, simple and rapid techniques for their genetic engineering is therefore a validated goal. In this article, we report the use of bacteriophage recombineering with electroporated DNA (BRED), for the first time in a coliphage. With the help of BRED, we removed a copy of mobile element IS1, shown to be active, from the genome of P1vir, a coliphage frequently used in genome engineering procedures. The engineered, IS‐free coliphage, P1virdeltaIS, displayed normal plaque morphology, phage titre, burst size and capacity for generalized transduction. When performing head‐to‐head competition experiments, P1vir could not outperform P1virdeltaIS, further indicating that the specific copy of IS1 plays no direct role in lytic replication. Overall, P1virdeltaIS provides a genome engineering vehicle free of IS contamination, and BRED is likely to serve as a generally applicable tool for engineering bacteriophage genomes in a wide range of taxa.
Introduction
The birth of molecular biology would have been inconceivable without the aid of bacteriophages. Not only were basic molecular processes, like replication, transcription regulation or recombination first studied in these systems, but they also provided inevitable tools for cloning and sequencing. Transduction, a technique once central to genetic mapping, is still widely used in bacterial genome engineering. Phage typing and phage display are two further important methods in microbiology and in the study of protein–protein interactions respectively.
Phage therapy, the use of hypervirulent (non‐lysogenizing) bacteriophages to eliminate bacterial infections preceded molecular biology by several decades. This technique seems to have found its place in veterinary sciences and the food industry (for review, see Atterbury, 2009), as the routine addition of antibiotics to livestock‐feed is becoming banned in an increasing number of countries. Phages used for such purposes are usually sought for in classical selection schemes, but with the exponential expansion of phage genome sequences, their targeted manipulation is also becoming a realistic demand. Altering host specificity, elimination of potential bacterial virulence genes and decreasing antigenicity are examples of tasks that would certainly benefit from a simple and rapid method for phage genome engineering in the lytic state.
A parallel need for engineering phages during their lytic cycle emerged in the course of our research. We have previously reduced the genome of Escherichia coli K‐12 strain MG1655 by sequentially deleting 42 genomic segments, a total of 14.3% of the genome (Pósfai et al., 2006). In the course of the project, we removed all mobile elements (ISes, prophages, recombination hot spots) from the genome, thereby significantly reducing the spontaneous mutation rate of the engineered strain, MDS42. The impact of the lack of IS elements on the rate of gene inactivation is especially pronounced under conditions of various stressors (Ashburner and Bonner, 1979; Kretschmer and Cohen, 1979; Pfeifer and Blaseio, 1990; Ratner et al., 1992; Levy et al., 1993; Eichenbaum and Livneh, 1998; Lamrani et al., 1999; Ilves et al., 2001; Christie‐Oleza et al., 2009) or when focusing on genetic systems that favour insertions over other mutational mechanisms (Reynolds et al., 1981; Kitamura et al., 1995; Hall, 1999; Stoebel et al., 2009). An example demonstrating both factors was recently described in an experimental system where the reintroduction of a single IS1 into MDS42 dramatically accelerated the mutational inactivation of a toxic cloned gene (Umenhoffer et al., 2010). Consequently, to keep genetic stability at its maximum, it is important to avoid the re‐entry of transposable elements in the course of genome engineering carried out in MDS42 and its derivates.
Phage P1 is one of the most widely used tools for general transduction when engineering enterobacterial genomes. Two of its strains have been sequenced to date (phage P1 mod749::IS5 c1‐100 and prophage P1mod1902::IS5 c1‐100 rev‐6 dmt_MB, GenBank Accession No. AF234172 and AF234173 respectively) (Lobocka et al., 2004). It was found that both carry single copies of IS1 and IS5 in their DNA. This raised the possibility that the use of P1 might result in the inadvertent transfer of IS sequences into the target genome. This would be undesirable and we decided to avoid it by constructing an IS free P1. As starting point, we decided to use P1vir, the hypervirulent, non‐lysogenizing version (Luria et al., 1960) to pursue this goal because it also eliminates the possibility of another type of genome contamination, the production of P1 lysogens. This introduced an additional complication, since phage genome engineering is most easily done in the lysogenic form. Bacteriophage recombineering using electroporated DNA (BRED), offered a possible solution by allowing the modification of phages in the lytic state (Marinelli et al., 2008). This method, described recently for mycobacteriophage engineering, employs bacterial overexpression of cloned phage recombinases to assemble phage genomes from purified phage DNA, and synthetic DNA fragments of choice.
In this article we report that P1vir contains a single IS1, demonstrate that it is capable of transposition, present the construction of an IS‐free P1vir phage using BRED, and verify its ability to carry out generalized transduction efficiently.
Results
Screening of the P1vir genome for ISes
To test which IS elements might be present in the genome of P1vir, test PCRs were applied to the P1vir phage lysate using the primer collection of the White Glove IS Detection Kit, targeting all ISes occurring in E. coli. Since phage lysates inevitably carry DNA contamination originating from the host bacteria, the phages to be screened were grown on MDS42+MD64 cells, which provided an IS‐free background. (Deletion MD64 removes a supposedly inactive IS609 that was identified after construction of IS‐free MDS42.) Besides the control reaction, only the PCR targeting IS1 yielded a strong band upon gel electrophoresis (Fig. S1A), indicating the presence of this element in P1vir. Although this experiment gave no information on the copy number or the location of IS1 in P1vir, we hypothesized that at least one copy is the ‘resident IS1’ (Iida et al., 1978) lying in the same position and orientation as in the sequenced P1 strains. This was confirmed by a PCR reaction carried out with primers IS1A1 and P1D (Fig. S2), and the specific IS1 was designated IS1P1vir (Fig. 1).
Figure 1.
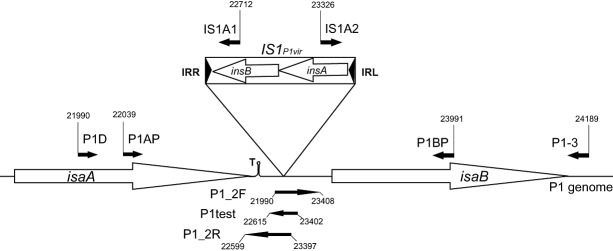
Map of the IS1P1vir region. Small filled arrows depict PCR primers, large open arrows mark genes. Numbers indicate primer coordinates, related to the P1mod749::IS5;c1‐100 sequence. IRR: right inverted repeat of IS1; IRL: left inverted repeat of IS1; T: Rho‐independent transcription terminator. Drawing is not to scale.
