

Summary
Pelotomaculum thermopropionicum is a syntrophic propionate‐oxidizing bacterium that catalyses the intermediate bottleneck step of the anaerobic‐biodegradation process. As it thrives on a very small energy conserved by propionate oxidation under syntrophic association with a methanogen, its catabolic pathways and regulatory mechanisms are of biological interest. In this study, we constructed high‐density oligonucleotide microarrays for P. thermopropionicum, and used them to analyse global transcriptional responses of this organism to different growth substrates (propionate, ethanol, propanol and lactate) in co‐culture with a hydrogenotrophic methanogenic archaeon, Methanothermobacter thermautotrophicus (by reference to fumarate monoculture). We found that a substantial number of genes were upregulated in the syntrophic co‐cultures irrespective of growth substrates (including those related to amino‐acid and cofactor metabolism), suggesting that these processes were influenced by the syntrophic partner. Expression of the central catabolic pathway (the propionate‐oxidizing methylmalonyl‐CoA pathway) was found to be substrate‐dependent and was largely stimulated when P. thermopropionicum was grown on propionate and lactate. This finding was supported by results of growth tests, revealing that syntrophic propionate oxidation was largely accelerated by supplementation with lactate. These results revealed that P. thermopropionicum has complex regulatory mechanisms that alter its metabolism in response to the syntrophic partner and growth substrates.
Introduction
Methanogenesis is an important process in the global carbon cycle and is widely distributed in various anaerobic environments, such as soil, sediment, and digestive tracts of animals. In addition, the methanogenic digestion has widely been used for the treatment of municipal and industrial organic waste (Lettinga, 1995), contributing to human activities.
Methanogenesis is a complex microbiological process that is accomplished by catabolic interactions among microorganisms in different niches, namely, primary fermenting bacteria, secondary fermenting bacteria, hydrogenotrophic methanogenic archaea and acetoclastic methanogenic archaea (Schink, 1997). Among them, secondary fermenting bacteria (also called syntrophic bacteria) catalyse the intermediate bottleneck step in the methanogenic‐digestion pathway, whereby volatile fatty acids (VFA, such as propionate and butyrate) and alcohols (such as propanol and ethanol) are converted to acetate, hydrogen and carbon dioxide under syntrophic association with methanogens (Kasper and Wuhrmann, 1978a,b; McInerney et al., 2008). Studies have revealed that the catabolic reaction catalysed by syntrophic bacteria is thermodynamically unfavorable (Schink, 1997; McInerney et al., 2008) and is feasible only when reducing equivalents (H2 and/or formate) are efficiently scavenged by methanogenic archaea (Harper and Pohland, 1986; Boone et al., 1989; Schmidt and Ahring, 1993;Stams 1994). As stagnation of this step causes the accumulation of unfavorable metabolites and whole process decay (van Lier et al., 1996), understanding of syntrophic bacteria is considered pivotal for the management of methanogenic‐digestion processes. Furthermore, given that syntrophic bacteria thrive on very small energy gained by the oxidation of VFAs (Schink, 1997; Jackson and McInerney, 2002; McInerney et al., 2008), it is reasonable to deduce that these bacteria should have extremely efficient catabolic systems, and their catabolic pathways and regulatory mechanisms are of biological interest.
Pelotomaculum thermopropionicum strain SI is a syntrophic propionate‐oxidizing bacterium isolated from a thermophilic methanogenic digester (Imachi et al., 2000; 2002). Pelotomaculum thermopropionicum can syntrophically oxidize lactate and various alcohols (e.g. ethanol, 1‐propanol and 1‐butanol) in addition to propionate, and can fermentatively grow on fumarate and pyruvate in monoculture (Imachi et al., 2002). The genome sequence of P. thermopropionicum has recently been completed (Kosaka et al., 2008), revealing interesting features of this organism as a syntrophy specialist. Reconstruction of its metabolic pathways has suggested that it has simple catabolic pathways, in which the propionate‐oxidizing methylmalonyl–CoA (MMC) pathway constitutes the backbone and is linked to several peripheral pathways (Kosaka et al., 2006). Another interesting finding in the genome analysis was that genes for important catabolic enzymes were physically linked to those for PAS‐domain‐containing regulators, suggesting that the catabolic pathways are regulated in response to environmental conditions and/or global cellular situations rather than specific substrates (Kosaka et al., 2006).
In the present study, high‐density oligonucleotide microarrays were constructed for P. thermopropionicum, and used to gain insights into transcriptional regulation mechanisms operating in this organism. In particular, in order to address the genomic perspective on the regulation of the catabolic pathways (Kosaka et al., 2006), we examined its transcriptomic responses to different growth substrates under syntrophic association with a methanogen.
