

Summary
In the recent years, the number of drug‐ and multi‐drug‐resistant microbial strains has increased rapidly. Therefore, the need to identify innovative approaches for development of novel anti‐infectives and new therapeutic targets is of high priority in global health care. The detection of small RNAs (sRNAs) in bacteria has attracted considerable attention as an emerging class of new gene expression regulators. Several experimental technologies to predict sRNA have been established for the Gram‐negative model organism Escherichia coli. In many respects, sRNA screens in this model system have set a blueprint for the global and functional identification of sRNAs for Gram‐positive microbes, but the functional role of sRNAs in colonization and pathogenicity for Listeria monocytogenes, Staphylococcus aureus, Streptococcus pyogenes, Enterococcus faecalis and Clostridium difficile is almost completely unknown. Here, we report the current knowledge about the sRNAs of these socioeconomically relevant Gram‐positive pathogens, overview the state‐of‐the‐art high‐throughput sRNA screening methods and summarize bioinformatics approaches for genome‐wide sRNA identification and target prediction. Finally, we discuss the use of modified peptide nucleic acids (PNAs) as a novel tool to inactivate potential sRNA and their applications in rapid and specific detection of pathogenic bacteria.
Introduction
Small non‐coding RNAs and especially microRNAs (miRNAs) have been recently identified as key regulators of several cellular processes in multicellular eukaryotes (Garzon et al., 2009). Massive resources are now allocated to understand this extra layer of gene regulation. Involvement of miRNA expression in different types of tumours (e.g. breast, colon or brain cancer) has been reported (Garzon et al., 2009; Negrini et al., 2009). Consequently, blocking of miRNAs has become of therapeutic interest for tumour treatment (Dalmay, 2008; Negrini et al., 2009). In addition, miRNA profiles are now considered as a new tool for diagnostics. Although miRNA profiles contain much less information than an mRNA profile, the potential value may be higher because of the regulatory role of miRNAs (Dalmay, 2008).
In bacteria, small RNAs (sRNAs) have attracted considerable attention as an emerging class of new gene expression regulators. Apart from open reading frames, the genome codes for a number of RNAs with non‐coding functions such as rRNAs, tRNAs, and some small non‐coding RNAs, which may act as regulators of transcription or translation and influence mRNA stability (Waters and Storz, 2009). Although the majority of sRNAs might indeed be non‐coding, there are sRNAs, which have both a coding and a non‐coding function (Ji et al., 1995).
Small RNAs interact by pairing with other RNAs, forming parts of RNA–protein complexes, or adopting structures of other nucleic acids (Storz et al., 2004). A toolbox of experimental technologies such as microarray detection, shotgun cloning (RNomics), co‐purification with proteins and algorithms to predict sRNAs have been established for Gram‐negative bacteria such as Escherichia coli (Sharma and Vogel, 2009). Investigation of the role of sRNAs for other Gram‐negative pathogens such as Salmonella typhimurium and Pseudomonas aeruginosa has recently been started (Sharma and Vogel, 2009). sRNAs were detected within genetic islands of S. typhimurium, which showed host‐induced expression in macrophages and thus contributed to virulence (Padalon‐Brauch et al., 2008).
Recently, an extended number of 103 sRNAs was described for Listeria monocytogenes (Christiansen et al., 2006; Mandin et al., 2007; Nielsen et al., 2008; Toledo‐Arana et al., 2009). Only two sRNAs have been identified and studied in some detail in Streptococcus pyogenes (Kreikemeyer et al., 2001; Mangold et al., 2004) and five sRNAs for Streptococcus pneumoniae (Halfmann et al., 2007). A first approach identified 12 sRNAs in Staphylococcus aureus, seven of which are localized on pathogenicity islands. Some of the sRNAs show remarkable variations of expression levels among pathogenic S. aureus strains, which suggest their involvement in the regulation of virulence factors (Pichon and Felden, 2005). Presently, there is no information available for sRNAs from Clostridium difficile and Enterococcus faecalis.
To date, sRNAs have been found to be implicated in stress response, iron homeostasis, outer membrane protein biogenesis, sugar metabolism and quorum sensing, suggesting that they might also play an essential and central role in the pathogenicity of many bacteria. A global approach to identify sRNAs in L. monocytogenes has been recently published (Toledo‐Arana et al., 2009), but genome‐wide approaches to elucidate the functional role of sRNAs for other high‐risk Gram‐positive pathogens in pathogenicity are limited.
To address this deficiency, we recently established a European consortium (http://www.pathogenomics‐era.net/2ndJointCall/) to perform comparative global analysis of sRNAs for L. monocytogenes, S. aureus, S. pyogenes, E. faecalis and C. difficile. In this project, we will utilize bioinformatics, novel high‐throughput sRNA screening methods, whole‐genome transcriptomics and proteomics, coupled with existing robust molecular characterization methods to provide new information regarding production, regulation and pathogenic implications of sRNAs in these five major high‐risk Gram‐positive pathogens on a global scale.
These analyses will help us to design novel potential therapeutics based on sRNA‐complementary peptide nucleic acids (PNAs). Furthermore, the knowledge about sRNA expression will be used to develop a novel ultra‐sensitive diagnostic system, which has been conceived for detection of small target samples at extremely low concentrations in a very short time.
Listeria monocytogenes
Listeria monocytogenes is an opportunistic facultative intracellular bacterium which is ubiquitously distributed in nature. This food‐borne pathogen belongs to the group of bacteria with low G + C DNA content which also includes other species of genera such as Streptococcus, Staphylococcus, Enterococcus and Clostridium. The genus Listeria consists of seven different species, namely Listeria monocytogenes, Listeria ivanovii, Listeria innocua, Listeria welshimeri, Listeria seeligeri, Listeria grayi and Listeria marthii (Hain et al., 2007; Graves et al., 2009), of which L. ivanovii is predominantly a serious animal pathogen while L. monocytogenes can cause fatal infections in humans and animals. With regard to its transmission, a majority of listerial cases have been documented through contaminated food products. The major clinical symptoms exhibited by this pathogen in humans include meningitis, septicaemia, abortion, prenatal infection and gastroenteritis. In spite of an appropriate antibiotic therapy, approximately 20–30% of deaths have been reported in patients suffering from listeriosis (Hof et al., 2007).
A hallmark of this human pathogen is its ability to invade and survive inside vertebrate and invertebrate host cells, wherein the bacterium can freely multiply within the cytosol and can induce actin‐based movement and cell‐to‐cell spreading. Prior to infection, Internalin A and B induce the first step of the infection process by interacting with the eukaryotic host cell and promote intracellular uptake of the pathogen after binding with E‐Cadherin and c‐Met as interacting receptors in mammals. However, the main virulence genes, responsible for the intracellular life cycle of L. monocytogenes, are clustered in a ∼9 kb chromosomal region (Hain et al., 2007; Cossart and Toledo‐Arana, 2008).
Over the last decade, regulatory RNA elements have gained increasing importance in the physiology and pathogenesis of prokaryotes. They are divided into two major groups based on their mode of action: cis (untranslated region, UTR) and trans acting regulatory RNAs (small non‐coding RNAs). Riboswitches and RNA thermometers (Narberhaus et al., 2006) belong to the class of cis acting regulatory RNAs located at the 5′UTR of their genes. In general, riboswitches modulate their regulatory structure in response to metabolite binding, which are available in their own environment whereas, RNA thermometers are involved in sensing global signals, e.g. the intracellular temperature. Both cis regulatory RNA structures are essential for fine regulatory tuning so as to ensure an immediate physiological response of the bacterial cell to a varying habitat.
The first cis regulatory RNA was described for the master virulence regulator PrfA (Johansson et al., 2002). This 5′UTR acts as RNA thermometer which is turned off under low‐temperature conditions. In addition, the importance of 5′UTRs has also been reported for other virulence genes (Wong et al., 2004; Shen and Higgins, 2005; Stritzker et al., 2005). In general, the effects of UTRs on the expression of ActA, Hly and InlA were analysed by deleting the 5′UTR, while their ribosome binding sites were retained. Further computational analysis has revealed the presence of putative UTRs in surface proteins encoding operons associated with virulence in L. monocytogenes and suggests a potential post transcriptional function for these long RNA sequences in pathogenesis (Loh et al., 2006).
The role of trans regulatory RNAs such as small non‐coding RNAs has been addressed recently. The housekeeping sRNA 4.5S which binds to the signal recognition particle (SRP) was the first sRNA identified in L. monocytogenes (Barry et al., 1999). SRPs are highly conserved in most bacteria and are involved in translation and targeting of proteins for cellular secretion.
In L. monocytogenes, the RNA chaperone Hfq is involved in stress tolerance and virulence (Christiansen et al., 2004). In order to investigate sRNA–Hfq interaction, a co‐immunoprecipitation approach identified three Hfq‐binding sRNAs (lhrA, lhrB and lhrC; lhrC reveals five copies in the chromosome), which are growth phase‐dependently expressed and induced during intracellular multiplication within HepG2 cells. In a later study, new algorithms were used to predict and functionally map novel sRNA for L. monocytogenes. This allowed the identification of 12 additional sRNA (rliA–I, ssrS, ssrA and rnpB), and for three of them (rliB, rilE and rliI), potential mRNA targets were predicted in silico and experimentally verified (Mandin et al., 2007).
Listeria monocytogenes is well known for its robust physiology because of its capability to grow under refrigeration temperature, low pH and also at high osmolarity. However, relatively little is known about the role of sRNA for the adaptive physiological response in promoting survival and growth of the bacterium in such hostile environments. Besides, the involvement of the alternative sigma factor σB in the regulation of sRNA was elucidated not until recently when a 70‐nt‐long sRNA (sbrA) was identified which was reported to be under control of this stress tolerance factor (Nielsen et al., 2008).
In order to study global transcriptional profiling in response to the changing environmental niche of the pathogen, several different stress and in vitro/in vivo infection conditions were exploited using genome‐wide microarray approaches (Joseph et al., 2006; van der Veen et al., 2007; Hain et al., 2008; Raengpradub et al., 2008; Camejo et al., 2009). We have used transcriptome profiling to examine the expression profile of L. monocytogenes inside the vacuolar and the cytosolic environments of the host cell using whole‐genome microarray and mutant analysis. We found that ∼17% of the total genome was mobilized to enable adaptation to intracellular growth (Chatterjee et al., 2006), but the role of sRNA during host cell infection was not addressed in this study.
A first genome‐wide approach has been recently reported by Toledo‐Arana and colleagues (2009) using tiling arrays to detect novel sRNA. Thus, the transcriptional profile of L. monocytogenes was analysed under several different conditions including bacteria growing in BHI exponentially as well as to the stationary phase, under low oxygen and temperature (30°C), in the murine intestine and human blood and compared with the transcriptome of ΔprfA, ΔsigB and Δhfq isogenic mutants. Under these conditions a complete operon map with 5′ and 3′ end boundaries was determined as well as 103 small regulatory RNA were identified (Table 1). Among the regulatory RNAs, 29 novel sRNAs, 13 cis regulatory RNAs (5′‐ and 3′UTR, putative riboswitches) and 40 cis regulatory RNAs including known riboswitches were identified. Isogenic mutant analyses of rliB and rli38 indicated their contribution in virulence in mice. This initial study describes the global transcriptional landscape in L. monocytogenes under various growth conditions and provides insights into strategies of extracellular survival of the human pathogen. Nevertheless, details on the precise role of sRNAs in the pathogenesis of L. monocytogenes remain limited and additional studies on physiological function are needed in order to understand the sRNA regulatory function.
Table 1.
Current overview of published Gram‐positive sRNAs of the genera Staphylococcus, Streptococcus, Enterococcus, Clostridium and Listeria.
| Genus | Species | SIPHTa | Experimentally verified | ||
|---|---|---|---|---|---|
| Chromosome | Plasmid | Chromosome | Plasmid | ||
| Staphylococcus | aureus | 32–79 (12) | 0–5 (9) | 23b | 1c |
| epidermidis | 116–127 (2) | 0–4 (7) | |||
| haemolyticus | 74 (1) | – | |||
| saprophyticus | 38 (1) | 1 (2) | |||
| Streptococcus | agalactiae | 29–34 (3) | – | ||
| mutans | 18 (1) | – | |||
| pneumoniae | 28–66 (3) | – | 5d | ||
| pyogenes | 18–29 (12) | – | 3e | ||
| thermophilus | 31–36 (3) | 0 (2) | |||
| sanguinis | 34 (1) | – | |||
| suis | 23–24 (2) | – | |||
| Enterococcus Clostridium | faecalis | 14 (1) | 0–2 (3) | 2f | |
| acetobutylicum | 18 (1) | 2 (1) | 1g | ||
| beijerinckii | 31 (1) | – | |||
| botulinum | 54–68 (4) | 0 (2) | |||
| difficile | 2 (1) | 0 (1) | |||
| kluyveri | 46 (1) | 0 (1) | |||
| novyi | 26 (1) | – | |||
| perfringens | 14–18 (3) | 0–2 (3) | 1h | ||
| tetani | 45 (1) | 0 (1) | |||
| thermocellum | 6 (1) | – | |||
| Listeria | innocua | 115 (1) | 2 (1) | ||
| monocytogenes | 94–124 (2) | – | 27i | ||
| welshimeri | 100 (1) | – | |||
Barry et al. (1999); Christiansen et al. (2006); Mandin et al. (2007); Nielsen et al. (2008); Toledo‐Arana et al. (2009).