Examining the transpositional activity of IS1P1vir
Activation of the cryptic bgl operon in E. coli K‐12, known to occur primarily via IS transposition events, allows growth on salicin‐minimal medium, providing a convenient test system for detecting transpositional activity. To test whether it is capable of transposition, IS1P1vir, along with its flanking sequences, was cloned into a low‐copy temperature‐sensitive vector (pST76‐AYeaJ), and propagated in E. coli MDS42 cells at 30°C. MDS42/pST76‐AYeaJP1IS was plated on salicin‐minimal medium to select for spontaneous mutations activating the cryptic bgl operon (Hall, 1998). At 30°C, when pST76‐AYeaJP1IS plasmid is able to replicate, PCR screening of the bgl region of the arising colonies indicated that 100% (48/48) of the tested bgl+ mutants harboured an insertion in the bglR region. Sequencing one of the relevant PCR fragments yielded a sequence 100% identical to IS1P1vir, embedded in bglR, indicating that IS1P1vir is capable of transposition. The mutation rate calculated from the number of colonies was approximately three times higher than for MDS42Yea : IS1, an E. coli harbouring a single genomic copy of wild‐type (wt) IS1 (Fig. 2). At 37°C, when pST76‐AYeaJP1IS is not able to replicate as a plasmid, and antibiotic selection results in its integration into the genome in a single copy, 93% (45/48) of the colonies were insertion mutants, and the mutation rate was similar to that of MDS42Yea : IS1 at 37°C.
Figure 2.
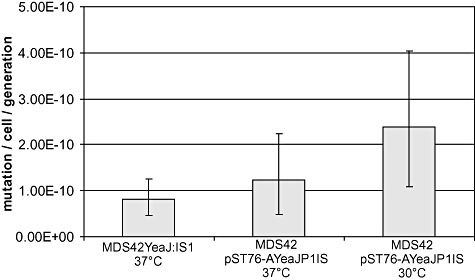
Mutation rates of E. coli strains harbouring a single copy of IS1 in the genome (MDS42Yea : IS1, 37°C), a single copy of IS1P1vir in the genome (MDS42 pST76‐AYeaJP1IS, 37°C) and IS1P1vir on a low‐copy plasmid (MDS42 pST76‐AYeaJP1IS, 30°C). Error bars indicate 95% confidence intervals.
Deletion of IS1P1vir from P1vir
P1virΔIS was constructed by deleting IS1P1vir, as described in Experimental procedures. Briefly, the genomic segment containing the deletion joint was assembled using overlapping PCR fragments. A mixture of this DNA fragment and the genomic DNA of P1vir was electroporated into a host expressing the λ‐red recombinases, allowing assembly of both wt and IS‐free P1 genomes. Host cells were plated in soft agar, and the emergence of plaques indicated successful assembly of phage genomes, despite the inevitable presence of double‐stranded breaks in the P1 chromosome (no plaques appeared when transforming λ‐red recombinase‐negative cells with P1 DNA). Plaques were picked, and the agarose plugs were digested with agarase to increase the number of released virions. Screening was done using a primer that spans the deletion (P1test), and a primer that anneals to the unaltered part of the phage genome (P1D) (Fig. 3A). The ratio of P1 DNA to the linear DNA fragment, the agarase treatment, as well as the incubation time after electroporation were all varied to optimize the BRED process (Table S1). In an optimal case, four out of 24 plaques tested positive. One mixed plaque, identified this way, was re‐plated on a bacterial lawn to obtain plaques that are either purely wt or purely recombinants. Four out of 15 plaques turned out to harbour recombinant P1virΔIS. Phage lysates generated from such pure recombinant plaques were confirmed to carry the deletion by PCR, using primers P1 BP and P1D (Fig. 3B). P1virΔIS was grown on MDS42+MD64 for another round of IS‐screening, performed using the White Glove IS Detection Kit. The test kit indicated no further copies of IS1, or any other transposable element of E. coli to be present in the phage genome (Fig. S1B).
Figure 3.
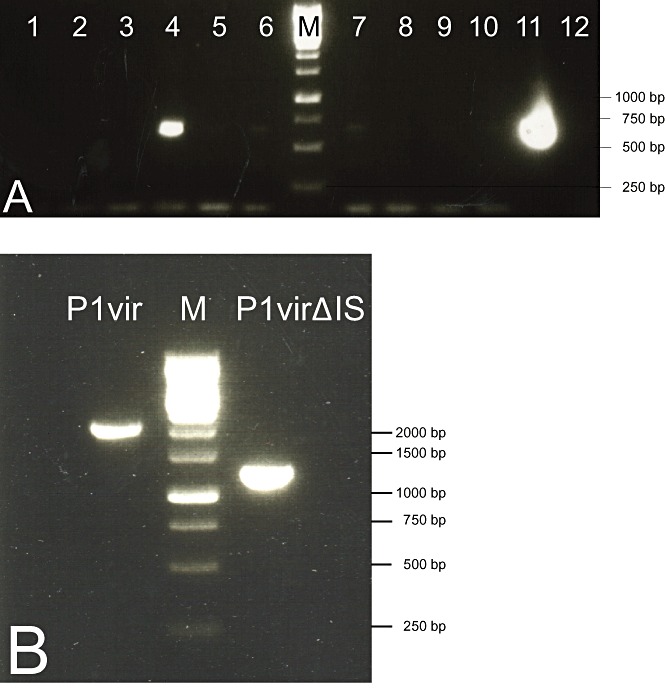
A. PCR screening, using primers P1D and P1test, of plaques obtained upon phage recombineering. Lanes 4 and 11 contain PCR products that indicate the presence of P1virΔIS within the plaques. B. PCR verification of the shortening of the IS‐deleted region in a pure P1virΔIS lysate using primers P1D and P1 BP. M: 1 kb DNA ladder (Fermentas).
Characterization of P1virΔIS
Phage P1virΔIS was grown on E. coli MG1655, and was titred as described in Experimental procedures. The mean phage titre was 1.16 × 1010 plaque forming units (pfu) ml−1, which falls in range of that of P1vir lysates, generated by the same protocol. Dependence of phage titre on multiplicity of infection (moi) yielded a similar tendency for both phages (Fig. S3). Moreover, based on plaque morphology, P1virΔIS is indistinguishable from P1vir (Fig. S4). The one‐step growth curves of P1virΔIS and P1vir are compared on Fig. 4. The latency time, rise period, and the burst size of the two phages are also basically identical (Fig. 4).
Figure 4.
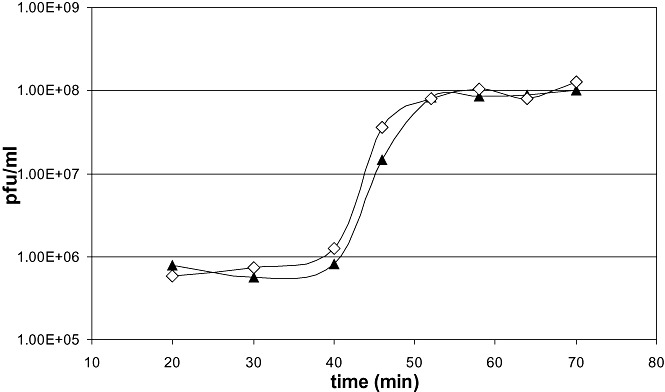
One‐step growth curves of P1vir (filled triangles) and P1virΔIS (open diamonds).
Next, competition experiments were carried out between P1vir and P1virΔIS to test whether removal of IS1P1vir from P1vir has a fitness cost. The mixture was grown on E. coli MG1655 for seven transfers, corresponding to 25–30 phage generations. The initial P1vir : P1virΔIS ratio of 11:27 displayed an insignificant change to 10:19 (P = 0.63).