Results and discussion
Growth properties of syntrophic co‐cultures with different substrates
Pelotomaculum thermopropionicum was grown in monoculture on fumarate or in co‐culture with Methanothermobacter thermautotrophicus on ethanol, 1‐propanol, lactate or propionate. Substrates degradation and products formation are shown in Fig. 1, and growth rates of P. thermopropionicum and M. thermautotrophicus are summarized in Table 1. As reported previously (Imachi et al., 2000; 2002), syntrophic propionate degradation was very slow; it took more than 40 days to utilize 20 mM propionate, and there was a long lag period before a detectable amount of methane was produced. In contrast, the other substrates were completely degraded within 3 days (Fig. 1). A specific growth rate of P. thermopropionicum in propionate culture was estimated to be less than one‐tenth of that in the monoculture on fumarate (Table 1).
Figure 1.
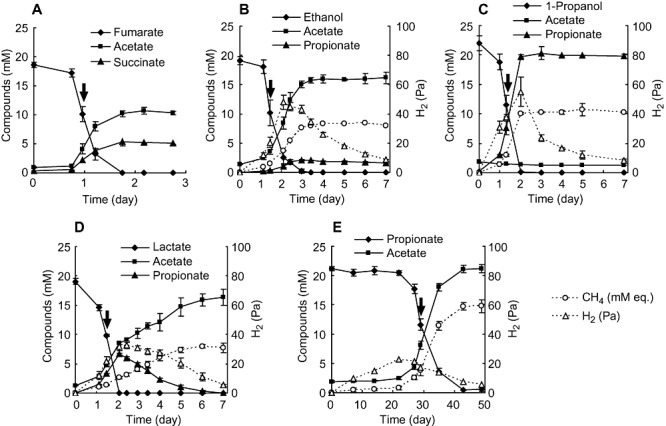
Substrate utilization and metabolite formation in monoculture grown on fumarate (A) and co‐cultures with M. thermautotrophicus on ethanol (B), 1‐propanol (C), lactate (D) and propionate (E). Methane concentrations were expressed as ‘mM equivalents’ (mM eq.) by assuming that all methane was present in the aqueous phase. The arrows indicate time points at which cells were harvested for RNA extraction. Values are means of three independent cultures. Error bars indicate standard deviations.
Table 1.
Summary of growth and transcriptome trends of P. thermopropionicum in monoculture and syntrophic co‐cultures with M. thermautotrophicus.
| Growth substrate | ΔG°a (kJ mol−1) | Specific growth rate (day−1) | P. thermopropionicum RNA ratiod (%) | No. of CDS differentially expressede | ||
|---|---|---|---|---|---|---|
| P. thermopropionicub | M. thermautotrophicusc | Up | Down | |||
| Monoculture | ||||||
| Fumarate | – | 1.83 ± 0.12 | – | – | – | – |
| Syntrophic co‐culture | ||||||
| Ethanol | 9.6 | 1.30 ± 0.03 | 1.39 ± 0.02 | 53.7 ± 5.1 | 167 (5.8%) | 128 (4.4%) |
| 1‐Propanol | 3.0 | 2.36 ± 0.07 | 2.31 ± 0.02 | 48.0 ± 6.3 | 212 (7.3%) | 171 (5.9%) |
| Lactate | −1.0 | 1.13 ± 0.02 | 0.88 ± 0.04 | 84.7 ± 9.2 | 176 (6.1%) | 182 (6.3%) |
| Propionate | 76.1 | 0.16 ± 0.02 | 0.22 ± 0.03 | 27.3 ± 3.2 | 432 (14.9%) | 534 (18.5%) |
Standard Gibbs‐free energy changes for anaerobic hydrogen‐producing reactions (Schink, 1997).
A growth rate of P. thermopropionicum was calculated from a substrate‐degradation rate.
A growth rate of M. thermautotrophicus was calculated from a methane‐production rate.
A ratio of P. thermopropionicum RNA to the total RNA extracted from a syntrophic co‐culture was determined by the RNase H method.
A number of differentially expressed coding sequences in P. thermopropionicum was determined by microarray analysis with criteria of fold > 2 (Up) or < 0.5 (Down), and P < 0.05.
Total RNA was extracted from cells in middle exponential‐growth phases (as indicated with arrows in Fig. 1) of the syntrophic co‐cultures, and ratios of P. thermopropionicum RNA to the total RNA were determined by the RNaseH method (Table 1). The RNA ratios indicate that P. thermopropionicum RNA was abundant, when it catalysed a substrate with which a large energy could be conserved (e.g. lactate). In contrast, the ratio of P. thermopropionicum RNA was low, when it was grown on propionate. The RNA preparations obtained from the syntrophic co‐cultures were subjected to the microarray analysis, in which the RNA extracted from the P. thermopropionicum monoculture was used as the reference.
Validation of microarray data
Analyses of the global gene expression were performed using six independent array experiments (i.e. two colour‐swap experiments in three biological replicates), and each microarray contained three sets of probes for each coding sequences (CDSs), facilitating a total of 18 expression measurements per CDS. The microarray datasets of technical replicates (colour‐swap samples) were correlated well (correlation coefficients r = 0.88 ± 0.04), and the correlation of biological replicates was also confirmed (r = 0.82 ± 0.11).