The SIPHT columns show the minimum and maximum number of annotated sRNAs of each species.
The quantity of analysed strains is depicted in brackets.
Staphylococcus aureus
Staphylococcus aureus is one main cause of nosocomial infections worldwide. Because of the high occurrence of multiple resistant strains in hospital settings, infections caused by this pathogen are often difficult to treat and new therapeutic strategies are urgently needed. The post genomic era of S. aureus started in 2001 with the publication of the genome sequences of two strains, N315 and Mu50 (Kuroda et al., 2001). In the S. aureus N315 genome, 110 non‐coding RNAs are annotated. Among them are 78 tRNAs and rRNAs and 32 putative sRNAs and riboswitches (Geissmann et al., 2009).
A large number of virulence factors are known to be involved in the pathogenesis of S. aureus whose expression is subject of temporal control and mainly affected by RNAIII. RNAIII was the first regulatory RNA shown to be involved in bacterial virulence and is part of the agr locus in S. aureus that acts as a quorum sensing system (Recsei et al., 1986; Novick et al., 1993; Ji et al., 1995) RNAIII is a 514 nt regulatory RNA and controls the switch between production of cell wall adhesins and that of extracellular proteins (Dunman et al., 2001; Ziebandt et al., 2004). The mechanism by which RNAIII regulates such a large number of genes has been unknown for a long time. In the meantime, a direct regulatory effect of RNAIII has been shown for hla expression (Morfeldt et al., 1995) and for spa expression (Huntzinger et al., 2005). However, RNAIII possibly affects the expression of most of the virulence genes in S. aureus indirectly via the pleiotropic regulator Rot (for repressor of toxins). RNAIII antagonizes Rot activity by blocking its synthesis, with RNAIII acting as an antisense RNA, pairing with complementary regions in the 5′ end of rot mRNA. Binding of RNAIII induces cleavage of the rot transcript (Geisinger et al., 2006; Boisset et al., 2007).
Pichon and Felden (2005) described the first global approach to identify sRNAs in S. aureus. They used a comparative genomic approach to characterize the RNome of S. aureus on the basis of the genome sequence of strain N315 and detected at least 12 sRNAs: five are encoded in the core genome and seven are localized on pathogenicity islands. Some of the sRNAs show remarkable variations of expression among pathogenic strains and it is postulated that they might be involved in the regulation of virulence factors as it is described for RNAIII.
Most recently, a more comprehensive search for sRNAs in S. aureus was performed by Geissmann and colleagues (2009). They identified 11 new sRNAs (RsaA to K). Transcriptional analyses using three reference strains of S. aureus (RN6390, Newman and COL) also showed different transcription profiles of these sRNAs between the strains. Most of the sRNAs were growth phase‐dependently expressed and transcribed in response to environmental changes such as oxidative stress, heat stress, osmotic stress and acidic pH. Transcription of three of them was mediated by the alternative sigma factor σB and one was regulated by the quorum sensing system agr.
Details on the regulatory function of sRNAs in physiology and pathogenesis of S. aureus are still limited. By using transcriptomic and proteomic analyses, Geissmann and colleagues (2009) have shown that RsaE which was expressed under various stress conditions is involved in the regulation of several genes involved in amino acid and peptide transport, cofactor synthesis, lipid and carbohydrate metabolism and the TCA cycle. RsaE binds to its target mRNAs via a specific sequence motif and inhibits the formation of the translational initiation complex. Direct interactions of RsaE with oppB, sucD and SA0873 mRNAs have been shown. The specific sequence motif seems to be highly conserved among sRNAs in S. aureus indicating a very similar mode of action (Geissmann et al., 2009).
These studies (Pichon and Felden, 2005; Geissmann et al., 2009), however, can only represent the blueprint for the prediction, detection and characterization of sRNAs in S. aureus. It can be postulated that the S. aureus genome likely codes for more regulatory sRNAs that affect gene expression than have been identified to date. By using tiling DNA arrays and new sequencing techniques, we will certainly get a more comprehensive picture of regulatory sRNAs in S. aureus expressed under certain conditions. Characterization of their regulatory role in global gene expression could shed more light on processes of S. aureus involved in adaptation to the host environment and pathogenicity and may provide new targets for therapeutic strategies to treat infections caused by this pathogen.
Streptococcus pyogenes
Streptococcus pyogenes (group A streptococci, GAS) is another important exclusively human bacterial pathogen. The annual global burden of GAS diseases, including 111 million cases of pyoderma, over 616 million cases of pharyngitis, and at least over 500 000 deaths due to severe invasive diseases, places this pathogen among the most important Gram‐positive bacterial species with major impact in global mortality and morbidity (Carapetis et al., 2005). GAS encodes a large number of virulence factors that are involved in adhesion, colonization, immune evasion and long‐term survival within the host (Courtney et al., 2002; Kreikemeyer et al., 2003; 2004). For successful adaptation and survival in various targeted host compartments, about 30 orphan transcriptional regulators (RR), together with 13 two‐component signal transduction systems (TCSs) (of which 11 are conserved across all GAS serotypes), sense and integrate the environmental information in GAS (Kreikemeyer et al., 2003; Beyer‐Sehlmeyer et al., 2005; Musser and DeLeo, 2005).
As in many other bacterial species, a novel level of potential virulence regulation has emerged from the discovery and activity of sRNAs. Two bioinformatics‐based approaches have identified candidate sRNAs in the S. pyogenes genome. Livny and colleagues developed SIPTH (sRNA identification protocol using high‐throughput technologies) to identify sRNAs in intergenic loci based on colocalization of intergenic conservation and the presence of Rho‐independent terminators (Livny et al., 2008). In order to integrate all available sRNA identification methods we have recently developed MOSES (modular sequence suit), a Java‐based framework, to integrate detection approaches for sRNAs. Using MOSES on the genome sequence of the S. pyogenes M49 NZ131 strain we identified four highly probable sRNA candidate genes which were verified by RT‐PCR (P. Raasch, U. Schmitz, N. Patenge, B. Kreikemeyer and O. Wolkenhauer, submitted).
Experimental approaches prior to the availability of tiling array technology allowed identification and partial characterization of three sRNAs (SLS, FasX and RivX) in S. pyogenes (Kreikemeyer et al., 2001; Mangold et al., 2004; Roberts and Scott, 2007).
A first GAS sRNA sagA was discovered during the identification and functional characterization of the major haemolysin of GAS, streptolysin S (SLS/SagA) (Li et al., 1999; Carr et al., 2001). Mangold and colleagues later reported that the mRNA transcribed from the sls gene most likely acts as sRNA and effectively regulates expression of emm (M protein), sic (streptococcal inhibitor of complement) and nga (NAD‐glycohydrolase) at the transcriptional level, and expression of SpeB (cystein protease) at the post‐transcriptional level (Mangold et al., 2004).
The second putative sRNA experimentally identified was fasX, a 300‐nucleotide‐long transcript, which is associated with the FasBCAX (fibronectin/fibrinogen binding/haemolytic activity/streptokinase regulator) system (Kreikemeyer et al., 2001).
Measurement of a luciferase–promotor fusion revealed a growth phase‐associated transcription of fasX with peak activities during late exponential phase. Mutation of fasX uncovered a reduced expression of the secreted virulence factors sls and streptokinase (ska), and simultaneously a prolonged expression of fibronectin‐ (fbp54) and fibrinogen‐binding (mrp) adhesins. Analysis of the role of fasX in S. pyogenes adherence to and internalization into HEp‐2 cells (a human laryngeal carcinoma cell line) revealed that this sRNA apparently promotes high adherence and internalization rates, leading to massive cytokine gene transcription and cytokine release, host cell apoptosis via a novel caspase 2 activation pathway and cytotoxicity (Klenk et al., 2005). It is evident that the Fas two‐component signal transduction system, with the fasX sRNA as its integral and main effector part, mediates virulence gene regulation and could be involved in GAS aggressiveness and local tissue destruction (Klenk et al., 2005).
Recently, Roberts and Scott showed that rivX encodes another sRNA and microarray analysis revealed that products of the rivRX locus exert positive control over transcription of members of the Mga regulon (Roberts and Scott, 2007).
The first genome‐wide and comprehensive approach to identify novel sRNAs in the M1T1 MGAS2221 S. pyogenes strain, using a combination of bioinformatics and intergenic region tiling arrays, was most recently performed by Sumby and colleagues (Perez et al., 2009). The tiling array approach identified 40 candidate sRNAs, of which only seven were also identified by the bioinformatics approach of Livny and co‐workers. This small number of cumulatively identified sRNAs stresses the necessity for multifaceted and integrated approaches. Most of the investigated sRNAs varied in their stability and inter‐ and/or intra‐serotype‐specific levels of abundance. Most interestingly, Sumby and colleagues could not confirm earlier results from Mangold and colleagues who postulated a regulatory function for sagA/Pel (Mangold et al., 2004).
In summary, three candidate sRNAs were identified experimentally, 29 candidate sRNAs were identified by Livny using a bioinformatic approach (Table 1), a further four appeared from the novel MOSES sRNA software and 40 (of which only seven matched candidates from the Livny paper) were found by the first tiling array approach performed by Sumby and co‐workers (Perez et al., 2009).
Enterococcus faecalis
Enterococcus faecalis is a human commensal and member of the lactic acid bacteria. It can be used as a starter in food industry and for some strains, probiotic effects have been claimed (Domann et al., 2007). However, E. faecalis is also used as an indicator of faecal contamination and represent one of the principal causes of nosocomial infections (Ogier and Serror, 2008). Despite the increasing number of infections due to E. faecalis, mechanisms of virulence remain poorly understood. These infections affect mainly young and immuno‐depressed subjects causing endocarditis, meningitis, pneumonias, peritonitis, visceral abscesses, urinary infections and septicaemias (Gilmore et al., 2002). This makes E. faecalis an ambiguous microorganism and, consequently, the use of enterococci in the food industry has been viewed more critically (Eaton and Gasson, 2001). Thus, efforts are urgently needed in order to get a better understanding of the molecular reasons why this normally harmless bacterium can transform into a dangerous pathogen. In this context, analysis of regulation of gene expression is of prime importance.
From the 3337 predicted protein‐encoding open reading frames in E. faecalis V583, 214 have or may have regulatory functions (Paulsen et al., 2003) but only few transcriptional regulators have been studied so far. Some of them have been shown to be correlated with stress response and/or virulence such as Fsr, EtaRS, CylR, HypR, PerR or Ers (Qin et al., 2001; Gilmore et al., 2002; Teng et al., 2002; Verneuil et al., 2004; Verneuil et al., 2005; Riboulet‐Bisson et al., 2008). However, in recent years it has been discovered that not only proteins but also small non‐coding RNAs are key players in the control of bacterial gene expression. At present, only one mechanism based on two sRNAs (RNA I and RNA II), required for the stable inheritance of the plasmid pAD1, has been reported for E. faecalis (Weaver, 2007). The two sRNAs are convergently transcribed towards a bidirectional intrinsic terminator. RNA I encodes the Fst toxin the translation of which is inhibited by the interaction with RNA II. RNA II acts then as the antitoxin and is less stable than RNA I. This suggests that in absence of continued transcription from the resident plasmid, RNA I is removed from the complex allowing the translation of fst that kills the cell. One recent study shows that the stability of the toxin‐encoding RNA (RNA I) is due to an intramolecular helix sequestering the 5′ end of the RNA (Shokeen et al., 2009).
Recently, an in silico study led to the prediction and annotation of 17 further putative sRNA‐encoding genes in E. faecalis V583 (Livny et al., 2008), 14 on the chromosome and three on plasmids (Table 1). However, in comparison with the number of sRNA identified in the same study in model bacteria such as E. coli and Bacillus subtilis, the number of potential candidates in E. faecalis is roughly 10‐fold lower.