To test the ability of P1virΔIS for general transduction, a cyan fluorescent protein gene, linked to a CmR marker on a 3 kb genomic segment, was transduced to a lacZΔM15 acceptor strain, using the protocol described in Experimental procedures. The obtained colonies were PCR‐screened for the transferred CFP gene as well as the recipient‐specific lac allele on a routine basis. In each transduction experiment, PCR fragments generated with primers ClaGFP/RihE or Zup/Zdn yielded fragments of the expected sizes (1700 bp and 400 bp respectively), verifying the presence of both markers within each tested colony. The efficiency of transduction for P1virΔIS was 3.04 × 10−6 transductants/phage (± 2.19 × 10−6), which is not significantly different from the value for P1vir (2.16 × 10−6 ± 1.51 × 10−6, P = 0.44).
Discussion
In our pursuit to reduce the chances of IS contamination during P1 transduction, our first question was whether P1vir had, similarly to its relatives, picked up and retained any ISes from the E. coli genome. Using a multi‐step PCR analysis, we localized a copy of IS1 in P1vir present in the same locus as in the sequenced P1 genomes. Sequencing of IS1P1vir revealed that, besides the two polymorphisms found in the IS1 of sequenced P1 strains, IS1P1vir harbours seven more point mutations (Table 1). Eight of the polymorphisms are present in various combinations in numerous IS1 elements of E. coli and its plasmids, possibly indicating that they do not inhibit transposition. The only mutation specific to IS1 elements in the three P1 genomes is the G > T transversion within the IR repeat. Since this latter mutation made annotators question its activity, we set out to test whether IS1P1vir is capable of transposition. In wt E. coli cells starving on salicin‐minimal medium, salicin breakdown is activated in 97% of the cases by an insertion sequence integrating upstream of the bgl operon (Hall, 1998). This system is such a sensitive indicator of active IS1 elements that a single copy reintroduced into the genome of MDS42 increases the frequency of bgl+ mutants ninefold (T. Fehér, B. Bogos, O. Méhi, G. Fekete, B. Csörgö, K. Kovács, G. Pósfai, B. Papp, L.D. Hurst, C. Pal, submitted). In our experiment, propagating IS1P1vir in MDS42 either on a low‐copy plasmid or on the genome, IS1P1vir was responsible for the majority of the bgl‐activating mutations. Quantitative analysis of its rate of insertion into bglR indicated a transposition rate comparable to that of other IS1 elements (Fig. 2).
Table 1.
Single‐nucleotide polymorphisms (SNPs) of IS1P1vir, as compared with IS1B (Accession No. X17345) of E. coli
| SNP | Locus (bp) |
|---|---|
| T > C | 128 |
| C > A | 262 |
| C > A | 298 |
| C > T | 301 |
| T > C | 304 |
| A > C | 393 |
| C > T | 396 |
| T > G | 486 |
| G > T | 757 |
The SNPs also found in the sequenced P1 genomes are in italics, the one in the IR repeat is in bold.
Insertion sequences have been reported on numerous occasions to dominate the mutations inactivating various genetic constructs propagated in E. coli (Rood et al., 1980; Nakamura and Inouye, 1981; Blumenthal et al., 1985; Muller et al., 1989; Prather et al., 2006; Rawat et al., 2009; Umenhoffer et al., 2010). In most of these cases, the selective advantage of the inactivated construct over the correct one was readily apparent. But can a transducing P1vir phage grown on an IS‐free donor strain serve as a source of ISes? Such an event requires that, upon P1 infection, the phage IS transposes into the host genome, followed by packaging of the IS‐carrying genomic segment into a P1 phage head during lytic growth. Transposition of various insertion sequences into the P1 prophage has been extensively studied by Werner Arber and colleagues (Iida et al., 1985). The group has also reported the transposition of IS1 from the prophage into the pBR325 plasmid (Meyer et al., 1980; Iida et al., 1981), therefore, transposition of IS1 from the circularized P1 DNA into the host chromosome upon lytic growth is also a realistic scenario. The chance of IS1 integrating within 110 kb (the maximum distance for co‐transduction) of a given selective marker in the genome is approximately 10−7 per cell (see Supporting information for details). This means that, upon lysis, only a minute fraction of the selectable transducing virions is expected to carry the IS, suggesting that contamination of the genome in routine marker‐driven genome manipulation processes using P1 transduction ought to be extremely rare. However, if P1 transduction is serially applied in combinatorial genome engineering and batch selection experiments, used for the transfer of multiple unknown alleles, emergence of IS1P1vir in the population becomes a realistic event. In such cases, the provided increase in evolutionary potential may facilitate escaping the burden of carrying certain genetic constructs, thereby allowing the rapid expansion of the small IS‐acquiring subpopulation. In light of these considerations, and to stay on the safe side of marker‐driven transduction, the elimination of ISes from P1 seemed worthwhile.
Deletion of IS1P1vir from the P1vir genome was accomplished using BRED, resulting in IS‐free P1virΔIS. Table S1 lists the details of our attempts to engineer P1virΔIS, along with the detected frequency of mixed plaques. It is apparent that liberation of the virions from the agarose plugs was necessary to detect the presence of recombinant phages. Melting the plugs was quite effective, but inactivated the phages, therefore we used agarase treatment for this purpose, with a minimal time span of 4 h. Increasing the incubation time to 40 min in‐between electroporation and plating, thereby allowing the amplification of phages, improved the success rate, presumably by increasing the quantity of recombinant phages above the threshold of detection. Electroporation of 100 ng of P1 DNA, along with 200–300 ng of linear DNA fragment, and applying the protocol described below resulted in straightforward and reproducible recombineering the P1vir genome.
To the best of our knowledge, this was the first example of using the λ phage recombinases for engineering a coliphage in the lytic state. λ‐red recombinases have been successfully applied previously for genome engineering in Salmonella enterica serovar Typhimurium (Stanley et al., 2000), S. enterica serovar Enteritidis (Lu et al., 2002), Shigella flexneri (Beloin et al., 2003; Zurawski et al., 2006), Shigella sonnei (Taniya et al., 2003), Klebsiella pneumoniae (Zheng et al., 2006), enterohaemorrhagic and enteropathogenic E. coli (Murphy and Campellone, 2003), uropathogenic E. coli (Eto et al., 2007), Yesinia pestis (Sun et al., 2008), Yersinia pseudotuberculosis (Derbise et al., 2003), Pseudomonas aeruginosa (Lesic and Rahme, 2008), Serratia marcescens (Rossi et al., 2003) and Vibrio anguillarum (Stork et al., 2004), suggesting that using the same enzymes for recombineering their bacteriophages should be effective. Most of the bacteria listed above generated interest due to their pathogenicity, and are consequently potential targets of phage therapy, as well. BRED could therefore become a valuable tool of ‘phage therapists’ for targeted engineering of phage genomes. Phage‐mediated biocontrol is already successfully used for the elimination of various Salmonella typhimurium (Berchieri et al., 1991; Fiorentin et al., 2005) and E. coli strains (Smith and Huggins, 1982; Barrow et al., 1998), as well as Pseudomonas (Greer, 1982) and Vibrio species (Cerveny et al., 2002) in agriculture and the food industry. Circumventing the need to generate temperate phage strains or to clone their genomes into various vectors could significantly speed up the engineering and development of therapeutic phages, generally classified as hypervirulent. The ongoing reduction in the cost of gene synthesis should enhance the use of BRED to engineer ever‐larger segments of phage genomes using synthetic genes.