Five genes (listed in the legend for Fig. 2) were selected for quantitative real‐time RT‐PCR (qRT‐PCR) analysis to examine if differential expression levels determined from the microarray data were supported by qRT‐PCR. Gene expression levels determined by qRT‐PCR (copies per ng of P. thermopropionicum RNA) in each syntrophic co‐culture sample were compared to those under the reference condition (P. thermopropionicum monoculture), and relative levels were plotted against the mean relative expression levels of corresponding genes determined by the microarray experiment (Fig. 2). This analysis demonstrated that qRT‐PCR and microarray data were consistent; the data were correlated well (r = 0.98) and had a slope that approached the unity (0.95).
Figure 2.
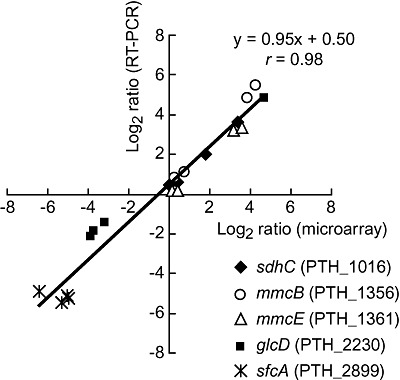
Comparison of qRT‐PCR and microarray data. Filled diamond, succinate dehydrogenase cytochrome b subunit (sdhC, PTH_1016); open circle, fumarase N‐terminal domain (mmcB, PTH_1356); open triangle, methylmalonyl‐CoA mutase N‐terminal domain (mmcE, PTH_1361); filled square, predicted lactate dehydrogenase (glcD, PTH_2230); asterisk, malic enzyme (sfcA, PTH_2899). The approximation curve and correlation coefficient (r) are given.
Overview of transcriptome data
Numbers of differentially expressed CDSs (at least twofold change with the P‐values of < 0.05) in the syntrophic co‐cultures by reference to those in the fumarate monoculture are summarized in Table 1. The numbers for the ethanol, 1‐propanol and lactate cultures [295 (10.2%), 383 (13.2%) and 358 (12.4%) CDSs, respectively, among the 2892 CDSs analysed] were not largely different, while substantially more CDSs were differentially expressed in the propionate culture [966 CDSs (33.4%)]. This indicates that the global cellular situation in the propionate co‐culture was largely different from that in the fumarate monoculture, which may have been related to the large difference in the growth rate. In order to systematically compare the global transcriptional responses in the syntrophic co‐cultures, a hierarchical cluster analysis was performed for the microarray data (Fig. 3). This analysis indicated that the expression profiles for ethanol and 1‐propanol culture were similar (r = 0.96), while those for the lactate and propionate cultures were relatively largely different.
Figure 3.
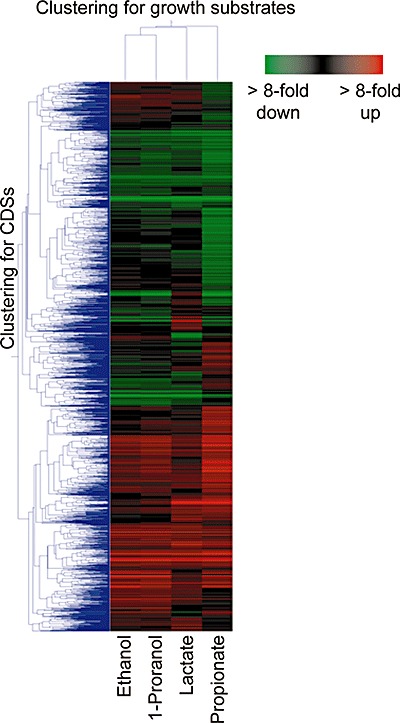
Hierarchical clustering of expression patterns of 2892 CDSs in P. thermopropionicum. Each row represents a CDS (red, upregulated; green, downregulated), while each column represents a growth substrate.
The cluster analysis (Fig. 3) also revealed that some CDSs were significantly upregulated [69 CDSs (2.4%)] or downregulated [48 CDSs (1.7%)] in all syntrophic co‐cultures (refer to Tables S1 and S2 for the lists); we consider that the expression of these CDSs was regulated in response to the syntrophic association with M. therautotrophicus. According to the classification in the Clusters of Orthologous Groups of proteins (COG) database (Tatusov et al., 2000; 2001), many CDSs differentially expressed under the syntrophic conditions were categorized into the ‘amino acid transport and metabolism (E)’, ‘coenzyme transport and metabolism (H)’ and ‘transcription (K)’ groups. These results suggest a possibility that amino acids and/or coenzymes are transferred between P. thermopropionicum and M. thermautotrophicus. This idea is supported by our finding that P. thermopropionicum obligatorily required yeast extract for its growth in monocultures, while it could grow in co‐cultures with M. therautotrophicus in the absence of yeast extract (our unpublished results).