Clostridium difficile
Clostridium difficile is currently one of the increasingly important nosocomial pathogens, but also rising are the numbers of human community‐associated infections and animal infections (Rupnik et al., 2009). The disease spectrum ranges from mild diarrhoea to severe forms of colitis, pseudomembranous colitis and bowel perforation. Most of the symptoms can be explained by effects of two large toxins produced by the bacterium, toxin A (TcdA) and toxin B (TcdB). While both toxins, TcdA and TcdB, are well characterized from molecular and biochemical perspective, our understanding on the regulation mechanisms involved in their expression is only partially elucidated (Dupuy et al., 2008). Even less is known about regulation of additional factors influencing the virulence such as binary toxin CDT, adhesion molecules, sporulation properties and antibiotic resistance.
The regulatory role of small non‐coding RNAs was so far not studied or reported in C. difficile. Only two candidate loci (Table 1) were found in a large computational study by Livny and colleagues (2008). In the same study candidate sRNAs were detected in some other pathogenic and non‐pathogenic Clostridia, e.g. Clostridium tetani, Clostridium botulinum, Clostridium perfringens, Clostridium novyi (Table 1). None of them is a closer relative to C. difficile; however, C. novyi has a toxin TcnA that is homologous to C. difficile large toxins.
So far, C. perfringens is the only pathogenic clostridial species where the role of sRNA was studied in detail (Shimizu et al., 2002). VR‐RNA is a region expressed under the control of the VirR/VirS system. It encodes an RNA molecule involved in regulation of some C. perfringens toxins and other virulence factors. This strongly suggests that regulatory mechanism involving sRNA could also be present in other toxigenic Clostridia and justifies an extensive search for and characterization of novel sRNAs in C. difficile.
Facultative requirement of Hfq for sRNA–mRNA interaction in Gram‐positive bacteria
The bacterial RNA chaperone Hfq facilitates pairing interactions between sRNAs and their mRNA targets in several bacteria. However, approximately only half of the sequenced bacterial genomes encode a Hfq homologue (Sun et al., 2002). Hfq modulates the stability, translation and polyadenylation of many mRNAs in E. coli (Folichon et al., 2003). In L. monocytogenes, Hfq is required for several sRNAs (LhrA–C) (Christiansen et al., 2006), whereas other identified sRNAs in the same bacterium are able to interact with their mRNA without the contribution of Hfq (Mandin et al., 2007; Toledo‐Arana et al., 2009). Similar examples from Vibrio cholerae (Qrr1–4) illustrate how heterogeneous the role of Hfq can be in sRNA–mRNA‐mediated regulations within the same bacterium (Lenz et al., 2004).
Although present in S. aureus, Hfq seems not to be required for sRNA–mRNA interactions (Bohn et al., 2007). For the sporogenic bacterium C. difficile less is known about the involvement of the clostridial Hfq homologue in the regulation of sRNAs.
Surprisingly, a gene encoding a homologue of the protein Hfq is not present in the in S. pyogenes and E. faecalis genome sequence (Sun et al., 2002). From this finding arose the question of how sRNAs are stabilized and annealed to their targets in this species? It is unlikely that non‐coding RNAs are ‘naked’ in a cellular environment and are more likely to be associated with RNA‐binding proteins throughout its lifetime (Vogel, 2009). Thus, an important step for the molecular characterization of sRNAs and identification of their targets will also need the discovery of these RNA‐binding proteins in S. pyogenes and E. faecalis.
Whole‐genome sRNA identification methods
Small RNAs have been hard to detect both computationally and experimentally. The first sRNAs were discovered four decades ago serendipitously by radiolabelling of total RNA and subsequent isolation via gel separation (Hindley, 1967; Griffin, 1971; Ikemura and Dahlberg, 1973). The sRNA discovery in bacteria over the last decade has shifted from the direct labelling and sequencing to genome‐wide technologies including, for example, the genomic SELEX approach (Lorenz et al., 2006), collecting of sRNA genes by shotgun cloning (RNomics) (Vogel et al., 2003), detection of sRNAs by DNA microarrays (Selinger et al., 2000) and next‐generation sequencing (NGS) technologies to provide new means to sRNA discovery (Meyers et al., 2006).
DNA microarrays are a powerful tool for screening sRNA genes on a genome‐wide scale. Employing the so‐called tiling array approach for sRNA gene discovery, probes specific to the region of interest have to be designed. These regions can span the whole genome or just the intergenic regions, where most of the currently known bacterial sRNAs are encoded. Critical design parameters are probe length and their overlap. As the probe sequence is defined by the sequence of the genome, options for probe optimization are limited. By varying the probe lengths, minor adjustments of melting temperatures can be obtained. Also incorporation of modified nucleotides, such as locked nucleic acids (LNAs), might result in more uniform melting temperatures. In any case, sensitive labelling and optimized hybridization conditions are essential. Bacterial sRNAs are highly heterogeneous in length (ranging from approximately 35 to several hundred nucleotides) and do not contain a common sequence such as the polyA tail of eukaryotic mRNAs. Therefore, careful size selection ensures that the complexity of the input RNA sample is reduced and excludes RNA species (e.g. mRNA, rRNA) for downstream microarray analysis. As amplification methods tend to be biased, direct labelling is preferred, even though lower signal intensities are obtained. Due to the fact that only a small number of sRNA genes (∼20–150) are expected to be discovered from bacteria, this results in a relatively low number of signals on the tiling microarrays. Therefore, it is vitally important to provide a range of controls upon design of the microarrays to discriminate between signals arising from other RNA species (e.g. mRNA, rRNA, tRNA) or truncated RNA fragments and existing sRNA genes.
Such tiling experiments can also be performed in more than one round, e.g. by running a first ‘rough’ mapping approach using long probes (around 50‐mer) and then taking promising regions for a second ‘fine‐mapping’ with shorter probes (around 25‐mer) to narrow down the 5′ and 3′ ends of the sRNA genes. Although such a second round of screening increases confidence, results always have to be verified by independent techniques, such as real‐time PCR, NGS, RACE‐PCR, parallelized gene knock‐outs or microtitre plate‐based phenotype screens (Bochner, 2003).
Microarrays for such tiling experiments are usually not available off the shelf, but need to be individually designed. Systems such as the flexible Febit® microarray technology (Baum et al., 2003) however provide cost‐efficient and easy‐to‐use solutions for sRNA genes discovery employing the aforementioned tiling approach.
Next‐generation sequencing technology provides new means in sRNA discovery and gives access to a screenshot of the global transcriptional landscape without complicated cloning (Sittka et al., 2008; Simon et al., 2009). Methods like sRNA sequencing allow for interrogation of the entire small, non‐coding RNA (sRNA) repertoire. It includes a novel treatment that depletes total RNA fractions of highly abundant tRNAs and small subunit rRNA, thereby enriching the starting pool for sRNA transcripts with novel functionality (Liu et al., 2009). An additional advantage of the NGS technology is the immediate information about 5′ and 3′ ends of the sRNAs. However, data analysis is still in its infancy and might make data interpretation difficult and the library preparation necessary for NGS might introduce bias. Thus, the combination of both array‐based and NGS approaches appears to be the most promising experimental procedure for genome‐wide identification of novel sRNAs in bacteria.
Computational approaches on genome‐wide sRNA prediction and target identification
Despite the new era of NGS technologies, the task to search for and predict candidate sRNA genes on genome level remains a challenge and an open problem. Over the last years tremendous progress has been made in this field and sRNA gene prediction has been implemented based on genome scanners or on classifiers. Sequence‐based classifiers use input information such as sequence, motif or alignment to assign a homology‐based label to the output. Genome scanning uses combinations of assumptions and a sliding window to analyse entire genome sequences.
These genome scanning and ab initio methods to predict new sRNA candidates are based on the assumptions that structured RNAs have a lower free energy than random sequences (Le et al., 1988). Rivas and Eddy (2000) demonstrated that minimum free energy (MFE) difference is not significant enough to be used as a general sRNA discriminator. Taken the fact that MFE calculation is based on base stacking, not only the nucleotide frequency but the di‐nucleotide frequency is of interest (Bonnet et al., 2004; Clote, 2005). A thermodynamic calculation to predict the MFE is highly affected by an additional or missing nucleotide related to the window size applied for scanning the genome. Combining all the information obtained from all individual windows decreases the problem (Bernhart et al., 2006).
A common way to analyse nucleotide sequences is in terms of its nucleotide composition. This ranges from mononucleotide, di‐nucleotide to n‐nucleotide analyses and leads to their frequencies. Applying compositional statistics to predict new candidates was driven by reports that sRNAs have an average of 50% GC content (Rivas and Eddy, 2000). This has been successfully tested when searching GC‐rich islands in some AT‐rich organisms (Klein et al., 2002) and applying di‐nucleotide statistics in other organisms (Schattner, 2002). Also machine learning techniques have been performed to classify a sequence either as a coding or non‐coding RNA sequence (Liu et al., 2006).
Comparative methods use genome sequences of two or more related species performing alignments in order to find maximum regions of similarity between them (Rivas and Eddy, 2001; di Bernardo et al., 2003; Coventry et al., 2004; Rivas, 2005; Washietl et al., 2005). Once the regions of similarity are identified, either thermodynamic information or covariate analysis can provide more evidence on sRNA assignment (Washietl et al., 2005; Uzilov et al., 2006).
Since the first experimentally identified sRNA in 1981 (Stougaard et al., 1981; Tomizawa et al., 1981), sRNA gene‐finders based on a set of known sequences have been applied to identify genes belonging to the same gene family. Rfam is a collection of multiple sequence alignments, consensus secondary structures and covariance models representing predicted sRNA families. Starting with 25 sRNA gene families in 2003, the Rfam 9.1 release covers 1372 families in January 2009 (Griffiths‐Jones et al., 2003; Gardner et al., 2009).
SIPHT is a high‐throughput computational tool focusing on genome‐wide bacterial sRNA prediction and annotation (Livny et al., 2008). Candidate sRNA‐encoding loci are identified based on the presence of putative Rho‐independent terminators downstream of conserved intergenic sequences and each locus is annotated for several features, including conservation in other species, association with one of several transcription factor binding sites and homology to any of over 300 previously identified sRNAs and cis regulatory RNA elements. The procedure is sufficiently automated to be applied to all available bacterial genomes; however, the requirement for a terminator motif introduces a bias against the significant fraction of RNA elements that are not followed by a detectable terminator.
Phylogenetic analysis is one of the most informative computational methods for the annotation of genes and identification of evolutionary modules of functionally related genes and sequences (Pellegrini et al., 1999). Nucleic acid phylogenetic profiling is successfully applied to protein annotation and has recently been applied for high‐throughput sRNA identification and their functional characterization to S. aureus (Marchais et al., 2009).
Given the scores of sRNAs of unknown function, the identification of their cellular targets has become essential and requires both experimental technologies and computational approaches (Vogel and Wagner, 2007). Mandin and colleagues described such a tool that has been applied successfully in the genome of L. monocytogenes, but it is not available as a webserver (Mandin et al., 2007). TargetRNA is a web tool which uses a dynamic programming algorithm to search each annotated transcript to generate base‐pair‐binding potential of the input sRNA sequence and rank list of candidate genes. Many existing target prediction programs except IntaRNA neglect the accessibility of target sites and intra‐molecular base pairs using free energy of the hybridized duplex to predict the potential target site (Busch et al., 2008).
There is no general toolset that provides a comprehensive solution to the prediction of sRNA. A good practice when starting with computational tools is to carefully choose a set of applicable methods with different approaches and compare the results. An example for such an integrative prediction framework and modular sequence suite is MOSES (P. Raasch, U. Schmitz, N. Patenge, B. Kreikemeyer and O. Wolkenhauer, submitted). Finally, we reiterate that while all of the outlined tools predict sRNAs, the planning of experiments and integration of predictions and experimental data is needed for a reliable validation of in silico sRNA and target prediction.
In order to streamline and support the iterative processes, the consortium uses a central data management solution for data integration provided by Genedata. The infrastructure platform, based on Genedata Phylosopher® for sharing and interpreting the experimental data, covers the required comparative genomics and cross‐technology approach. Furthermore, the consortium reconstructs genome‐wide regulatory networks and pathogenesis to identify and validate targets for diagnostics and novel anti‐infective therapies within the Genedata platform.
Antisense reagents as an alternative treatment of microbial infections
The growing knowledge on sRNAs in bacteria, their regulatory role and function and their possible involvement in pathogen infection makes this class of molecules extremely interesting as new targets for antisense therapy and drug development.