A potential by‐product of this work was the accumulation of information regarding the function of the genes flanking IS1, and on the role of IS1 in their transcriptional regulation. Gene isaA encodes a 36 kDa protein of unknown function, which is absent from the closely related phage P7. It has no separate promoter, but lies within an operon with the upstream ssb gene (encoding a single‐stranded DNA‐binding protein), regulated by a σ70‐dependent promoter and a cI‐binding operator (Lobocka et al., 2004). Transcription of the negative DNA strand, originating from IS1P1vir is unlikely to interfere with isaA expression due to the presence of a Rho‐independent terminator located in‐between the two elements, abolishing transcription from the direction of insB (Fig. 1).
isaB encodes a 23 kDa protein, the function of which is also unknown. Contrary to isaA, isaB has its own σ70‐dependent promoter, and is not controlled by the main phage repressors, cI or Lpa. Its extensive richness in AT, as well as the presence of the rare ATA codon and CTAG (the most infrequent tetramer of P1), indicates that it is a recent acquisition of the P1 genome (Lobocka et al., 2004). The TTGGCA within the left inverted repeat (IRL) of IS1P1vir, together with the genomic AATATGC found 18 bp downstream, may act as ‘−35’ and ‘−10’ regions, respectively, composing an outward pointing promoter (Prentki et al., 1986). This, as well as the inward pointing promoter activity of the right inverted repeat (IRR) (Machida et al., 1984), may increase the expression level of isaB. This facilitation of transcription was not investigated in our work, but even if present, it confers the phage no measurable fitness advantage in the lytic cycle, as seen in our competition experiments.
In the course of our rapid characterization, we found no particular changes in phenotype caused by the engineering process, and therefore gained no further information of the roles of isaA and isaB. We conclude that IS1P1vir plays no detectable function in the lytic state of P1vir. One cannot rule out the possibility, however, that the resident IS1 elements of temperate P1 phages do provide the prophages, or their host bacteria a primary fitness gain [similarly to the interruption of the restriction‐modification genes of P1 by an IS5 (Lobocka et al., 2004)], or possibly a second‐order selective advantage by increasing their evolutionary potential (Woods et al., 2011). From the practical point of view, P1virΔIS maintains its capacity of general transduction, a property we have taken advantage of in eight further genome engineering steps carried out in E. coli to date (data not shown).
In the work described above, we have engineered an IS‐free P1 phage for generalized transduction. We have also shown that the λ‐red recombinase system can be successfully used for recombineering coliphages in their lytic state.
Experimental procedures
Buffers and media
Buffer Φ80+, containing 0.1 M NaCl, 0.01 M Tris (pH 7.9), 0.01 M CaCl2 and 0.01 M MgCl2, was used for making all phage dilutions (Blattner et al., 1974). TBE buffer contained 45 mM Tris, 45 mM boric acid and 1 mM EDTA (Sambrook et al., 1987). Bacteria were grown in Luria–Bertani (LB) medium (Sambrook et al., 1987) or in minimal salts (MS) medium (Hall, 1998). Agar was used in a concentration of 1.5% in plates, and 0.75% in soft agar. In some cases (see below), 0.5% Seakem LE agarose (Lonza, Basel, Switzerland) was used instead of soft agar. Soft agar and soft agarose were always supplemented with 5 mM CaCl2 and 5 mM MgSO4. Antibiotics (Sigma‐Aldrich, St. Louis, MO, USA) were used in the following end‐concentrations: Chloramphenicol (Cm): 25 µg ml−1, Ampicillin (Ap): 50 µg ml−1.
Strains and plasmids
Phage P1vir was a kind gift of Tamás Gaál. Escherichia coli K‐12 MG1655 (Blattner et al., 1997) and its derivative, MDS42+MD64, were used for phage propagation. Strain MDS42, harbouring a genome reduced by 14.3%, and lacking all active mobile DNA elements, has been described elsewhere (Pósfai et al., 2006). Strain MDS42lacZΔM15 has the 5′ end of the lacZ gene deleted (MG1655 coordinates: 365406 to 365497). In strain MDS42+MD64, the genomic region 1501205–1515026 (MG1655 coordinates) has also been deleted using the suicide plasmid method (Pósfai et al., 1999). This deletion removes IS609, an insertion sequence that is inactive due to a premature stop codon in its transposase gene, yncK. Strain MDS42rihB::(CFP tetR CmR) carries a tetracycline‐inducible cyan fluorescent protein gene, along with a repressor and a Cm‐resistance marker within the rihB pseudogene ( T. Fehér, B. Bogos, O. Méhi, G. Fekete, B. Csörgö, K. Kovács, G. Pósfai, B. Papp, L.D. Hurst, C. Pal, submitted). Plasmid pKD46 (Datsenko and Wanner, 2000) was a kind gift of Barry Wanner. Plasmid pST76‐A (GenBank Accession No. Y09895.1) has been described previously (Pósfai et al., 1997). Its derivative, pST76‐AyeaJ is a temperature‐sensitive suicide plasmid that was constructed by cloning genomic regions of E. coli up‐ and downstream of yeaJ (Pósfai et al., 2006). Plasmid pST76‐AyeaJP1IS was constructed by PCR‐amplifying the P1AP30–P1BP30 segment of the P1vir genome (Fig. 1, Table S2), and cloning the phosphorylated fragment into the Klenow‐treated EcoRI site of pST76‐AyeaJ. Strain MDS42Yea : IS1 harbours a single copy of IS1 in its genome, integrated into the yeaJ gene (T. Fehér, B. Bogos, O. Méhi, G. Fekete, B. Csörgö, K. Kovács, G. Pósfai, B. Papp, L.D. Hurst, C. Pal, submitted).
Phage propagation and titreing
Bacteriophages were grown as described by Miller (1977). Host bacteria were grown to 0.5 optical density (OD540) in LB medium supplemented with 5 mM CaCl2 and 5 mM MgSO4. Nine hundred microlitres of the culture was mixed with 100 µl of phage suspension, containing 107–108 pfu ml−1. After a 10 min still incubation at 37°C, the mixture was added to 10 ml of fresh LB medium, supplemented with 5 mM CaCl2 and 5 mM MgSO4, and incubated in a shaker at 37°C. A gradual increase in OD540, followed by a sudden drop, usually after 3–4 h, indicated cell lysis. At this point, 50 µl of chloroform was added, and the mixture was vortexed to lyse remaining cells. The lysate was centrifuged at 10 000 g for 2 min, and the supernatant was aspirated and stored at 4°C. To measure the titre of the phage lysates, they were serially 10‐fold diluted in Φ80+ buffer, and 100 µl of each dilution was mixed with 100 µl of mid‐log phase (OD540 = 0.5) bacteria. After a 10 min still incubation at 37°C, the mixture was added to 3 ml of molten soft agar (kept at 42°C), vortexed and poured onto pre‐warmed LB plates. Plaques appearing on the plates were counted after 16 h of incubation at 37°C, and the titre of the original lysate was calculated in units of pfu ml−1.