Regulation of the central catabolic pathway
Gene‐expression patterns for the central catabolic‐pathway (the MMC pathway) enzymes in P. thermopropionicum under the syntrophic conditions are summarized in Fig. 4. Most of the genes encoding enzymes in the MMC pathway form an operon‐like cluster [namely the mmc cluster (PTH_1356‐1369)], and these are indicated with blue letters in Fig. 4 (Kosaka et al., 2006). As presented in Fig. 4, the mmc cluster genes and genes for succinate dehydrogenase I (SdhI) showed similar expression patterns; they were highly expressed in the propionate and lactate cultures. Our previous proteome data have shown that products of genes in the mmc cluster were abundantly expressed in the presence of propionate (Kosaka et al., 2006). The present finding that these genes were also upregulated in the lactate culture was unexpected, as the amounts of energy conserved by the oxidation, growth trends and global gene‐expression patterns were largely different between the propionate and lactate cultures. The growth test (Fig. 1D), however, supported this finding in the microarray analyses; namely, in the lactate culture, nearly half of lactate was initially converted to propionate, and propionate was subsequently consumed rapidly, indicating that the MMC pathway was expressed in the presence of lactate. On the other hand, in the ethanol culture (Fig. 1B), only a small amount of propionate was produced, and it was not consumed to the end.
Figure 4.
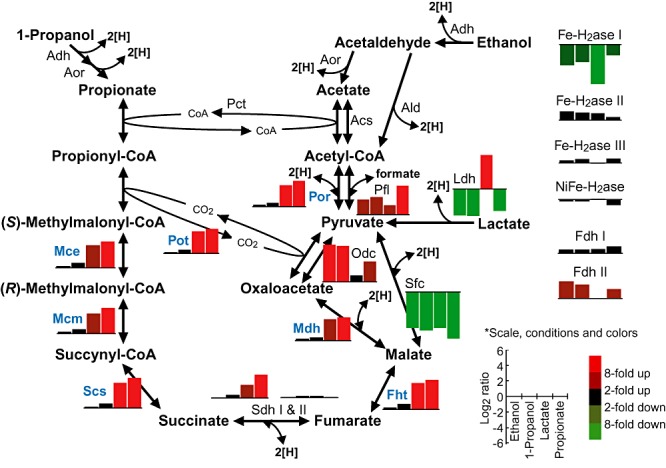
Differential expression of the central catabolic‐pathway (the MMC pathway) enzymes, hydrogenases, and formate dehydrogenases in P. thermopropionicum. Enzymes encoded by the genes in the mmc cluster are indicated with blue letters. Bar graphs represent the log2 expression ratios of enzymes in a syntrophic co‐culture by reference to the monoculture. For enzymes with multiple genes, mean values are shown. Refer to bottom right of this figure for the x‐ and y‐axes and colours of the bar graphs. Multiple candidate genes are present for Pct (propionate CoA transferase), Adh (alcohol dehydrogenase), Aor (aldehyde oxidereductase) Ald (aldehyde dehydrogenase) and Acs (acetyl‐CoA synthase), and expression profiles for these genes are listed separately (Table S3). Pot, propionyl‐CoA : oxaloacetate transcarboxylase (PTH_1364‐1366, 1368); Mce, methylmalonyl‐CoA epimerase (PTH_1363); Mcm, methylmalonyl‐CoA mutase (PTH_1361‐1362); Scs, succinyl‐CoA synthase (PTH_1358‐1360); Sdh, succinate dehydrogenase (I, PTH_1016‐1018; II, PTH_1492‐1490); Fht, fumarase (PTH_1356‐1357); Mdh, malate dehydrogenase (PTH_1367); Odc, oxaloacetate decarboxylase (PTH_1189‐1190); Ldh, lactate dehydrogenase (PTH_2230), Pfl, pyruvate formate lyase (PTH_2436); Por, pyruvate : ferredoxin oxidoreductase (PTH_1369); Fe‐H2ase, Fe‐hydrogenase (I, PTH_0668‐0670; II, PTH_1377‐1379; III, PTH_2010‐2012); NiFe‐H2ase, NiFe‐hydrogenase (PTH_1701‐1704); Fdh, formate dehydrogenase (I, PTH_1711‐1714; II, PTH_2645‐2649).
Previous studies have suggested that pathways similar to the MMC pathway of P. thermopropionicum are used for propionate degradation in mesophilic syntrophic bacteria [e.g. Syntrophobacter fumaroxidans (Plugge et al., 1993) and Syntrophobacter wolinii (Houwen et al., 1990)] and sulfate‐reducing bacteria [e.g. Desulfobulbus propionicus (Kremer and Hansen, 1988)], and for propionate production in fermentative bacteria [e.g. Propionibacterium acnes (Brüggemann et al., 2005) and Pelobacter propionicus (Schink et al., 1987)]. Genomes of S. fumaroxidans, P. acnes and P. propionicus have been sequenced, and it was found that genes for the propionate‐metabolizing pathways of these organisms do not form clusters as the mmc cluster in the P. thermopropionicum genome. Gene clustering is considered to be beneficial for P. thermopropionicum to coordinately express the series of enzymes with limited transcriptional machineries. Our genomic analysis has also revealed that the mmc cluster is associated with a putative transcriptional regulator containing a PAS domain (PTH_1355). In addition, putative σL (σ54)‐dependent promoter sequences were found immediately upstream the mmc cluster and the SdhI operon (data not shown). Based on these findings, the genome analyses have suggested that the mmc‐cluster genes are expressed in response to environmental conditions and/or global cellular situations rather than specific substrates (Kosaka et al., 2008). On the contrary, the transcriptome analysis reported herein revealed that the expression of the mmc genes was substrate‐dependent. Several possibilities can be considered for this observation. First, the mmc cluster is regulated by an unknown transcriptional regulator but not by the PTH_1355 product. Second, the PTH_1355 product is an unusual PAS domain‐containing regulator that can sense specific substrates. Finally (and the most likely), P. thermopropionicum has an unknown regulator to sense propionate and lactate that cooperatively work with the PAS domain‐containing regulator. The genome analyses have revealed that P. thermopropionicum has a relatively large number of signal‐transduction machineries compared to other bacteria (Kosaka et al., 2008), suggesting that this organism has complex regulatory mechanisms; it is likely that some of them are involved in the substrate‐dependent regulation. We will focus our studies on genetic analyses of this organism to reveal how it regulates the expression of the MMC pathway in response to the presence of these substrates.