Antisense therapy is a form of treatment where genes which are known to be causative for a particular disease are inactivated (‘turned off’) by a small synthetic oligonucleotide analogue (RNA or DNA, 14–25 nt in length). This analogue is designed to bind either to the DNA of the gene and inhibit transcription or to the respective mRNA produced by this gene, thereby inhibiting translation. For eukaryotes, the strand might also be targeted to bind a splicing site on pre‐mRNA (Morcos, 2007). Recently, miRNAs with their regulatory role have gained interest as targets, too (Fabani and Gait, 2008). Here, in most cases the oligonucleotide is designed to inhibit micro‐RNA function essentially by a steric block, RNase H‐ and RISC‐independent antisense mechanism through complementary binding to the micro‐RNA sequence.
To date, antisense drugs are studied to treat diseases such as diabetes, different types of cancer, amyothrophic lateral sclerosis, Duchenne muscular dystrophy as well as asthma and arthritis.
Apart from finding suitable and promising targets, the design of such drugs themselves represents another challenge. Issues like in vivo stability and efficient cell delivery have high impact on the dose–response relationship and need to be addressed. In order to avoid enzymatic in vivo degradation, modified oligonucleotides or completely synthetic analogues are used. For the antiviral Fomivirsen (marketed as Vitravene) (Roush, 1997; Mulamba et al., 1998), for example, which is the first antisense reagent approved by the FDA (US Food and Drug Administration), a 21‐mer oligonucleotide with phosphorothioate linkages is used. Phosphorothioates are also employed for a few other promising antisense reagents, some of which already entered preclinical trials, such as AP12009 (Hau et al., 2007). Nevertheless, this class of compounds exhibits several disadvantages: they have a comparable low binding affinity to complementary nucleic acids and show non‐specific binding to proteins (Brown et al., 1994; Guvakova et al., 1995), causing toxic side‐effects that limit many applications. The toxicity is reduced but not absent in second generation antisense agents with mixed backbone oligonucleotides.
Meeting the challenge to find modifications that provide efficient and specific antisense activity in vivo without being toxic, a third generation of antisense agents have emerged (Fig. 1). Analogues such as LNAs, phosphorodiamidate morpholino oligomers (PMOs) and PNAs are altogether no substrates for enzymatic degradation and hybridize with exceptional affinity and target specificity, thereby forming duplexes which are even more stable than the respective RNA:RNA duplexes themselves. A short general overview on PNAs, LNAs and morpholinos was published recently (Karkare and Bhatnagar, 2006).
Figure 1.
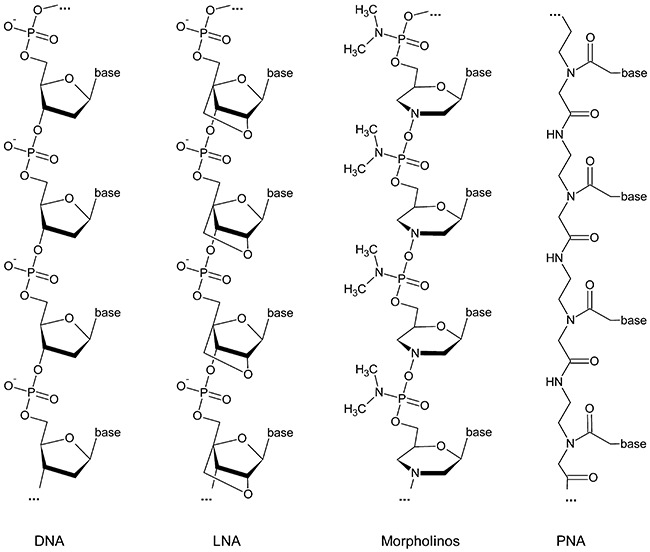
Overview of chemical structures of DNA, LNA (locked nucleic acid), PMO (phosphorodiamidate morpholino oligomer) and PNA (peptide nucleic acid).
As shown in Fig. 1, LNAs are RNA nucleotide analogues in which the ribose ring is constrained by a methylene linkage between the 2′‐oxygen and the 4′‐carbon, resulting in the locked C3′‐endo conformation. The introduction of LNA residues in oligonucleotides stabilizes the LNA:RNA duplex by either pre‐organization or increased base stacking (increasing Tm by 1–8°C against DNA and 2–10°C against RNA by one LNA substitution in an oligonucleotide). This results in increased RNA target accessibility and higher selectivity. In general, for efficient gene silencing in vitro and in vivo, fully modified or chimeric LNA oligonucleotides (LNA mixmers) have been applied.
Locked nucleic acids and LNA mixmers can be transfected using standard transfection reagents based on ionic interactions of the negatively charged backbone. However, due to the very strong base pairing in LNA:LNA duplexes it is important to design LNAs without extensive self‐complementary sequences or to apply chimeric LNAs (LNA mixmers), thereby diminishing biological stability to some degree. Furthermore, unless very short probes are required, stretches of more than four LNAs should be avoided.
Contrary to LNAs, PMOs and PNAs have a neutral backbone and thereby different physiochemical properties. Here, high specificity and increased duplex stability with RNAs and DNAs are due to the missing interstrand repulsion between the neutral PMOs (or PNAs) and the negatively charged RNA or DNA target. Thus, hybridization is nearly independent of salt concentration because no counter‐ions are needed to stabilize the duplex.
Phosphorodiamidate morpholino oligomers represent the closest relatives to the previously mentioned first‐generation phosphorothioates. But instead of the negatively charged sulfur, they comprise a neutral dimethylamino‐group bound to the phosphor. In addition to their excellent base stacking, standard PMOs are highly soluble in water, but their thermodynamic properties are not well defined, yet.
For PNAs it is the other way round, thermodynamics are well defined, but water solubility is limited, at least for longer purine‐rich oligomers which tend to aggregate. However, water solubility can be adjusted by introduction of one or more lysines which can be easily employed, as PNA synthesis strictly follows Fmoc‐peptide synthesis. PNA is a fully synthetic DNA analogue, which consists of the four nucleobases attached to a neutral peptide‐like amide backbone (Fig. 1). Despite the totally different backbone, PNA hybridizes with very high specificity to RNA and DNA via Watson‐Crick as well as Hoogsteen base pairing. In vitro studies indicate that PNAs could inhibit both transcription and translation of genes to which it has been targeted (Good and Nielsen, 1998; Nekhotiaeva et al., 2004). Due to higher duplex stability, probes can be much shorter in length than DNA probes. Very high chemical as well as biological stability make them excellent probes for antisense drug development and diagnostics.
In summary, LNAs, PMOs and PNAs have great potential as antisense drugs, each of them having its own advantages and draw‐backs. Even though there are some recent reports comparing these analogues in experiments (Gruegelsiepe et al., 2006; Fabani and Gait, 2008), at the time being, the choice of analogue still depends on the experimental conditions and detailed applications.
For all analogues, cell‐wall permeability is very limited, but cellular uptake can be improved by shortening the length of the antisense oligomer and/or attaching cell‐penetrating peptides (CPPs) (Tan et al., 2005; Gruegelsiepe et al., 2006; Kurupati et al., 2007) or other modifications which increase uptake (Koppelhus and Nielsen, 2003; Koppelhus et al., 2008). The CPP approach is currently a major avenue in engineering delivery systems that are hoped to mediate the non‐invasive import of cargos into cells. The large number of cargo molecules that have been efficiently delivered by CPPs ranges from small molecules to proteins, oligonucleotide analogues and even liposomes and particles.
To date, the most common reported CPP for bacteria is (KFF)3K. It is mainly used in combination with PNAs or PMOs. These compounds show efficient inhibition of gene expression in a sequence specific and dose‐dependent manner at low micromolar concentrations in different bacteria (Nekhotiaeva et al., 2004; Tan et al., 2005; Gruegelsiepe et al., 2006; Kurupati et al., 2007; Goh et al., 2009).
The search of efficient CPPs, which may improve cell delivery even further, is still an emerging field of research and has high potential for optimization. As demonstrated recently by Mellbye and colleagues variations in amino acid compositions of CPPs and linker molecules show clear effects on the efficiency of cell delivery (Mellbye et al., 2009). Broader systematic studies like this are still missing, but may open the road to a toolbox of different CPPs with optimized and well‐characterized efficiencies to transfer drugs specifically into different cell types.
Due to similar synthesis methods, PNA–peptide conjugates can easily be produced in microwell plates in a parallel manner (Brandt et al., 2003), thereby allowing for a fast optimization of the antisense as well as the cell‐delivery sequences for antisense drug design. A proof of principle that these compounds can be used as therapeutic drugs against bacterial infection was demonstrated by Tan and colleagues (2005) in a mouse model. In combination with promising targets such as sRNAs, which are suggested to play a major regulatory role in bacterial pathogenesis, the development of highly efficient antisense drugs becomes feasible as an excellent alternative to classical antibiotic treatment.
sRNAs for microbial diagnosis
Newly discovered sRNA sequences that were verified to be involved in virulence mechanisms of the different Gram‐positive bacteria may not only be used as potential antisense drugs but also exploited as targets for diagnostic systems.
In this respect sRNAs represent a useful alternative to mRNA or DNA oligonucleotides, because of his putative regulatory function. The potential of this class of compounds as diagnostic markers is currently a very active field of research (Hartmann & Consorten).
Several different formats for rapid diagnostic tests based on nucleic acid interactions have been described in the literature. Many of them are PCR‐based with the Genexpert system produced by Cepheid (Sunnyvale, CA) being the most advanced as it integrates sample preparation, amplification and detection (Chandler et al., 2001). These methods require experienced personnel and need 30–60 min for an assay to be completed. Therefore they are not suitable for point‐of‐care (POC) testing, the ultimate goal in the development of rapid diagnostic screening tests. An alternative and more cost‐effective approach to nucleic acid testing is the use of a lateral‐flow platform. Lateral‐flow assays (LFAs) were originally developed as immunoassays and combine chromatographic purification with immunodetection at high speed (Fig. 2A). They are very simple in handling and represent the only true POC test up to now (Seal et al., 2006).
Figure 2.
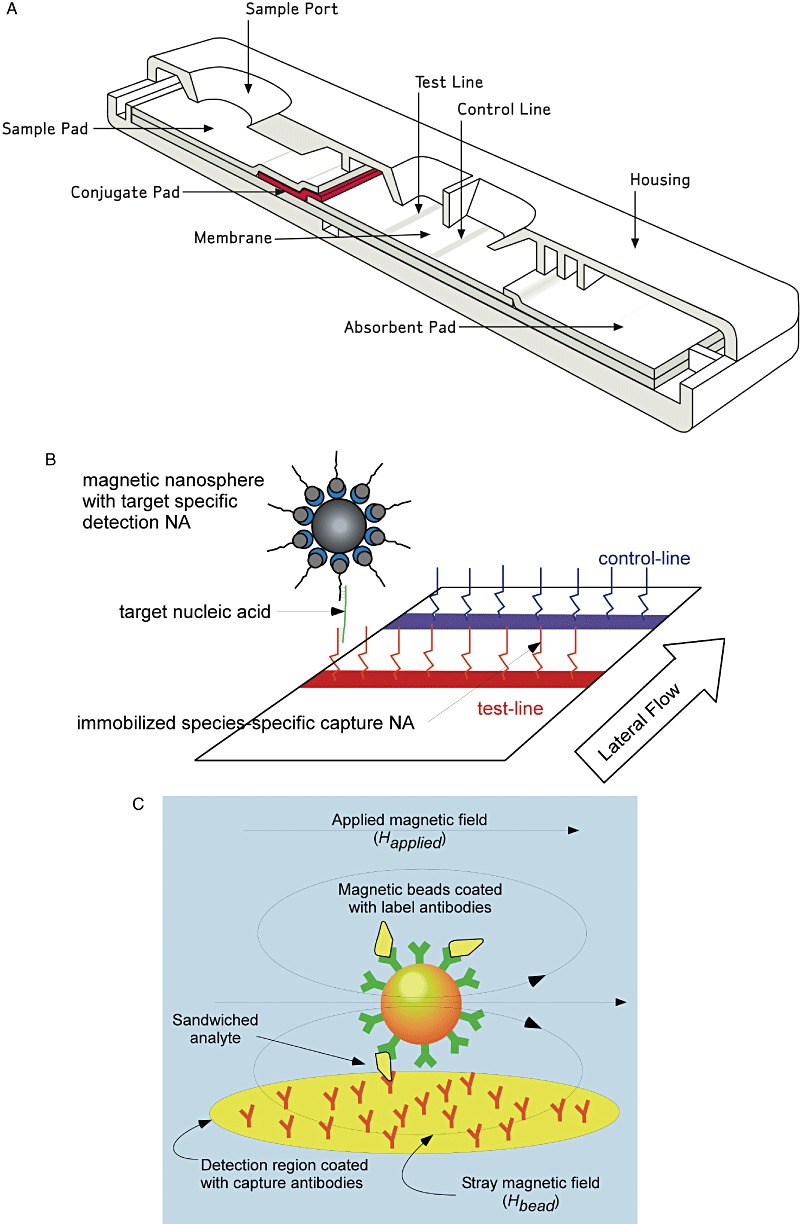
A. Schematic outline of a standard lateral‐flow assay device (Carney et al., 2006). B. Lateral‐flow assay of sRNA captured by PNAs using superparamagnetic magnet nanoparticles. C. Magnetoresistive detection of captured magnet nanoparticles (Tondra, 2007).