Screening of the P1vir genome for ISes
The White Glove IS Detection Kit (Scarab Genomics, Madison, WI, USA) was used as described in its User Protocol. Briefly, the kit provides primer pairs for PCR detection of IS1, IS2, IS3 (ISEc17), IS4, IS5, IS10, IS30D, IS150, IS186, IS600 (ISsd1), IS609, IS911, ISEc1,3,5, ISEc4, RhsA,B,C and RhsD,E. PCR reactions were carried out using Taq Polymerase (Fermentas, Vilnius, Lithuania) in 25 µl reaction volumes, each containing 0.5 µl of 100‐fold diluted phage lysate (grown on MDS42+MD64) as a template. Primers P1C1A and P1C1B, amplifying the cI repressor gene, were used for the positive control reaction.
Assaying IS1 transpositional activity
Cells to be assayed (MDS42/pST76‐AyeaJP1IS) were grown to saturation in 20 parallel 1 ml cultures at 30°C, using MS+Ap medium. Cells of each tube were pelleted, and plated onto MS plates supplemented with 0.4% salicin (Alpha Aesar, Ward Hill, MA, USA) and Ap, and incubated at 30°C. To allow for the appearance of slow‐growing pre‐exposure mutants, the appearing colonies were counted and logged daily for 4 days. To calculate the mutation rate, a fluctuation analysis was performed on the colony numbers on day four, using the Ma‐Sandri‐Sarkar maximum likelihood method (Sarkar et al., 1992). Total cell counts were obtained by plating appropriate dilutions onto LB+Ap plates. The ratio of insertion mutants was calculated using colony‐PCR screens amplifying the bglR1–bglR2 region of 48 colonies. Insertion mutants were called when PCR fragments were longer than amplicons obtained from wt cells. One such PCR fragment was sequenced using primers bglR1 and bglR2. The assay measuring IS1 transpositional activity was repeated at 37°C.
Phage DNA preparation
Phage DNA was prepared from phage lysate as described earlier (Lech, 1987). Briefly, cell debris was removed from 1 l of phage lysate by centrifugation at 10 000 g for 10 min. Phages were precipitated with 5.3% (m/v) PEG6000 overnight at 4°C, followed by pelleting at 3000 g. The pellet was resuspended in 30 ml of Suspension Medium (50 mM Tris‐HCl pH 7.5, 0.58% NaCl, 0.1% MgSO4 and 0.1% gelatin), and the phage was released from PEG by the slow addition of KCl to a final concentration of 1 M at 4°C. After centrifugation at 10 000 g for 10 min, the supernatant (containing the phages) was purified by step gradient centrifugation at 65 000 g for 2 h using CsCl solutions of 1.7, 1.5 and 1.3 g ml−1. The lowest blue band was recovered from the 1.5 g ml−1 layer, and resuspended in 1.5 g ml−1 CsCl for equilibrium gradient centrifugation at 100 000 g for 24 h. The blue band was removed, and dialysed twice for 4 h against 500 ml of Low Salt Buffer (50 mM Tris‐HCl pH 7.5, 50 mM NaCl, 10 mM MgSO4). Phage capsids were removed by three phenol extractions and two chloroform extractions (20 min each), using the gentle agitation of a shaker to minimize the mechanical damage of the DNA. Finally, we dialysed the DNA against 500 ml of Tris‐EDTA buffer (10 mM Tris‐HCl pH 7.5, 1 mM EDTA) at 4°C for 8 h.
Construction of a linear DNA fragment targeting the phage genome
The P1AP–P1 BP PCR fragment targeting the phage genome was constructed in two steps. First, the P1AP–P1_2R and the P1 BP–P1_2F segments of the P1vir genome were PCR‐amplified using the respective primers. PCR reactions were carried out in volumes of 100 µl using Phusion High Fidelity DNA Polymerase (New England Biolabs, Ipswich, MA, USA) at an annealing temperature of 57°C. The PCR products were purified using the PCR Advanced™ PCR Cleanup System (Viogene, Taiwan, China), and were resuspended in 50 µl of the kit's elution buffer. In the second step, 2 µl of both purified PCR fragments were mixed and used as templates in a PCR carried out with primers P1AP and P1 BP using HotMaster DNA Polymerase (Eppendorf AG, Hamburg, Germany). The product of the correct size (1200 bp) was isolated from a 1% agarose gel using the Wizard SV Gel and PCR Clean‐up System (Promega, Madison, WI, USA), and was concentrated with SureClean Plus DNA Purification Kit (BioLine, London, UK) to approximately 150 ng µl−1.
Bacteriophage recombineering
Engineering the genome of P1vir was based on the method described by Marinelli and colleagues (2008). MG1655/pKD46 cells were grown to mid‐log phase at 30°C in the presence of 0.1% arabinose in 100 ml of LB+Ap, and were harvested by centrifugation at OD540 = 0.5. In repeated suspension–centrifugation cycles, cells were washed twice in ice‐cold water, and once in ice‐cold 10% glycerol, then suspended in 0.2 ml of ice‐cold 10% glycerol, and divided into 40 µl aliquots (Sambrook et al., 1987). A mixture containing 100 ng of phage DNA and 300 ng of the P1AP–P1 BP PCR fragment was electroporated into an aliquot of MG1655/pKD46 cells at a voltage of 1800 V in a 0.1 cm electroporation cuvette using a Bio‐Rad MicroPulser Electroporator. Next, cells were added to 1 ml of LB medium, and were shaken for 40 min at 37°C. After mixing the suspension with 3 ml of soft agarose, it was poured onto LB plates, and incubated overnight at 37°C. The following day, plaques were picked with a micropipette using cut‐off 200 µl tips. Each aspirated agarose plug was suspended in 20 µl of TBE buffer, supplemented with 0.25 units of agarase (Fermentas, Vilnius, Lithuania). After 4 h of incubation at 42°C, 1 µl of each sample was used directly as a template in 25 µl of PCR reactions screening for recombinants with primers P1test‐P1D, at an annealing temperature of 58°C. Five microlitres of plaque‐suspensions yielding a PCR fragment were mixed with 100 µl of mid‐log‐phase MG1655 cells, and plated in soft agarose. Plaques arising were suspended and PCR screened again, and 5 µl of PCR‐positive plaque suspensions were used to grow pure phage lysates, as described above.
One‐step phage growth experiments
Growth characteristics of wt and engineered phages were measured by the modified protocol of Chow and colleagues (1988). Briefly, E. coli MDS42 was grown to an OD540 of 0.6 in LB medium supplemented with 5 mM CaCl2 and 5 mM MgSO4. One hundred microlitres of bacteria were mixed with 100 µl of phage lysate (108 pfu ml−1) to adjust the moi to approximately 0.2. After 1 min incubation at 37°C, the mixture was centrifuged for 40 s at 10 000 g. The supernatant, containing unadsorbed bacteriophages, was discarded, and the pellet was resuspended in 30 ml of LB medium supplemented with 5 mM CaCl2 and 5 mM MgSO4, and was shaken at 37°C at 220 r.p.m. Aliquots of the culture were taken at regular intervals and were diluted 103 and 105‐fold respectively. Each dilution was mixed with 100 µl of mid‐log‐phase MDS42 cells, and was plated in soft agar, as described above. The plaque counts obtained the next day were multiplied by the dilution factor and plotted against the elapsed time to obtain the one‐step growth curves.