In the MMC pathway, bypaths are present at several steps (Fig. 4). Pelotomaculum thermopropionicum and the other propionate‐oxidizing bacteria are proposed to use malate dehydrogenase (Mdh) and propionyl‐CoA : oxaloacetate transcarboxylase (Pot) for the conversion between malate and pyruvate, whereas P. acnes is proposed to use malic enzyme (Sfc) for this conversion. Transcriptome analyses disclosed the gene for Sfc of P. thermopropionicum was highly expressed in the fumarate monoculture and downregulated in the syntrophic co‐cultures (Fig. 4). This indicates that P. thermopropionicum uses the propionate producer‐like pathway in the fumarate‐fermenting life style. In addition to the genes for Pot, P. thermopropionicum also possesses the genes for oxaloacetate decarboxylase (Odc). This study found that the odc genes were highly upregulated in the ethanol and 1‐propanol cultures, suggesting that Odc is used to catalyse ATP‐dependent carboxylation of pyruvete to synthesize the precursors for cellular components in the alcohol‐metabolizing conditions.
Regulation of the peripheral pathways
Hydrogen and formate mediate interspecies transfer of reducing equivalents between syntrophic bacteria and methanogens. Pelotomaculum thermopropionicum has four hydrogenases (H2ases) and two formate dehydrogenases (Fdhs) (Kosaka et al., 2008) that may work in concert with the central catabolic pathway. Expression patterns of genes for these enzymes in P. thermopropionicum under the syntrophic conditions are also summarized in Fig. 4. The microarray data revealed that genes for one hydrogenase (Fe‐H2ase I) were downregulated in the syntrophic co‐cultures, while genes for one of formate dehydrogenases (FdhII) were upregulated in the alcohol and propionate cultures. Comparison in absolute fluorescence intensities in the microarray data for the four H2ase genes suggests a possibility that Fe‐H2ase III was the major hydrogenase in all the culture conditions (Table S3). In addition, the gene for pyruvate : fomate lyase (Pfl) that catalyses the conversion between pyruvate and acetyl‐CoA (pyruvate + CoA = Acetyl‐CoA + formate) were upregulated in the syntrophic co‐cultures (Fig. 4).
It has been suggested that the first step of propionate oxidation is catalysed by propionate CoA transferase (Pct) (Kosaka et al., 2006; 2008). Pelotomaculum thermopropionicum encodes several candidate genes for acyl‐CoA transferases with unknown substrate specificities (Table S4). Although the transcriptome analyses disclosed that some of them were upregulated in the propionate culture (PTH_0268, 1541, 1577 and 1771), further biochemical studies are needed for the identification of the genes for the first step of propionate oxidation.
The P. thermopropionicum genome encodes multiple putative genes for alchohol metabolism; 12 alcohol dehydrogenases (Adhs) and 10 aldehyde oxidereductases (Aors) (Table S4). Transcriptome analyses disclosed that some of them were upregulated in both of the ethanol and 1‐propanol cultures. Although these are candidates for the key genes for alcohol metabolism in P. thermopropionicum, further biochemical studies are needed to confirm it.
The microarray analyses revealed that a gene cluster (PTH_2229‐2237) was specifically upregulated in the presence of lactate (Table S5). PTH_2229 encodes l‐lactate permease and PTH_2230 encodes FAD/FMN‐containing dehydrogenase. Conserved domain search (Marchler‐Bauer and Bryant, 2004) revealed that this gene is related to GlcD‐like 2‐hydroxy‐acid oxidase (Pellicer et al., 1996), indicating that the gene product works as lactate dehydrogenase. This gene cluster contains two GntR family, FadR‐type transcriptional regulators (PTH_2232, 2233), which contain effecter binding C‐terminal domains as other GntR‐family transcriptional regulators (Xu et al., 2001; Rigali et al., 2002). FadR‐type transcriptional regulators have been reported to sense various effecter molecules, including lactate (Georgi et al., 2008). The two transcriptional regulators are predicted to sense lactate and regulate the expression of this gene cluster. Although roles of the other genes are unclear, the gene‐expression data imply that products of these genes are relevant to lactate sensing and metabolism.