Nucleic acid lateral flow uses nucleic acid hybridization to capture and detect the nucleic acid analyte in a manner akin to lateral‐flow immunoassays. The most favourable approach is to immobilize oligonucleotide probes directly onto the nitrocellulose membrane used for the chromatographic step of the LFA. This may be done by covalent chemical bonding or by passive adsorption of an albumin–oligonucleotide conjugate to the membrane.
In principle any detection chemistry used in traditional LFA systems may also be applied for nucleic acid detection. Reverse hybridization enzymatic strip assays are the most advanced methods for the detection of nucleic acid interactions. In these tests, enzyme‐labelled probes are hybridized to complementary nucleic acid target species on the surface of the nitrocellulose membrane. The result is a hapten–antibody–enzyme complex such as for example biotin–streptavidine–alkaline phosphatase. But complex wash and substrate incubation steps are necessary to develop a readable colorimetric signal. This vastly limits the speed and simplicity of the assay and makes the POC use of such a system very difficult to achieve.
As an alternative, bead technologies mainly in the form of nanoparticles were successfully applied to nucleic acid LFAs. In this context, gold nanoparticles are the beads of choice, mainly because of their small size, good visibility and robustness in manufacturing (Carney et al., 2006). They can be conjugated with oligonucleotides and labelled with small binding moieties such as biotin. Gold NA‐LFA systems generally use 30–80 nm particles conjugated to an anti‐biotin antibody. This conjugate is complexed with a biotin‐oligonucleotide detection probe and forms a visible red line on the membrane strip when captured by a complementary NA sequence. Typical detection limits of such assay formats are about 2.5 mg DNA ml−1 (Mao et al., 2009).
A number of methods for improving the sensitivity of nanoparticle NA‐LFA have been investigated. Detection probes may be labelled with multiple hapten moieties to form large signal‐enhancing complexes or make use of DNA dendrimeres, which may result in a detection limit up to 200‐fold lower compared with standard assay conditions (Dineva et al., 2005). Other approaches use fluorescence labels to achieve a better detection. For example, a dual fluorescein‐biotin‐labelled oligonucleotide probe has been used to detect single‐stranded nucleic acids. However the utility of common fluorophores is limited by high background and the need for a complex reader. These problems may be overcome by the application of quantum dots (Lambert and Fischer, 2005). They consist of nanometre‐sized semiconductor nanocrystals and have superior fluorescence properties that enable them to emit about 1000‐fold brighter light than conventional dyes. They have narrow, symmetrical emission profiles, but have to be excited with UV light. Especially the development of water‐soluble quantum dots has enabled their use in LFAs. However, the price of these fluorescent nanocrystals is quite high, so that no commercial application has been developed so far.
In summary, specificity, sensitivity and cost‐effectiveness are key issues in the development of new nucleic acid‐based diagnostic systems.
To overcome these problems, we use PNA as capture molecules to increase the specificity of the interaction. The peptide backbone of the nucleic acid derivatives makes them much more stable compared with RNA or DNA (Fig. 1). Therefore they are much easier to handle, a great advantage for the development of an appropriate assay as well as for the shelf half life of the reagents in such a diagnostic kit. Equally important, the loss of the negative charge in the phosphate diester of the standard nucleic acid in PNAs led to a higher binding energy, mainly because electrostatic repulsion between the two strands does not exist in PNA/NA duplexes. This effect largely increases the specificity of the interaction. In fact, the melting temperature of a PNA/DNA duplex was measured to be about 15°C higher compared with an isosequential DNA/DNA duplex (Sen and Nielsen, 2007).
To increase the sensitivity in the detection of the sRNAs captured by the complementary PNA, we use superparamagnetic nanoparticles combined with a suitable magnetoelectronic device (Fig. 2B). This set‐up offers a number of advantages for LFA‐based POC diagnostic measurements. A comprehensive review on magnetic labelling, detection and system integration has appeared recently (Tamanaha et al., 2008). A huge benefit of magnetic labelling and detection over fluorescence labelling and optical detection is that there is usually very little magnetic background present in most samples, thereby making single magnetic particle detection both possible and technically feasible (Perez et al., 2002). Nanoscale magnetic beads have been extensively developed over the last decade and are commercially available from a number of sources and in different varieties of sizes and surface chemistry. A prime requirement for use as diagnostic labels is that beads must be paramagnetic or non‐remanent to avoid clustering caused by residual magnetic moment in the absence of magnetic fields. Magnetic nanoparticles used in LFA‐based diagnostic devices are preferred to be single domain and small enough to be superparamagnetic, i.e. the thermal fluctuations at room temperature overcome magnetic anisotropy forces spontaneously randomizing magnetic directions. The beads consist of iron oxide cores coated by organic polymers.
Magnetic detection can be established with a number of different sensor types. The simplest devices are based on Maxwell‐Wien bridges or frequency dependent magnetometers made of LC circuits. With these devices, to our best knowledge, single magnetic particle detection has not yet been accomplished. This goal has however been reached with devices based on superconducting quantum interference devices (SQUID), magnetoresistance or Hall sensors. For an excellent review on magnetic sensing see for example Tamanaha and colleagues (2008). SQUID‐based sensors, although being the most sensitive ones, are not apt for POC applications because they need extensive cooling down to at least N2 temperatures (77 K).
A schematic illustration of the concept of magnetic detection in traditional lateral‐flow immunoassays is shown in Fig. 2C. Magnetic beads coated with antibodies bind the analyte and were captured by an appropriate antibody immobilized on a magnetoresistive detector. This way magnetic beads loaded with analyte accumulate on the sensor area. Stray magnetic fields from the beads change the electric resistance of the sensor. This change is used to determine the amount of analyte present in the sample. This detection principle is adapted to PNA as capture molecule and sRNA analyte without great difficulties.
Conclusion
The current knowledge about sRNAs of S. aureus, S. pyogenes, E. faecalis, C. difficile and L. monocytogenes represents only the blueprint for the prediction, detection and characterization of this group of RNAs and will be extended by the inclusion of additional strains of these five Gram‐positive genomes. Currently (October 2009) genome sequences of 39 S. aureus, 14 S. pyogenes, 23 E. faecalis, 12 C. difficile and 24 L. monocytogenes strains have become available in the NCBI databases, and more serotype‐ and strain‐specific genome sequences will be analysed in future studies.
In silico prediction of sRNA showed that the genus of Listeria bears a high number of putative sRNA (Livny et al., 2008) compared with other genera such as Streptococcus, Staphylococcus, Enterococcus and Clostridium (Table 1), which might reflect their potential ubiquitous adaption ability in nature and mammals. Comparative analyses of the recently reported 103 regulatory RNAs of L. monocytogenes among the genomes of S. aureus, S. pyogenes, E. faecalis and C. difficile revealed that riboswitches seem to be more conserved among these Gram‐positive pathogens than sRNA (Table 2). Thus it could be speculated that a common ancient mechanism of cis acting RNA regulation might exist in Gram‐positive bacteria, while trans acting RNAs might be involved in speciation of bacteria in response to their host adaptation processes.
Table 2.
Comparative genome analysis of 21 of 100 published listerial sRNAs having orthologues (Toledo‐Arana et al., 2009) in Staphylococcus aureus, Streptococcus pyogenes, Enterococcus faecalis and Clostridium difficile.
| Regulatory RNA | Length (bp) | Position in Listeria monocytogenes EGD‐e (bp) | Clostridium difficile 630 | Enterococcus faecalis V583 | Staphylococcus aureus COL | Streptococcus pyogenes M1 GAS |
|---|---|---|---|---|---|---|
| FMN | 123 | 2020609–2020487 | + (2) | + | + (2) | + |
| Glycine | 92 | 1372840–1372931 | + | + | + | |
| L13 | 53 | 2685233–2685181 | + | + | ||
| L19 | 39 | 1862308–1862270 | + (4) | + | + (9) | + |
| L21 | 79 | 1577146–1577068 | + | |||
| M‐box | 165 | 2766104–2765940 | + (5) | |||
| PreQ1 | 48 | 907926–907973 | + | |||
| SAM2 | 120 | 309385–309266 | + (5) | |||
| SAM3 | 101 | 637926–637826 | + (4) | |||
| SAM5 | 109 | 1716651–1716543 | + | + | ||
| SAM6 | 107 | 1739597–1739491 | + (5) | |||
| SAM7 | 119 | 2491176–2491058 | + | |||
| SRP | 333 | 2784604–2784272 | + | + | ||
| glmS | 195 | 756458–756652 | + | |||
| rli31 | 115 | 597812–597926 | + | |||
| rli36 | 84 | 859527–859444 | + | |||
| rli45 | 78 | 2154775–2154852 | + | + | ||
| rli50 | 177 | 2783274–2783098 | + | |||
| rliD | 328 | 1359529–1359202 | + | + | ||
| rnpB | 385 | 1965188–1961804 | + | + | + | |
| ssrA | 367 | 2510220–2509854 | + |
Comparative analysis of five genomes: L. monocytogenes EGD‐e, C. difficile 630, E. faecalis V583, S. aureus COL and S. pyogenes M1 GAS.
Genome sequences were downloaded from NCBI http://www.ncbi.nlm.nih.gov/genomes/lproks.cgi.
One hundred sRNAs from L. monocytogenes EGD‐e (Toledo‐Arana et al., 2009) were used as reference and compared against the other four genomes using blast.
Default settings for blast were used except reward match penalty (flag‐r), which was set to 2 to accommodate the distant relation among the genomes.
The blast results were filtered by identity = 30% and query coverage = 50–150%.
Query coverage is defined as the percentage of the sRNA that is covered by the alignment [(alignment length * 100)/query length].
The number of hits is depicted in brackets.
Combating of emerging antibiotic‐resistant microbial strains remains an urgent and continuous task worldwide. Thus the search for novel sRNAs on a global scale is another facet for a better and systematic understanding of the pathogenesis of Gram‐positive bacteria and for the identification of potential novel drug targets and intervention strategies. Newly identified sRNAs are also potential markers and will be used to establish diagnostic tests that are fast, sensitive and suitable for POC applications at low cost.
The most urgent questions for the future will be: (i) what is the mode of action of many of the candidate sRNAs, (ii) how many of them are relevant for pathogenesis and regulatory networks, and most importantly (iii) are some of them targets for novel anti‐infective therapy by PNA technology and (iv) can they be used for the development of novel diagnostic systems.
Acknowledgments
The authors want to thank Alexandra Amend for excellent technical assistance and Deepak Rawool for critical reading of the manuscript. The work was supported by grants from the Bundesministerium für Bildung und Forschung (BMBF), the Agence National de Recherche (ANR) and the ERA‐NET Pathogenomics Network to the sRNAomics project.