P1 transduction
Transducing phage was grown on the donor cell MDS42rihB::(CFP tetR Cmr) as described above. For transduction, 0.1 ml of fresh overnight culture of the acceptor strain MDS42lacZΔM15, grown in the presence of 5 mM CaCl2 and 5 mM MgSO4, was mixed with 0.1 ml of the transducing phage. After incubating the mixture at 37°C for 15 min, 0.5 ml of LB and 35 µl of 1 M Na3 citrate were added, and incubation continued for 1 h. Finally, cells were pelleted at 10 000 g, plated on LB+Cm plates, and incubated for 36 h at 37°C. Ten colonies from each transduction were checked by colony‐PCR for genetic markers of both the donor and the acceptor cell, using primer pairs ClaGFP5+RihE and Zup+Zdn respectively (Table S2).
To calculate the efficiency of transduction, the number of obtained colonies was divided by the titre of the phage lysate used. A total of six transductions were carried out using three distinct lysates for both P1vir and P1virΔIS, and replicating each experiment.
Phage competition experiments
Mixtures containing virions of P1vir and P1virΔIS in 1:1 ratios were prepared. The phage mixtures were grown on MG1655 cells for a total of seven rounds of lysis and re‐infection. The exact ratios of the phages in the initial and the final lysate were measured by plating appropriate dilutions onto MG1655 lawns (as described under Phage propagation and titreing), and PCR‐screening the obtained plaques using primers P1‐3 and P1D. The significance of the difference in the initial and final ratios was checked with a χ2‐test.
Acknowledgments
This work was supported by the Hungarian Research Fund (OTKA K43260 and OTKA PD72719).
We thank Gabriella Balikó, Bálint Csörgö, Gábor Draskovits, Zsuzsanna Györfy, Erzsébet Magyaródi, Ágnes Szalkanovics, Attila Szvetnik and Edit Tímár for technical assistance and discussions.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Results of PCR reactions screening for ISes within the P1vir (A) and P1virΔIS (B) genomes, using the White Glove IS Detection Kit. In both panels, lanes contain primers specific for the following elements: lane 1: IS1, lane 2: IS2, lane 3: IS3 (ISEc17), lane 4: IS4, lane 5: IS5, lane 6: IS10, lane 7: IS30D, lane 8: IS150, lane 9: IS186, lane 10: IS600 (ISsd1), lane 11: IS609, lane 12: IS911, lane 13: ISEc1,3,5, lane 14: ISEc4, lane 15: RhsA,B,C, lane 16: RhsD,E. Lane 17 contained primers specific for P1 C1 gene. M: 1 kb DNA ladder (Fermentas).
PCR verification of the position and orientation of IS1P1vir using primers IS1A1 and P1D.
Dependence of phage titre on multiplicity of infection (moi).
{kind=link}
Comparison of plaque morphology. Left: P1vir. Right: P1virΔIS.
Table S1. Details of attempts to engineer P1virΔIS using BRED. Note that multiple batches of arabinose‐induced electrocompetent MG1655/pKD46 cells were used during the optimization, which could cause inconsistencies.
Table S2. Primers used in this study.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Ashburner M., Bonner J.J. The induction of gene activity in drosophilia by heat shock. Cell. 1979;17:241–254. doi: 10.1016/0092-8674(79)90150-8. [DOI] [PubMed] [Google Scholar]
- Atterbury R.J. Bacteriophage biocontrol in animals and meat products. Microb Biotechnol. 2009;2:601–612. doi: 10.1111/j.1751-7915.2009.00089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrow P., Lovell M., Berchieri A., Jr Use of lytic bacteriophage for control of experimental Escherichia coli septicemia and meningitis in chickens and calves. Clin Diagn Lab Immunol. 1998;5:294–298. doi: 10.1128/cdli.5.3.294-298.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beloin C., Deighan P., Doyle M., Dorman C. Shigella flexneri 2a strain 2457T expresses three members of the H‐NS‐like protein family: characterization of the Sfh protein. Mol Genet Genomics. 2003;270:66–77. doi: 10.1007/s00438-003-0897-0. [DOI] [PubMed] [Google Scholar]
- Berchieri A., Jr, Lovell M.A., Barrow P.A. The activity in the chicken alimentary tract of bacteriophages lytic for Salmonella typhimurium. Res Microbiol. 1991;142:541–549. doi: 10.1016/0923-2508(91)90187-f. [DOI] [PubMed] [Google Scholar]
- Blattner F.R., Fiandt M., Hass K.K., Twose P.A., Szybalski W. Deletions and insertions in the immunity region of coliphage lambda: revised measurement of the promoter‐startpoint distance. Virology. 1974;62:458–471. doi: 10.1016/0042-6822(74)90407-3. [DOI] [PubMed] [Google Scholar]
- Blattner F.R., Plunkett G., 3rd, Bloch C.A., Perna N.T., Burland V., Riley M. The complete genome sequence of Escherichia coli K‐12. Science. 1997;277:1453. doi: 10.1126/science.277.5331.1453. et al. [DOI] [PubMed] [Google Scholar]
- Blumenthal R.M., Gregory S.A., Cooperider J.S. Cloning of a restriction‐modification system from Proteus vulgaris and its use in analyzing a methylase‐sensitive phenotype in Escherichia coli. J Bacteriol. 1985;164:501–509. doi: 10.1128/jb.164.2.501-509.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerveny K.E., DePaola A., Duckworth D.H., Gulig P.A. Phage therapy of local and systemic disease caused by Vibrio vulnificus in iron‐dextran‐treated mice. Infect Immun. 2002;70:6251–6262. doi: 10.1128/IAI.70.11.6251-6262.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow J.J., Batt C.A., Sinskey A.J. Characterization of Lactobacillus bulgaricus Bacteriophage ch2. Appl Environ Microbiol. 1988;54:1138–1142. doi: 10.1128/aem.54.5.1138-1142.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie‐Oleza J.A., Lanfranconi M.P., Nogales B., Lalucat J., Bosch R. Conjugative interaction induces transposition of ISPst9 in Pseudomonas stutzeri AN10. J Bacteriol. 2009;191:1239–1247. doi: 10.1128/JB.01071-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko K.A., Wanner B.L. One‐step inactivation of chromosomal genes in Escherichia coli K‐12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derbise A., Lesic B., Dacheux D., Ghigo J.M., Carniel E. A rapid and simple method for inactivating chromosomal genes in Yersinia. FEMS Immunol Med Microbiol. 2003;38:113–116. doi: 10.1016/S0928-8244(03)00181-0. [DOI] [PubMed] [Google Scholar]