Lactate accelerated syntrophic propionate oxidation
Syntrophic propionate oxidation is a thermodynamically unfavorable reaction, and syntrophic propionate‐oxidizing cultures need long cultivation periods and are unstable (Boone et al., 1989; Harmsen et al., 1998; Liu et al., 1999; Imachi et al., 2000; 2002; Plugge et al., 2002). In the syntrophic co‐culture of P. thermopropionicum and M. thermautotrophicus, the long lag period was observed before the onset of propionate oxidation (Fig. 1E). In the microarray analyses, we found that the genes for propionate‐oxidizing pathway were highly upregulated in the lactate culture (Fig. 4). In addition, the growth test (Fig. 1D) showed that propionate temporally accumulated in the lactate culture and was rapidly degraded afterward. From these observations, we hypothesized that lactate may have been able to accelerate syntrophic propionate oxidation. In order to address this idea, co‐cultures of P. thermopropionicum and M. thermautotrophicus were grown on propionate, after they were pre‐grown on either ethanol or lactate (Fig. 5A). This figure shows that the lactate‐grown culture started to grow on propionate more rapidly than the ethanol‐grown culture. We also evaluated the above idea in a lactate‐supplemented experiment. In this experiment, the propionate medium was inoculated with the co‐culture pre‐grown on ethanol and supplemented with either 4 mM lactate or 4 mM ethanol (Fig. 5B). It was found that lactate showed larger effect on shortening the lag period before the onset of syntrophic propionate oxidation than ethanol. As the stimulation of the microbial growth by lactate or ethanol was same extent, the growth stimulation was not simply ascribable to an increase in cell concentrations. It has been considered that a long lag period is necessary for P. thermopropionicum to accumulate the substrates needed for the propionate activation (i.e. acetyl‐CoA), to increase cell numbers to an adequate level, and/or to express the catabolic enzymes under the energy‐limited condition (Kosaka et al., 2006). The results of the present study suggest that lactate stimulates the expression of the propionate‐oxidizing pathway, resulting in the rapid onset of syntrophic propionate oxidation.
Figure 5.
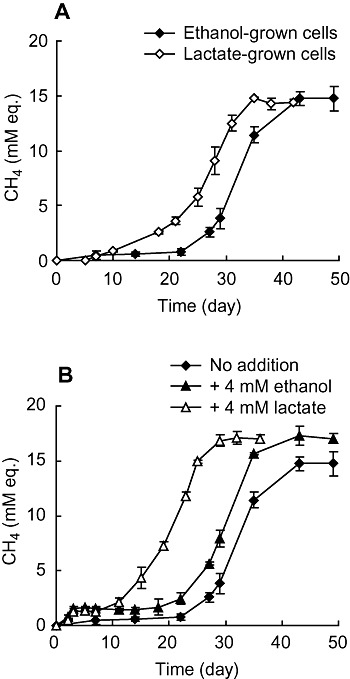
Effects of lactate on syntrophic methanogenesis from propionate. (A) The medium containing 20 mM propionate was inoculated with either ethanol‐ or lactate‐grown syntrophic co‐culture. (B) The medium containing 20 mM propionate was inoculated with ethanol‐grown syntrophic co‐culture and supplemented with either 4 mM ethanol or 4 mM lactate. Methane concentrations were expressed as ‘mM equivalents’ (mM eq.) by assuming that all methane was present in the aqueous phase. Values are means of three independent cultures. Error bars indicate standard deviations.
Conclusions
In the present study, we have successfully constructed and validated the transcriptome method for P. thermopropionicum. It was revealed that P. thermopropionicum vigorously regulated the expression of catabolic genes in response to growth substrates and the presence of the syntrophic partner. This finding suggests that understanding of its transcriptional regulation mechanisms will be the key to elucidate survival strategies of this organism under energy‐limited conditions. In addition, the microarray results are also useful to identify an important gene from several candidate genes for each catabolic step (e.g. alcohol dehydrogenease, propionate CoA transferase and hydrogenase). After all, our transcriptome analysis takes a step forward for global understanding of the lifestyle of P. thermopropionicum, and the findings reported herein will direct subsequent biochemical and molecular studies that can provide molecular bases for metabolism capacities of P. thermopropionicum.
Experimental procedures
Strain and culture conditions
Pelotomaculum thermopropionicum strain SI (DSM13744) was grown in monoculture or co‐culture with M. thermautotrophicus type II (Imachi et al., 2000) in a fresh water basal (FWB) medium (Schönheit et al., 1979) as described previously (Ishii et al., 2005). The medium was supplemented with 0.01% (w/v) Bacto yeast extract (Difco) and a growth substrate at 20 mM. Cultivation was conducted at 55°C under an atmosphere of N2 plus CO2[80/20 (v/v)] without shaking.
RNA isolation
Cells for RNA isolation were collected in the middle exponential‐growth phases by centrifugation at 10 000 g and 4°C. Total RNA was isolated by the bead‐beating method as described previously (Kato et al., 2008). The quality of total RNA was evaluated using an Agilent 2100 Bioanalyzer, RNA 6000 Pico reagents and RNA Pico Chips (Agilent Technologies) according to the manufacturer's instruction.