References
- Barry T., Kelly M., Glynn B., Peden J. Molecular cloning and phylogenetic analysis of the small cytoplasmic RNA from Listeria monocytogenes. FEMS Microbiol Lett. 1999;173:47–53. doi: 10.1111/j.1574-6968.1999.tb13483.x. [DOI] [PubMed] [Google Scholar]
- Baum M., Bielau S., Rittner N., Schmid K., Eggelbusch K., Dahms M. Validation of a novel, fully integrated and flexible microarray benchtop facility for gene expression profiling. Nucleic Acids Res. 2003;31:e151. doi: 10.1093/nar/gng151. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhart S.H., Hofacker I.L., Stadler P.F. Local RNA base pairing probabilities in large sequences. Bioinformatics. 2006;22:614–615. doi: 10.1093/bioinformatics/btk014. [DOI] [PubMed] [Google Scholar]
- Beyer‐Sehlmeyer G., Kreikemeyer B., Horster A., Podbielski A. Analysis of the growth phase‐associated transcriptome of Streptococcus pyogenes. Int J Med Microbiol. 2005;295:161–177. doi: 10.1016/j.ijmm.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Bochner B.R. New technologies to assess genotype–phenotype relationships. Nat Rev Genet. 2003;4:309–314. doi: 10.1038/nrg1046. [DOI] [PubMed] [Google Scholar]
- Bohn C., Rigoulay C., Bouloc P. No detectable effect of RNA‐binding protein Hfq absence in Staphylococcus aureus. BMC Microbiol. 2007;7:10. doi: 10.1186/1471-2180-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisset S., Geissmann T., Huntzinger E., Fechter P., Bendridi N., Possedko M. Staphylococcus aureus RNAIII coordinately represses the synthesis of virulence factors and the transcription regulator Rot by an antisense mechanism. Genes Dev. 2007;21:1353–1366. doi: 10.1101/gad.423507. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet E., Wuyts J., Rouze P., Van de P.Y. Evidence that microRNA precursors, unlike other non‐coding RNAs, have lower folding free energies than random sequences. Bioinformatics. 2004;20:2911–2917. doi: 10.1093/bioinformatics/bth374. [DOI] [PubMed] [Google Scholar]
- Brandt O., Feldner J., Stephan A., Schroder M., Schnolzer M., Arlinghaus H.F. PNA microarrays for hybridisation of unlabelled DNA samples. Nucleic Acids Res. 2003;31:e119. doi: 10.1093/nar/gng120. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D.A., Kang S.H., Gryaznov S.M., DeDionisio L., Heidenreich O., Sullivan S. Effect of phosphorothioate modification of oligodeoxynucleotides on specific protein binding. J Biol Chem. 1994;269:26801–26805. et al. [PubMed] [Google Scholar]
- Busch A., Richter A.S., Backofen R. IntaRNA: efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions. Bioinformatics. 2008;24:2849–2856. doi: 10.1093/bioinformatics/btn544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camejo A., Buchrieser C., Couve E., Carvalho F., Reis O., Ferreira P. In vivo transcriptional profiling of Listeria monocytogenes and mutagenesis identify new virulence factors involved in infection. PLoS Pathog. 2009;5:e1000449. doi: 10.1371/journal.ppat.1000449. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carapetis J.R., Steer A.C., Mulholland E.K., Weber M. The global burden of group A streptococcal diseases. Lancet Infect Dis. 2005;5:685–694. doi: 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- Carney J., Braven H., Seal J., Whitworth E. Present and future applications of gold in rapid assays. IVD Technol. 2006;12:41–50. [Google Scholar]
- Carr A., Sledjeski D.D., Podbielski A., Boyle M.D., Kreikemeyer B. Similarities between complement‐mediated and streptolysin S‐mediated hemolysis. J Biol Chem. 2001;276:41790–41796. doi: 10.1074/jbc.M107401200. [DOI] [PubMed] [Google Scholar]
- Chandler D.P., Brown J., Bruckner‐Lea C.J., Olson L., Posakony G.J., Stults J.R. Continuous spore disruption using radially focused, high‐frequency ultrasound. Anal Chem. 2001;73:3784–3789. doi: 10.1021/ac010264j. et al. [DOI] [PubMed] [Google Scholar]
- Chatterjee S.S., Hossain H., Otten S., Kuenne C., Kuchmina K., Machata S. Intracellular gene expression profile of Listeria monocytogenes. Infect Immun. 2006;74:1323–1338. doi: 10.1128/IAI.74.2.1323-1338.2006. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen J.K., Larsen M.H., Ingmer H., Sogaard‐Andersen L., Kallipolitis B.H. The RNA‐binding protein Hfq of Listeria monocytogenes: role in stress tolerance and virulence. J Bacteriol. 2004;186:3355–3362. doi: 10.1128/JB.186.11.3355-3362.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen J.K., Nielsen J.S., Ebersbach T., Valentin‐Hansen P., Sogaard‐Andersen L., Kallipolitis B.H. Identification of small Hfq‐binding RNAs in Listeria monocytogenes. RNA. 2006;12:1383–1396. doi: 10.1261/rna.49706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clote P. An efficient algorithm to compute the landscape of locally optimal RNA secondary structures with respect to the Nussinov‐Jacobson energy model. J Comput Biol. 2005;12:83–101. doi: 10.1089/cmb.2005.12.83. [DOI] [PubMed] [Google Scholar]
- Cossart P., Toledo‐Arana A. Listeria monocytogenes, a unique model in infection biology: an overview. Microbes Infect. 2008;10:1041–1050. doi: 10.1016/j.micinf.2008.07.043. [DOI] [PubMed] [Google Scholar]
- Courtney H.S., Hasty D.L., Dale J.B. Molecular mechanisms of adhesion, colonization, and invasion of group A streptococci. Ann Med. 2002;34:77–87. doi: 10.1080/07853890252953464. [DOI] [PubMed] [Google Scholar]
- Coventry A., Kleitman D.J., Berger B. MSARI: multiple sequence alignments for statistical detection of RNA secondary structure. Proc Natl Acad Sci USA. 2004;101:12102–12107. doi: 10.1073/pnas.0404193101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalmay T. MicroRNAs and cancer. J Intern Med. 2008;263:366–375. doi: 10.1111/j.1365-2796.2008.01926.x. [DOI] [PubMed] [Google Scholar]
- Di Bernardo D., Down T., Hubbard T. ddbRNA: detection of conserved secondary structures in multiple alignments. Bioinformatics. 2003;19:1606–1611. doi: 10.1093/bioinformatics/btg229. [DOI] [PubMed] [Google Scholar]
- Dineva M.A., Candotti D., Fletcher‐Brown F., Allain J.P., Lee H. Simultaneous visual detection of multiple viral amplicons by dipstick assay. J Clin Microbiol. 2005;43:4015–4021. doi: 10.1128/JCM.43.8.4015-4021.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domann E., Hain T., Ghai R., Billion A., Kuenne C., Zimmermann K., Chakraborty T. Comparative genomic analysis for the presence of potential enterococcal virulence factors in the probiotic Enterococcus faecalis strain Symbioflor 1. Int J Med Microbiol. 2007;297:533–539. doi: 10.1016/j.ijmm.2007.02.008. [DOI] [PubMed] [Google Scholar]
- Dunman P.M., Murphy E., Haney S., Palacios D., Tucker‐Kellogg G., Wu S. Transcription profiling‐based identification of Staphylococcus aureus genes regulated by the agr and/or sarA loci. J Bacteriol. 2001;183:7341–7353. doi: 10.1128/JB.183.24.7341-7353.2001. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuy B., Govind R., Antunes A., Matamouros S. Clostridium difficile toxin synthesis is negatively regulated by TcdC. J Med Microbiol. 2008;57:685–689. doi: 10.1099/jmm.0.47775-0. [DOI] [PubMed] [Google Scholar]
- Eaton T.J., Gasson M.J. Molecular screening of Enterococcus virulence determinants and potential for genetic exchange between food and medical isolates. Appl Environ Microbiol. 2001;67:1628–1635. doi: 10.1128/AEM.67.4.1628-1635.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabani M.M., Gait M.J. miR‐122 targeting with LNA/2′‐O‐methyl oligonucleotide mixmers, peptide nucleic acids (PNA), and PNA–peptide conjugates. RNA. 2008;14:336–346. doi: 10.1261/rna.844108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierro‐Monti I.P., Reid S.J., Woods D.R. Differential expression of a Clostridium acetobutylicum antisense RNA: implications for regulation of glutamine synthetase. J Bacteriol. 1992;174:7642–7647. doi: 10.1128/jb.174.23.7642-7647.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folichon M., Arluison V., Pellegrini O., Huntzinger E., Regnier P., Hajnsdorf E. The poly(A) binding protein Hfq protects RNA from RNase E and exoribonucleolytic degradation. Nucleic Acids Res. 2003;31:7302–7310. doi: 10.1093/nar/gkg915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner P.P., Daub J., Tate J.G., Nawrocki E.P., Kolbe D.L., Lindgreen S. Rfam: updates to the RNA families database. Nucleic Acids Res. 2009;37:D136–D140. doi: 10.1093/nar/gkn766. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzon R., Calin G.A., Croce C.M. MicroRNAs in cancer. Annu Rev Med. 2009;60:167–179. doi: 10.1146/annurev.med.59.053006.104707. [DOI] [PubMed] [Google Scholar]
- Geisinger E., Adhikari R.P., Jin R., Ross H.F., Novick R.P. Inhibition of rot translation by RNAIII, a key feature of agr function. Mol Microbiol. 2006;61:1038–1048. doi: 10.1111/j.1365-2958.2006.05292.x. [DOI] [PubMed] [Google Scholar]
- Geissmann T., Chevalier C., Cros M.J., Boisset S., Fechter P., Noirot C. A search for small noncoding RNAs in Staphylococcus aureus reveals a conserved sequence motif for regulation. Nucleic Acids Res. 2009;37:7239–7257. doi: 10.1093/nar/gkp668. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore M.S., Coburn P.S., Nallapareddy R.S., Murray B.E. Enterococcal virulence. In: Clewell D.B., Courvalin P., Dunny G.M., Murray B.E., Rice L.B., editors. American Society for Microbiology; 2002. pp. 301–354. [Google Scholar]
- Goh S., Boberek J.M., Nakashima N., Stach J., Good L. Concurrent growth rate and transcript analyses reveal essential gene stringency in Escherichia coli. PLoS ONE. 2009;4:e6061. doi: 10.1371/journal.pone.0006061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good L., Nielsen P.E. Inhibition of translation and bacterial growth by peptide nucleic acid targeted to ribosomal RNA. Proc Natl Acad Sci USA. 1998;95:2073–2076. doi: 10.1073/pnas.95.5.2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves L.M., Helsel L.O., Steigerwalt A.G., Morey R.E., Daneshvar M.I., Roof S.E. Listeria marthii sp. nov., isolated from the natural environment, Finger Lakes National Forest. Int J Syst Evol Microbiol. 2009 doi: 10.1099/ijs.0.014118-0. et al (in press): doi: ijs.0.014118‐0. [DOI] [PubMed] [Google Scholar]
- Griffin B.E. Separation of 32P‐labelled ribonucleic acid components. The use of polyethylenimine‐cellulose (TLC) as a second dimension in separating oligoribonucleotides of ‘4.5 S’ and 5 S from E. coli. FEBS Lett. 1971;15:165–168. doi: 10.1016/0014-5793(71)80304-6. [DOI] [PubMed] [Google Scholar]
- Griffiths‐Jones S., Bateman A., Marshall M., Khanna A., Eddy S.R. Rfam: an RNA family database. Nucleic Acids Res. 2003;31:439–441. doi: 10.1093/nar/gkg006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruegelsiepe H., Brandt O., Hartmann R.K. Antisense inhibition of RNase P: mechanistic aspects and application to live bacteria. J Biol Chem. 2006;281:30613–30620. doi: 10.1074/jbc.M603346200. [DOI] [PubMed] [Google Scholar]
- Guvakova M.A., Yakubov L.A., Vlodavsky I., Tonkinson J.L., Stein C.A. Phosphorothioate oligodeoxynucleotides bind to basic fibroblast growth factor, inhibit its binding to cell surface receptors, and remove it from low affinity binding sites on extracellular matrix. J Biol Chem. 1995;270:2620–2627. doi: 10.1074/jbc.270.6.2620. [DOI] [PubMed] [Google Scholar]
- Hain T., Chatterjee S.S., Ghai R., Kuenne C.T., Billion A., Steinweg C. Pathogenomics of Listeria spp. Int J Med Microbiol. 2007;297:541–557. doi: 10.1016/j.ijmm.2007.03.016. et al. [DOI] [PubMed] [Google Scholar]