- Eichenbaum Z., Livneh Z. UV light induces IS10 transposition in Escherichia coli. Genetics. 1998;149:1173–1181. doi: 10.1093/genetics/149.3.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eto D.S., Jones T.A., Sundsbak J.L., Mulvey M.A. Integrin‐mediated host cell invasion by type 1‐piliated uropathogenic Escherichia coli. PLoS Pathog. 2007;3:e100. doi: 10.1371/journal.ppat.0030100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorentin L., Vieira N.D., Barioni W., Jr Oral treatment with bacteriophages reduces the concentration of Salmonella Enteritidis PT4 in caecal contents of broilers. Avian Pathol. 2005;34:258–263. doi: 10.1080/01445340500112157. [DOI] [PubMed] [Google Scholar]
- Greer G.G. Psychrotrophic bacteriophages for beef spoilage pseudomonads. J Food Prot. 1982;45:1318–1325. doi: 10.4315/0362-028X-45.14.1318. [DOI] [PubMed] [Google Scholar]
- Hall B.G. Activation of the bgl operon by adaptive mutation. Mol Biol Evol. 1998;15:1–5. doi: 10.1093/oxfordjournals.molbev.a025842. [DOI] [PubMed] [Google Scholar]
- Hall B.G. Spectra of spontaneous growth‐dependent and adaptive mutations at ebgR. J Bacteriol. 1999;181:1149–1155. doi: 10.1128/jb.181.4.1149-1155.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida S., Meyer J., Arber W. The insertion element IS1 is a natural constituent of coliphage P1 DNA. Plasmid. 1978;1:357–365. doi: 10.1016/0147-619x(78)90051-3. [DOI] [PubMed] [Google Scholar]
- Iida S., Marcoli R., Bickle T.A. Variant insertion element IS1 generates 8‐base pair duplications of the target sequence. Nature. 1981;294:374–376. doi: 10.1038/294374a0. [DOI] [PubMed] [Google Scholar]
- Iida S., Meyer J., Arber W. Bacteriophage P1 derivatives unaffected in their growth by a large inversion or by IS insertions at various locations. J Gen Microbiol. 1985;131:129–134. doi: 10.1099/00221287-131-1-129. [DOI] [PubMed] [Google Scholar]
- Ilves H., Horak R., Kivisaar M. Involvement of sigma(S) in starvation‐induced transposition of Pseudomonas putida transposon Tn4652. J Bacteriol. 2001;183:5445–5448. doi: 10.1128/JB.183.18.5445-5448.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura K., Torii Y., Matsuoka C., Yamamoto K. DNA sequence changes in mutations in the tonB gene on the chromosome of Escherichia coli K12: insertion elements dominate the spontaneous spectra. Jpn J Genet. 1995;70:35–46. doi: 10.1266/jjg.70.35. [DOI] [PubMed] [Google Scholar]
- Kretschmer P.J., Cohen S.N. Effect of temperature on translocation frequency of the Tn3 element. J Bacteriol. 1979;139:515–519. doi: 10.1128/jb.139.2.515-519.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamrani S., Ranquet C., Gama M.J., Nakai H., Shapiro J.A., Toussaint A., Maenhaut‐Michel G. Starvation‐induced Mucts62‐mediated coding sequence fusion: a role for ClpXP, Lon, RpoS and Crp. Mol Microbiol. 1999;32:327–343. doi: 10.1046/j.1365-2958.1999.01352.x. [DOI] [PubMed] [Google Scholar]
- Lech K. Making phage DNA from liquid lysates. In: Ausubel F.M., Brent R., Kingston R.E., Moore D.D., Seidman J.G., Smith J.A., Struhl K., editors. Greene Publishing Associates and Wiley‐Interscience; 1987. pp. 1.13.1–1.13.6. [Google Scholar]
- Lesic B., Rahme L.G. Use of the lambda Red recombinase system to rapidly generate mutants in Pseudomonas aeruginosa. BMC Mol Biol. 2008;9:20. doi: 10.1186/1471-2199-9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy M.S., Balbinder E., Nagel R. Effect of mutations in SOS genes on UV‐induced precise excision of Tn10 in Escherichia coli. Mutat Res. 1993;293:241–247. doi: 10.1016/0921-8777(93)90075-r. [DOI] [PubMed] [Google Scholar]
- Lobocka M.B., Rose D.J., Plunkett G., 3rd, Rusin M., Samojedny A., Lehnherr H. Genome of bacteriophage P1. J Bacteriol. 2004;186:7032–7068. doi: 10.1128/JB.186.21.7032-7068.2004. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S., Killoran P.B., Fang F.C., Riley L.W. The global regulator ArcA controls resistance to reactive nitrogen and oxygen intermediates in Salmonella enterica serovar Enteritidis. Infect Immun. 2002;70:451–461. doi: 10.1128/IAI.70.2.451-461.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luria S.E., Adams J.N., Ting R.C. Transduction of lactose‐utilizing ability among strains of E. coli and S. dysenteriae and the properties of the transducing phage particles. Virology. 1960;12:348–390. doi: 10.1016/0042-6822(60)90161-6. [DOI] [PubMed] [Google Scholar]
- Machida C., Machida Y., Ohtsubo E. Both inverted repeat sequences located at the ends of IS1 provide promoter functions. J Mol Biol. 1984;177:247–267. doi: 10.1016/0022-2836(84)90455-8. [DOI] [PubMed] [Google Scholar]
- Marinelli L.J., Piuri M., Swigonova Z., Balachandran A., Oldfield L.M., van Kessel J.C., Hatfull G.F. BRED: a simple and powerful tool for constructing mutant and recombinant bacteriophage genomes. PLoS ONE. 2008;3:e3957. doi: 10.1371/journal.pone.0003957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer J., Iida S., Arber W. Does the insertion element IS1 transpose preferentially into A+T‐rich DNA segments? Mol Gen Genet. 1980;178:471–473. doi: 10.1007/BF00270502. [DOI] [PubMed] [Google Scholar]
- Miller J.H. Cold Spring Harbor Laboratory Press; 1977. Generalized transduction; use of P1 in strain construction; pp. 201–205. [Google Scholar]
- Muller J., Reinert H., Malke H. Streptokinase mutations relieving Escherichia coli K‐12 (prlA4) of detriments caused by the wild‐type skc gene. J Bacteriol. 1989;171:2202–2208. doi: 10.1128/jb.171.4.2202-2208.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy K.C., Campellone K.G. Lambda Red‐mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic E. coli. BMC Mol Biol. 2003;4:11. doi: 10.1186/1471-2199-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K., Inouye M. Inactivation of the Serratia marcescens gene for the lipoprotein in Escherichia coli by insertion sequences, IS1 and IS5; sequence analysis of junction points. Mol Gen Genet. 1981;183:107–114. doi: 10.1007/BF00270147. [DOI] [PubMed] [Google Scholar]
- Pfeifer F., Blaseio U. Transposition burst of the ISH27 insertion element family in Halobacterium halobium. Nucleic Acids Res. 1990;18:6921–6925. doi: 10.1093/nar/18.23.6921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pósfai G., Koob M.D., Kirkpatrick H.A., Blattner F.R. Versatile insertion plasmids for targeted genome manipulations in bacteria: isolation, deletion, and rescue of the pathogenicity island LEE of the Escherichia coli O157:H7 genome. J Bacteriol. 1997;179:4426–4428. doi: 10.1128/jb.179.13.4426-4428.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pósfai G., Kolisnychenko V., Bereczki Z., Blattner F.R. Markerless gene replacement in Escherichia coli stimulated by a double‐strand break in the chromosome. Nucleic Acids Res. 1999;27:4409–4415. doi: 10.1093/nar/27.22.4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pósfai G., Plunkett G., 3rd, Fehér T., Frisch D., Keil G.M., Umenhoffer K. Emergent properties of reduced‐genome Escherichia coli. Science. 2006;312:1044–1046. doi: 10.1126/science.1126439. et al. [DOI] [PubMed] [Google Scholar]