Determination of RNA ratio using RNase H
Sequence‐specific cleavage of rRNA using RNase H (Uyeno et al., 2004) was applied to determine a ratio of P. thermopropionicum RNA in total RNA extracted from a syntrophic co‐culture. Sequence‐dependent scission of rRNA was performed by a method described elsewhere (Sekiguchi et al., 2005). Probes used in this study were GIh821m (5′‐ACC TCC TAC ACC TAG CAC CC‐3′) specific for the Desulfotomaculum Ih group (including P. thermopropionicum) and CMB1175 (5′‐CCG TCG TCC ACT CCT TCC TC‐3′) specific for the family Methanobacteriaceae (including M. thermautotrophicus) (Y. Sekiguchi, pers. comm.). The resultant RNA fragments were analysed using an Agilent 2100 Bioanalyzer, and a ratio of P. thermopropionicum RNA was estimated based on peak areas in a chromatograph.
Construction of P. thermopropionicum microarrays
Specific oigonucleotides (60mer) were designed for 2892 CDSs [corresponding to 99.1% of the total predicted CDSs in the annotated genome of P. thermopropionicum (Kosaka et al. (2008); ribosomal and transfer RNA genes were not included] by the eArray protocol (Agilent Technologies) and fabricated on slide glasses by the SurePrint technology (Agilent Technologies). For each CDS, 3 spots (with 3 different specific sequences) were printed in one array. A mean value of the normalized signal intensities of the 3 spots was used for expression analysis. Two arrays were fabricated on one slide glass, which were used for a colour‐swap assay as described below.
Transcriptome analysis using the P. thermopropionicum microarray
Fluorescence labelling of cDNA, hybridization and scanning of a hybridized array were performed according to the method of the two‐colour microarray‐based gene‐expression analysis as described previously (Kato et al., 2008). Total RNA isolated from cells in the middle exponential‐growth phase of P. thermopropionicum monoculture grown on fumarate was used as the reference sample. Based on a ratio of P. thermopropionicum RNA to the total RNA determined by the RNase H method, an amount of total syntrophic co‐culture RNA used for the cDNA probe preparation were determined; an amount of P. thermopropionicum RNA used for the cDNA probe preparation was always 3 µg. Fluorescence‐dye labelled cDNA for a syntrophic co‐culture was mixed with oppositely labelled cDNA for the reference monoculture and hybridized with the probes on a microarray. To minimize dye biases, dyes (Cy3 and Cy5) were swapped for each replicate.
Microarray‐data analysis
Microarray data analysis was conducted by methods described previously (Kato et al., 2008). Microarray spot‐intensity data were estimated by subtracting a background signal‐intensity value from a raw signal‐intensity value and normalized by the linear‐LOWESS method (Yang et al., 2002). An expression ratio was calculated by dividing a normalized signal‐intensity value of each CDS in syntrophic co‐culture samples by that in the reference monoculture sample. Statistical analysis (by the Student's t‐test) and data visualization were carried out using GeneSpring GX ver. 7.3.1 (Agilent Technologies).
Quantitative real‐time RT‐PCR
All PCR primers used for real‐time RT‐PCR were designed by Nihon Gene Research Laboratories. Target genes and respective primer sets were as follows; succinate dehydrogenase cytochrome b subunit (sdhC, PTH_1016), sdhC‐F 5′‐TCC CTT GGA TGA TGG TAT TT‐3′ and sdhC‐R 5′‐ACA ATA ATC ACG GTG ATG GC‐3′; fumarase N‐terminal domain (mmcB, PTH_1356), mmcB‐F 5′‐CGA TCT TTA CGA GGC AAT‐3′ and mmcB‐R 5′‐CGA TCT TGG TGT GGA TAA CT‐3′; methylmalonyl‐CoA mutase N‐terminal domain (mmcE, PTH_1361), mmcE‐F 5′‐CAT CAG CGG CTA TCA TAT‐3′ and mmcE‐R 5′‐TCA GGT GAG CAT TGA AGA‐3′; predicted lactate dehydrogenase (glcD, PTH_2230), ldh‐F 5′‐GGG CTG GTT CTT TCC ACT C‐3′ and ldh‐R 5′‐GCT ACG GCC TCA TTG AGG T‐3′; and malic enzyme (sfcA, PTH_2899), sfcA‐F 5′‐TGC TGA ACC GCC AGG ATG T‐3′ and sfcA‐R 5′‐GGT TGG GGT AAT CCG ATC TGC‐3′. Quantitative gene expression analysis was performed by real‐time RT‐PCR using a LightCycler system and a LightCycler RNA Master SYBR Green I kit (Roche Applied Science) as described previously (Kato et al., 2008).