- Hain T., Hossain H., Chatterjee S.S., Machata S., Volk U., Wagner S. Temporal transcriptomic analysis of the Listeria monocytogenes EGD‐e sigmaB regulon. BMC Microbiol. 2008;8:20. doi: 10.1186/1471-2180-8-20. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halfmann A., Kovacs M., Hakenbeck R., Bruckner R. Identification of the genes directly controlled by the response regulator CiaR in Streptococcus pneumoniae: five out of 15 promoters drive expression of small non‐coding RNAs. Mol Microbiol. 2007;66:110–126. doi: 10.1111/j.1365-2958.2007.05900.x. [DOI] [PubMed] [Google Scholar]
- Hau P., Jachimczak P., Schlingensiepen R., Schulmeyer F., Jauch T., Steinbrecher A. Inhibition of TGF‐beta2 with AP 12009 in recurrent malignant gliomas: from preclinical to phase I/II studies. Oligonucleotides. 2007;17:201–212. doi: 10.1089/oli.2006.0053. et al. [DOI] [PubMed] [Google Scholar]
- Hindley J. Fractionation of 32P‐labelled ribonucleic acids on polyacrylamide gels and their characterization by fingerprinting. J Mol Biol. 1967;30:125–136. doi: 10.1016/0022-2836(67)90248-3. [DOI] [PubMed] [Google Scholar]
- Hof H., Szabo K., Becker B. [Epidemiology of listeriosis in Germany: a changing but ignored pattern] Dtsch Med Wochenschr. 2007;132:1343–1348. doi: 10.1055/s-2007-982034. [DOI] [PubMed] [Google Scholar]
- Huntzinger E., Boisset S., Saveanu C., Benito Y., Geissmann T., Namane A. Staphylococcus aureus RNAIII and the endoribonuclease III coordinately regulate spa gene expression. EMBO J. 2005;24:824–835. doi: 10.1038/sj.emboj.7600572. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemura T., Dahlberg J.E. Small ribonucleic acids of Escherichia coli. I. Characterization by polyacrylamide gel electrophoresis and fingerprint analysis. J Biol Chem. 1973;248:5024–5032. [PubMed] [Google Scholar]
- Ji G., Beavis R.C., Novick R.P. Cell density control of staphylococcal virulence mediated by an octapeptide pheromone. Proc Natl Acad Sci USA. 1995;92:12055–12059. doi: 10.1073/pnas.92.26.12055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson J., Mandin P., Renzoni A., Chiaruttini C., Springer M., Cossart P. An RNA thermosensor controls expression of virulence genes in Listeria monocytogenes. Cell. 2002;110:551–561. doi: 10.1016/s0092-8674(02)00905-4. [DOI] [PubMed] [Google Scholar]
- Joseph B., Przybilla K., Stuhler C., Schauer K., Slaghuis J., Fuchs T.M., Goebel W. Identification of Listeria monocytogenes genes contributing to intracellular replication by expression profiling and mutant screening. J Bacteriol. 2006;188:556–568. doi: 10.1128/JB.188.2.556-568.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karkare S., Bhatnagar D. Promising nucleic acid analogs and mimics: characteristic features and applications of PNA, LNA, and morpholino. Appl Microbiol Biotechnol. 2006;71:575–586. doi: 10.1007/s00253-006-0434-2. [DOI] [PubMed] [Google Scholar]
- Klein R.J., Misulovin Z., Eddy S.R. Noncoding RNA genes identified in AT‐rich hyperthermophiles. Proc Natl Acad Sci USA. 2002;99:7542–7547. doi: 10.1073/pnas.112063799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenk M., Koczan D., Guthke R., Nakata M., Thiesen H.J., Podbielski A., Kreikemeyer B. Global epithelial cell transcriptional responses reveal Streptococcus pyogenes Fas regulator activity association with bacterial aggressiveness. Cell Microbiol. 2005;7:1237–1250. doi: 10.1111/j.1462-5822.2005.00548.x. [DOI] [PubMed] [Google Scholar]
- Koppelhus U., Nielsen P.E. Cellular delivery of peptide nucleic acid (PNA) Adv Drug Deliv Rev. 2003;55:267–280. doi: 10.1016/s0169-409x(02)00182-5. [DOI] [PubMed] [Google Scholar]
- Koppelhus U., Shiraishi T., Zachar V., Pankratova S., Nielsen P.E. Improved cellular activity of antisense peptide nucleic acids by conjugation to a cationic peptide‐lipid (CatLip) domain. Bioconjug Chem. 2008;19:1526–1534. doi: 10.1021/bc800068h. [DOI] [PubMed] [Google Scholar]
- Kreikemeyer B., Boyle M.D., Buttaro B.A., Heinemann M., Podbielski A. Group A streptococcal growth phase‐associated virulence factor regulation by a novel operon (Fas) with homologies to two‐component‐type regulators requires a small RNA molecule. Mol Microbiol. 2001;39:392–406. doi: 10.1046/j.1365-2958.2001.02226.x. [DOI] [PubMed] [Google Scholar]
- Kreikemeyer B., McIver K.S., Podbielski A. Virulence factor regulation and regulatory networks in Streptococcus pyogenes and their impact on pathogen‐host interactions. Trends Microbiol. 2003;11:224–232. doi: 10.1016/s0966-842x(03)00098-2. [DOI] [PubMed] [Google Scholar]
- Kreikemeyer B., Klenk M., Podbielski A. The intracellular status of Streptococcus pyogenes: role of extracellular matrix‐binding proteins and their regulation. Int J Med Microbiol. 2004;294:177–188. doi: 10.1016/j.ijmm.2004.06.017. [DOI] [PubMed] [Google Scholar]
- Kuroda M., Ohta T., Uchiyama I., Baba T., Yuzawa H., Kobayashi I. Whole genome sequencing of meticillin‐resistant Staphylococcus aureus. Lancet. 2001;357:1225–1240. doi: 10.1016/s0140-6736(00)04403-2. et al. [DOI] [PubMed] [Google Scholar]
- Kurupati P., Tan K.S., Kumarasinghe G., Poh C.L. Inhibition of gene expression and growth by antisense peptide nucleic acids in a multiresistant beta‐lactamase‐producing Klebsiella pneumoniae strain. Antimicrob Agents Chemother. 2007;51:805–811. doi: 10.1128/AAC.00709-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong S.M., Skurray R.A., Firth N. Staphylococcus aureus multiresistance plasmid pSK41: analysis of the replication region, initiator protein binding and antisense RNA regulation. Mol Microbiol. 2004;51:497–509. doi: 10.1046/j.1365-2958.2003.03843.x. [DOI] [PubMed] [Google Scholar]
- Kwong S.M., Skurray R.A., Firth N. Replication control of staphylococcal multiresistance plasmid pSK41: an antisense RNA mediates dual‐level regulation of Rep expression. J Bacteriol. 2006;188:4404–4412. doi: 10.1128/JB.00030-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert J.L., Fischer A.L. 2005.
- Le S.V., Chen J.H., Currey K.M., Maizel J.V., Jr A program for predicting significant RNA secondary structures. Comput Appl Biosci. 1988;4:153–159. doi: 10.1093/bioinformatics/4.1.153. [DOI] [PubMed] [Google Scholar]
- Lenz D.H., Mok K.C., Lilley B.N., Kulkarni R.V., Wingreen N.S., Bassler B.L. The small RNA chaperone Hfq and multiple small RNAs control quorum sensing in Vibrio harveyi and Vibrio cholerae. Cell. 2004;118:69–82. doi: 10.1016/j.cell.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Li Z., Sledjeski D.D., Kreikemeyer B., Podbielski A., Boyle M.D. Identification of pel, a Streptococcus pyogenes locus that affects both surface and secreted proteins. J Bacteriol. 1999;181:6019–6027. doi: 10.1128/jb.181.19.6019-6027.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Gough J., Rost B. Distinguishing protein‐coding from non‐coding RNAs through support vector machines. PLoS Genet. 2006;2:e29. doi: 10.1371/journal.pgen.0020029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.M., Livny J., Lawrence M.S., Kimball M.D., Waldor M.K., Camilli A. Experimental discovery of sRNAs in Vibrio cholerae by direct cloning, 5S/tRNA depletion and parallel sequencing. Nucleic Acids Res. 2009;37:e46. doi: 10.1093/nar/gkp080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livny J., Teonadi H., Livny M., Waldor M.K. High‐throughput, kingdom‐wide prediction and annotation of bacterial non‐coding RNAs. PLoS ONE. 2008;3:e3197. doi: 10.1371/journal.pone.0003197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh E., Gripenland J., Johansson J. Control of Listeria monocytogenes virulence by 5′‐untranslated RNA. Trends Microbiol. 2006;14:294–298. doi: 10.1016/j.tim.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Lorenz C., Von Pelchrzim F., Schroeder R. Genomic systematic evolution of ligands by exponential enrichment (Genomic SELEX) for the identification of protein‐binding RNAs independent of their expression levels. Nat Protoc. 2006;1:2204–2212. doi: 10.1038/nprot.2006.372. [DOI] [PubMed] [Google Scholar]
- Mandin P., Repoila F., Vergassola M., Geissmann T., Cossart P. Identification of new noncoding RNAs in Listeria monocytogenes and prediction of mRNA targets. Nucleic Acids Res. 2007;35:962–974. doi: 10.1093/nar/gkl1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangold M., Siller M., Roppenser B., Vlaminckx B.J., Penfound T.A., Klein R. Synthesis of group A streptococcal virulence factors is controlled by a regulatory RNA molecule. Mol Microbiol. 2004;53:1515–1527. doi: 10.1111/j.1365-2958.2004.04222.x. et al. [DOI] [PubMed] [Google Scholar]
- Mao X., Ma Y., Zhang A., Zhang L., Zeng L., Liu G. Disposable nucleic acid biosensors based on gold nanoparticle probes and lateral flow strip. Anal Chem. 2009;81:1660–1668. doi: 10.1021/ac8024653. [DOI] [PubMed] [Google Scholar]
- Marchais A., Naville M., Bohn C., Bouloc P., Gautheret D. Single‐pass classification of all noncoding sequences in a bacterial genome using phylogenetic profiles. Genome Res. 2009;19:1084–1092. doi: 10.1101/gr.089714.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellbye B.L., Puckett S.E., Tilley L.D., Iversen P.L., Geller B.L. Variations in amino acid composition of antisense peptide‐phosphorodiamidate morpholino oligomer affect potency against Escherichia coli in vitro and in vivo. Antimicrob Agents Chemother. 2009;53:525–530. doi: 10.1128/AAC.00917-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers B.C., Souret F.F., Lu C., Green P.J. Sweating the small stuff: microRNA discovery in plants. Curr Opin Biotechnol. 2006;17:139–146. doi: 10.1016/j.copbio.2006.01.008. [DOI] [PubMed] [Google Scholar]
- Morcos P.A. Achieving targeted and quantifiable alteration of mRNA splicing with Morpholino oligos. Biochem Biophys Res Commun. 2007;358:521–527. doi: 10.1016/j.bbrc.2007.04.172. [DOI] [PubMed] [Google Scholar]
- Morfeldt E., Taylor D., Von Gabain A., Arvidson S. Activation of alpha‐toxin translation in Staphylococcus aureus by the trans‐encoded antisense RNA, RNAIII. EMBO J. 1995;14:4569–4577. doi: 10.1002/j.1460-2075.1995.tb00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulamba G.B., Hu A., Azad R.F., Anderson K.P., Coen D.M. Human cytomegalovirus mutant with sequence‐dependent resistance to the phosphorothioate oligonucleotide fomivirsen (ISIS 2922) Antimicrob Agents Chemother. 1998;42:971–973. doi: 10.1128/aac.42.4.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musser J.M., DeLeo F.R. Toward a genome‐wide systems biology analysis of host‐pathogen interactions in group A Streptococcus. Am J Pathol. 2005;167:1461–1472. doi: 10.1016/S0002-9440(10)61232-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narberhaus F., Waldminghaus T., Chowdhury S. RNA thermometers. FEMS Microbiol Rev. 2006;30:3–16. doi: 10.1111/j.1574-6976.2005.004.x. [DOI] [PubMed] [Google Scholar]
- Negrini M., Nicoloso M.S., Calin G.A. MicroRNAs and cancer – new paradigms in molecular oncology. Curr Opin Cell Biol. 2009;21:470–479. doi: 10.1016/j.ceb.2009.03.002. [DOI] [PubMed] [Google Scholar]
- Nekhotiaeva N., Awasthi S.K., Nielsen P.E., Good L. Inhibition of Staphylococcus aureus gene expression and growth using antisense peptide nucleic acids. Mol Ther. 2004;10:652–659. doi: 10.1016/j.ymthe.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Nielsen J.S., Olsen A.S., Bonde M., Valentin‐Hansen P., Kallipolitis B.H. Identification of a sigma B‐dependent small noncoding RNA in Listeria monocytogenes. J Bacteriol. 2008;190:6264–6270. doi: 10.1128/JB.00740-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick R.P., Ross H.F., Projan S.J., Kornblum J., Kreiswirth B., Moghazeh S. Synthesis of staphylococcal virulence factors is controlled by a regulatory RNA molecule. EMBO J. 1993;12:3967–3975. doi: 10.1002/j.1460-2075.1993.tb06074.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogier J.C., Serror P. Safety assessment of dairy microorganisms: the Enterococcus genus. Int J Food Microbiol. 2008;126:291–301. doi: 10.1016/j.ijfoodmicro.2007.08.017. [DOI] [PubMed] [Google Scholar]