- Prather K.L., Edmonds M.C., Herod J.W. Identification and characterization of IS1 transposition in plasmid amplification mutants of E. coli clones producing DNA vaccines. Appl Microbiol Biotechnol. 2006;73:815–826. doi: 10.1007/s00253-006-0532-1. [DOI] [PubMed] [Google Scholar]
- Prentki P., Teter B., Chandler M., Galas D.J. Functional promoters created by the insertion of transposable element IS1. J Mol Biol. 1986;191:383–393. doi: 10.1016/0022-2836(86)90134-8. [DOI] [PubMed] [Google Scholar]
- Ratner V.A., Zabanov S.A., Kolesnikova O.V., Vasilyeva L.A. Induction of the mobile genetic element Dm‐412 transpositions in the Drosophila genome by heat shock treatment. Proc Natl Acad Sci USA. 1992;89:5650–5654. doi: 10.1073/pnas.89.12.5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawat P., Kumar S., Pental D., Burma P.K. Inactivation of a transgene due to transposition of insertion sequence (IS136) of Agrobacterium tumefaciens. J Biosci. 2009;34:199–202. doi: 10.1007/s12038-009-0023-5. [DOI] [PubMed] [Google Scholar]
- Reynolds A.E., Felton J., Wright A. Insertion of DNA activates the cryptic bgl operon in E. coli K‐12. Nature. 1981;293:625–629. doi: 10.1038/293625a0. [DOI] [PubMed] [Google Scholar]
- Rood J.I., Sneddon M.K., Morrison J.F. Instability in tyrR strains of plasmids carrying the tyrosine operon: isolation and characterization of plasmid derivatives with insertions or deletions. J Bacteriol. 1980;144:552–559. doi: 10.1128/jb.144.2.552-559.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi M., Paquelin A., Ghigo J., Wandersman C. Haemophore‐mediated signal transduction across the bacterial cell envelope in Serratia marcescens: the inducer and the transported substrate are different molecules. Mol Microbiol. 2003;48:1467–1480. doi: 10.1046/j.1365-2958.2003.03516.x. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritch E.F., Maniatis T. Cold Spring Harbor Laboratory Press; 1987. [Google Scholar]
- Sarkar S., Ma W.T., Sandri G.H. On fluctuation analysis: a new, simple and efficient method for computing the expected number of mutants. Genetica. 1992;85:173–179. doi: 10.1007/BF00120324. [DOI] [PubMed] [Google Scholar]
- Smith H.W., Huggins M.B. Successful treatment of experimental Escherichia coli infections in mice using phage: its general superiority over antibiotics. J Gen Microbiol. 1982;128:307–318. doi: 10.1099/00221287-128-2-307. [DOI] [PubMed] [Google Scholar]
- Stanley T.L., Ellermeier C.D., Slauch J.M. Tissue‐specific gene expression identifies a gene in the lysogenic phage Gifsy‐1 that affects Salmonella enterica serovar Typhimurium survival in Peyer's patches. J Bacteriol. 2000;186:4406–4413. doi: 10.1128/jb.182.16.4406-4413.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoebel D.M., Hokamp K., Last M.S., Dorman C.J. Compensatory evolution of gene regulation in response to stress by Escherichia coli lacking RpoS. PLoS Genet. 2009;5:e1000671. doi: 10.1371/journal.pgen.1000671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stork M., Di Lorenzo M., Mourino S., Osorio C.R., Lemos M.L., Crosa J.H. Two tonB systems function in iron transport in Vibrio anguillarum, but only one is essential for virulence. Infect Immun. 2004;72:7326–7329. doi: 10.1128/IAI.72.12.7326-7329.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W., Wang S., Curtiss R., 3rd Highly efficient method for introducing successive multiple scarless gene deletions and markerless gene insertions into the Yersinia pestis chromosome. Appl Environ Microbiol. 2008;74:4241–4245. doi: 10.1128/AEM.00940-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniya T., Mitobe J., Nakayama S., Mingshan Q., Okuda K., Watanabe H. Determination of the InvE binding site required for expression of IpaB of the Shigella sonnei virulence plasmid: involvement of a ParB BoxA‐like sequence. J Bacteriol. 2003;185:5158–5165. doi: 10.1128/JB.185.17.5158-5165.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umenhoffer K., Feher T., Baliko G., Ayaydin F., Posfai J., Blattner F.R., Posfai G. Reduced evolvability of Escherichia coli MDS42, an IS‐less cellular chassis for molecular and synthetic biology applications. Microb Cell Fact. 2010;9:38. doi: 10.1186/1475-2859-9-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods R.J., Barrick J.E., Cooper T.F., Shrestha U., Kauth M.R., Lenski R.E. Second‐order selection for evolvability in a large Escherichia coli population. Science. 2011;331:1433–1436. doi: 10.1126/science.1198914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng P., Sun J., van den Heuvel J., Zeng A.‐P. Discovery and investigation of a new, second triose phosphate isomerase in Klebsiella pneumoniae. J Biotechnol. 2006;125:462–473. doi: 10.1016/j.jbiotec.2006.03.034. [DOI] [PubMed] [Google Scholar]
- Zurawski D.V., Mitsuhata C., Mumy K.L., McCormick B.A., Maurelli A.T. OspF and OspC1 are Shigella flexneri type III secretion system effectors that are required for postinvasion aspects of virulence. Infect Immun. 2006;74:5964–5976. doi: 10.1128/IAI.00594-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Results of PCR reactions screening for ISes within the P1vir (A) and P1virΔIS (B) genomes, using the White Glove IS Detection Kit. In both panels, lanes contain primers specific for the following elements: lane 1: IS1, lane 2: IS2, lane 3: IS3 (ISEc17), lane 4: IS4, lane 5: IS5, lane 6: IS10, lane 7: IS30D, lane 8: IS150, lane 9: IS186, lane 10: IS600 (ISsd1), lane 11: IS609, lane 12: IS911, lane 13: ISEc1,3,5, lane 14: ISEc4, lane 15: RhsA,B,C, lane 16: RhsD,E. Lane 17 contained primers specific for P1 C1 gene. M: 1 kb DNA ladder (Fermentas).
PCR verification of the position and orientation of IS1P1vir using primers IS1A1 and P1D.
Dependence of phage titre on multiplicity of infection (moi).
Comparison of plaque morphology. Left: P1vir. Right: P1virΔIS.
Table S1. Details of attempts to engineer P1virΔIS using BRED. Note that multiple batches of arabinose‐induced electrocompetent MG1655/pKD46 cells were used during the optimization, which could cause inconsistencies.
Table S2. Primers used in this study.