- Padalon‐Brauch G., Hershberg R., Elgrably‐Weiss M., Baruch K., Rosenshine I., Margalit H., Altuvia S. Small RNAs encoded within genetic islands of Salmonella typhimurium show host‐induced expression and role in virulence. Nucleic Acids Res. 2008;36:1913–1927. doi: 10.1093/nar/gkn050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen I.T., Banerjei L., Myers G.S., Nelson K.E., Seshadri R., Read T.D. Role of mobile DNA in the evolution of vancomycin‐resistant Enterococcus faecalis. Science. 2003;299:2071–2074. doi: 10.1126/science.1080613. et al. [DOI] [PubMed] [Google Scholar]
- Pellegrini M., Marcotte E.M., Thompson M.J., Eisenberg D., Yeates T.O. Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc Natl Acad Sci USA. 1999;96:4285–4288. doi: 10.1073/pnas.96.8.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez J.M., Josephson L., O'Loughlin T., Hogemann D., Weissleder R. Magnetic relaxation switches capable of sensing molecular interactions. Nat Biotechnol. 2002;20:816–820. doi: 10.1038/nbt720. [DOI] [PubMed] [Google Scholar]
- Perez N., Trevino J., Liu Z., Ho S.C., Babitzke P., Sumby P. A genome‐wide analysis of small regulatory RNAs in the human pathogen group A Streptococcus. PLoS ONE. 2009;4:e7668. doi: 10.1371/journal.pone.0007668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichon C., Felden B. Small RNA genes expressed from Staphylococcus aureus genomic and pathogenicity islands with specific expression among pathogenic strains. Proc Natl Acad Sci USA. 2005;102:14249–14254. doi: 10.1073/pnas.0503838102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin X., Singh K.V., Weinstock G.M., Murray B.E. Characterization of fsr, a regulator controlling expression of gelatinase and serine protease in Enterococcus faecalis OG1RF. J Bacteriol. 2001;183:3372–3382. doi: 10.1128/JB.183.11.3372-3382.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raengpradub S., Wiedmann M., Boor K.J. Comparative analysis of the sigma B‐dependent stress responses in Listeria monocytogenes and Listeria innocua strains exposed to selected stress conditions. Appl Environ Microbiol. 2008;74:158–171. doi: 10.1128/AEM.00951-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recsei P., Kreiswirth B., O'Reilly M., Schlievert P., Gruss A., Novick R.P. Regulation of exoprotein gene expression in Staphylococcus aureus by agrA. Mol Gen Genet. 1986;202:58–61. doi: 10.1007/BF00330517. [DOI] [PubMed] [Google Scholar]
- Riboulet‐Bisson E., Sanguinetti M., Budin‐Verneuil A., Auffray Y., Hartke A., Giard J.C. Characterization of the Ers regulon of Enterococcus faecalis. Infect Immun. 2008;76:3064–3074. doi: 10.1128/IAI.00161-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas E. Evolutionary models for insertions and deletions in a probabilistic modeling framework. BMC Bioinformatics. 2005;6:63. doi: 10.1186/1471-2105-6-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas E., Eddy S.R. Secondary structure alone is generally not statistically significant for the detection of noncoding RNAs. Bioinformatics. 2000;16:583–605. doi: 10.1093/bioinformatics/16.7.583. [DOI] [PubMed] [Google Scholar]
- Rivas E., Eddy S.R. Noncoding RNA gene detection using comparative sequence analysis. BMC Bioinformatics. 2001;2:8. doi: 10.1186/1471-2105-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts S.A., Scott J.R. RivR and the small RNA RivX: the missing links between the CovR regulatory cascade and the Mga regulon. Mol Microbiol. 2007;66:1506–1522. doi: 10.1111/j.1365-2958.2007.06015.x. [DOI] [PubMed] [Google Scholar]
- Roush W. Antisense aims for a renaissance. Science. 1997;276:1192–1193. doi: 10.1126/science.276.5316.1192. [DOI] [PubMed] [Google Scholar]
- Rupnik M., Wilcox M.H., Gerding D.N. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol. 2009;7:526–536. doi: 10.1038/nrmicro2164. [DOI] [PubMed] [Google Scholar]
- Schattner P. Searching for RNA genes using base‐composition statistics. Nucleic Acids Res. 2002;30:2076–2082. doi: 10.1093/nar/30.9.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seal J., Braven H., Wallace P. Point‐of‐care nucleic acid lateral‐flow tests. In Vitro Diagn Technol. 2006:41–54. [Google Scholar]
- Selinger D.W., Cheung K.J., Mei R., Johansson E.M., Richmond C.S., Blattner F.R. RNA expression analysis using a 30 base pair resolution Escherichia coli genome array. Nat Biotechnol. 2000;18:1262–1268. doi: 10.1038/82367. et al. [DOI] [PubMed] [Google Scholar]
- Sen A., Nielsen P.E. On the stability of peptide nucleic acid duplexes in the presence of organic solvents. Nucleic Acids Res. 2007;35:3367–3374. doi: 10.1093/nar/gkm210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma C.M., Vogel J. Experimental approaches for the discovery and characterization of regulatory small. RNA. Curr Opin Microbiol. 2009;12:536–546. doi: 10.1016/j.mib.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Shen A., Higgins D.E. The 5′ untranslated region‐mediated enhancement of intracellular listeriolysin O production is required for Listeria monocytogenes pathogenicity. Mol Microbiol. 2005;57:1460–1473. doi: 10.1111/j.1365-2958.2005.04780.x. [DOI] [PubMed] [Google Scholar]
- Shimizu T., Yaguchi H., Ohtani K., Banu S., Hayashi H. Clostridial VirR/VirS regulon involves a regulatory RNA molecule for expression of toxins. Mol Microbiol. 2002;43:257–265. doi: 10.1046/j.1365-2958.2002.02743.x. [DOI] [PubMed] [Google Scholar]
- Shokeen S., Greenfield T.J., Ehli E.A., Rasmussen J., Perrault B.E., Weaver K.E. An intramolecular upstream helix ensures the stability of a toxin‐encoding RNA in Enterococcus faecalis. J Bacteriol. 2009;191:1528–1536. doi: 10.1128/JB.01316-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon S.A., Zhai J., Nandety R.S., McCormick K.P., Zeng J., Mejia D., Meyers B.C. Short‐read sequencing technologies for transcriptional analyses. Annu Rev Plant Biol. 2009;60:305–333. doi: 10.1146/annurev.arplant.043008.092032. [DOI] [PubMed] [Google Scholar]
- Sittka A., Lucchini S., Papenfort K., Sharma C.M., Rolle K., Binnewies T.T. Deep sequencing analysis of small noncoding RNA and mRNA targets of the global post‐transcriptional regulator, Hfq. PLoS Genet. 2008;4:e1000163. doi: 10.1371/journal.pgen.1000163. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz G., Opdyke J.A., Zhang A. Controlling mRNA stability and translation with small, noncoding RNAs. Curr Opin Microbiol. 2004;7:140–144. doi: 10.1016/j.mib.2004.02.015. [DOI] [PubMed] [Google Scholar]
- Stougaard P., Molin S., Nordstrom K. RNAs involved in copy‐number control and incompatibility of plasmid R1. Proc Natl Acad Sci USA. 1981;78:6008–6012. doi: 10.1073/pnas.78.10.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stritzker J., Schoen C., Goebel W. Enhanced synthesis of internalin A in aro mutants of Listeria monocytogenes indicates posttranscriptional control of the inlAB mRNA. J Bacteriol. 2005;187:2836–2845. doi: 10.1128/JB.187.8.2836-2845.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X., Zhulin I., Wartell R.M. Predicted structure and phyletic distribution of the RNA‐binding protein Hfq. Nucleic Acids Res. 2002;30:3662–3671. doi: 10.1093/nar/gkf508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamanaha C.R., Mulvaney S.P., Rife J.C., Whitman L.J. Magnetic labeling, detection, and system integration. Biosens Bioelectron. 2008;24:1–13. doi: 10.1016/j.bios.2008.02.009. [DOI] [PubMed] [Google Scholar]
- Tan X.X., Actor J.K., Chen Y. Peptide nucleic acid antisense oligomer as a therapeutic strategy against bacterial infection: proof of principle using mouse intraperitoneal infection. Antimicrob Agents Chemother. 2005;49:3203–3207. doi: 10.1128/AAC.49.8.3203-3207.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng F., Wang L., Singh K.V., Murray B.E., Weinstock G.M. Involvement of PhoP–PhoS homologs in Enterococcus faecalis virulence. Infect Immun. 2002;70:1991–1996. doi: 10.1128/IAI.70.4.1991-1996.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo‐Arana A., Dussurget O., Nikitas G., Sesto N., Guet‐Revillet H., Balestrino D. The Listeria transcriptional landscape from saprophytism to virulence. Nature. 2009;459:950–956. doi: 10.1038/nature08080. et al. [DOI] [PubMed] [Google Scholar]
- Tomizawa J., Itoh T., Selzer G., Som T. Inhibition of ColE1 RNA primer formation by a plasmid‐specified small RNA. Proc Natl Acad Sci USA. 1981;78:1421–1425. doi: 10.1073/pnas.78.3.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tondra M. Using integrated magnetic microchip devices in IVDs. IVD Technol. 2007;13:33–38. [Google Scholar]
- Uzilov A.V., Keegan J.M., Mathews D.H. Detection of non‐coding RNAs on the basis of predicted secondary structure formation free energy change. BMC Bioinformatics. 2006;7:173. doi: 10.1186/1471-2105-7-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Veen S., Hain T., Wouters J.A., Hossain H., De Vos W.M., Abee T. The heat‐shock response of Listeria monocytogenes comprises genes involved in heat shock, cell division, cell wall synthesis, and the SOS response. Microbiology. 2007;153:3593–3607. doi: 10.1099/mic.0.2007/006361-0. et al. [DOI] [PubMed] [Google Scholar]
- Verneuil N., Sanguinetti M., Le Breton Y., Posteraro B., Fadda G., Auffray Y. Effects of the Enterococcus faecalis hypR gene encoding a new transcriptional regulator on oxidative stress response and intracellular survival within macrophages. Infect Immun. 2004;72:4424–4431. doi: 10.1128/IAI.72.8.4424-4431.2004. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verneuil N., Rince A., Sanguinetti M., Posteraro B., Fadda G., Auffray Y. Contribution of a PerR‐like regulator to the oxidative‐stress response and virulence of Enterococcus faecalis. Microbiology. 2005;151:3997–4004. doi: 10.1099/mic.0.28325-0. et al. [DOI] [PubMed] [Google Scholar]
- Vogel J. A rough guide to the non‐coding RNA world of Salmonella. Mol Microbiol. 2009;71:1–11. doi: 10.1111/j.1365-2958.2008.06505.x. [DOI] [PubMed] [Google Scholar]
- Vogel J., Wagner E.G. Target identification of small noncoding RNAs in bacteria. Curr Opin Microbiol. 2007;10:262–270. doi: 10.1016/j.mib.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Vogel J., Bartels V., Tang T.H., Churakov G., Slagter‐Jager J.G., Huttenhofer A., Wagner E.G. RNomics in Escherichia coli detects new sRNA species and indicates parallel transcriptional output in bacteria. Nucleic Acids Res. 2003;31:6435–6443. doi: 10.1093/nar/gkg867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washietl S., Hofacker I.L., Stadler P.F. Fast and reliable prediction of noncoding RNAs. Proc Natl Acad Sci USA. 2005;102:2454–2459. doi: 10.1073/pnas.0409169102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters L.S., Storz G. Regulatory RNAs in bacteria. Cell. 2009;136:615–628. doi: 10.1016/j.cell.2009.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver K.E. Emerging plasmid‐encoded antisense RNA regulated systems. Curr Opin Microbiol. 2007;10:110–116. doi: 10.1016/j.mib.2007.03.002. [DOI] [PubMed] [Google Scholar]
- Wong K.K., Bouwer H.G., Freitag N.E. Evidence implicating the 5′ untranslated region of Listeria monocytogenes actA in the regulation of bacterial actin‐based motility. Cell Microbiol. 2004;6:155–166. doi: 10.1046/j.1462-5822.2003.00348.x. [DOI] [PubMed] [Google Scholar]
- Ziebandt A.K., Becher D., Ohlsen K., Hacker J., Hecker M., Engelmann S. The influence of agr and sigmaB in growth phase dependent regulation of virulence factors in Staphylococcus aureus. Proteomics. 2004;4:3034–3047. doi: 10.1002/pmic.200400937. [DOI] [PubMed] [Google Scholar]