Analytical methods
The gas composition (methane and hydrogen) was analysed using a gas chromatograph (GC‐14A, Shimadzu) equipped with a thermal conductivity detector and a molecular sieve 5A 60‐80/Porapack Q 80‐100 column (Shimadzu) as described elsewhere (Ishii et al., 2005). VFAs (acetate and propionate) and alcohols (ethanol and propanol) were analysed using a gas chromatograph (GC‐2010, Shimadzu) with a flame ionization detector and a DB‐FFAP column (Shimadzu) as described elsewhere (Ishii et al., 2005). Lactate, fumarate and succinate were analysed using a high performance liquid chromatograph (Organic acid analysis system, Shimadzu) with a CDD detector and dual packed columns (Shim‐Pack SCR102‐H, Shimadzu) as described elsewhere (Ishii et al., 2008).
Transcriptome data accession number
The transcriptome data have been deposited in CIBEX (Center for Information Biology gene EXpression) database (http://cibex.nig.ac.jp/index.jsp) under accession number CBX56.
Acknowledgments
We thank Fusako Numazaki, Midori Sato and Reiko Hirano for technical support, and Greg Newton for critical reading of the manuscript. This work was supported by New Energy Development Organization (NEDO) and Japan Society for the Promotion of Science (JSPS).
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Table S1. List of genes upregulated under all syntrophic conditions.
Table S2. List of genes downregulated under all syntrophic conditions.
Table S3. Absolute fluorescence intensities in the microarray data for genes for hydrogenases and formate dehydrogenases.
Table S4. Differential expression of genes putatively related to propionate and alcohol metabolism.
Table S5. Differential expression of genes in the putative lactate operon.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Boone D.R., Johnson R.L., Liu Y. Diffusion of the interspecies electron carriers H2 and formate in methanogenic ecosystems and its implications in the measurement of Km for H2 or formate uptake. Appl Environ Microbiol. 1989;55:1735–1741. doi: 10.1128/aem.55.7.1735-1741.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brüggemann H., Henne A., Hoster F., Liesegang H., Wiezer A., Strittmatter A. The complete genome sequence of Propionibacterium acnes, a commensal of human skin. Science. 2005;305:671–673. doi: 10.1126/science.1100330. et al. [DOI] [PubMed] [Google Scholar]
- Georgi T., Engels V., Wendisch V.F. Regulation of l‐lactate utilization by the FadR‐type regulator LldR of Corynebacterium glutamicum. J Bacteriol. 2008;190:963–971. doi: 10.1128/JB.01147-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmsen H.J., Van Kuijk B.L., Plugge C.M., Akkermans A.D., De Vos W.M., Stams A.J. Syntrophobacter fumaroxidans sp. nov., a syntrophic propionate‐degrading sulfate‐reducing bacteria. Int J Syst Bacteriol. 1998;48:1383–1387. doi: 10.1099/00207713-48-4-1383. [DOI] [PubMed] [Google Scholar]
- Harper S.R., Pohland F.G. Recent developments in hydrogen management during anaerobic biological wastewater treatment. Biotechnol Bioeng. 1986;28:585–602. doi: 10.1002/bit.260280416. [DOI] [PubMed] [Google Scholar]
- Houwen F.P., Plokker J., Stams A.J.M., Zehnder A.J.B. Enzymatic evidence for involvement of the methyl‐malonyl‐CoA pathway in propionate oxidation by Syntrophobacter wolinii. Arch Microbiol. 1990;155:52–55. [Google Scholar]
- Imachi H., Sekiguchi Y., Kamagata Y., Ohashi A., Harada H. Cultivation and in situ detection of a thermophilic bacterium capable of oxidizing propionate in syntrophic association with hydrogenotrophic methanogens in a thermophilic methanogenic granular sludge. Appl Environ Microbiol. 2000;66:3608–3615. doi: 10.1128/aem.66.8.3608-3615.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imachi H., Sekiguchi Y., Kamagata Y., Ohashi A., Hanada S., Harada H. Pelotomaculum thermopropionicum gen. nov., sp. nov., an anaerobic, thermophilic, syntrophic propionate‐oxidizing bacterium. Int J Syst Evol Microbiol. 2002;52:1729–1735. doi: 10.1099/00207713-52-5-1729. [DOI] [PubMed] [Google Scholar]
- Ishii S., Kosaka T., Hori K., Hotta Y., Watanabe K. Coaggregation facilitates interspecies hydrogen transfer between Pelotomaculum thermopropionicum and Methanothermobacter thermautotrophicus. Appl Environ Microbiol. 2005;71:7838–7845. doi: 10.1128/AEM.71.12.7838-7845.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii S., Shimoyama T., Hotta Y., Watanabe K. Characterization of a filamentous biofilm community established in a cellulose‐fed microbial fuel cell. BMC Microbiol. 2008;8:6. doi: 10.1186/1471-2180-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson B.E., McInerney M.J. Anaerobic microbial metabolism can proceed close to thermodynamic limits. Nature. 2002;415:454–456. doi: 10.1038/415454a. [DOI] [PubMed] [Google Scholar]
- Kasper H.F., Wuhrmann K. Product inhibition in sludge digestion. Microb Ecol. 1978a;4:241–248. doi: 10.1007/BF02015080. [DOI] [PubMed] [Google Scholar]
- Kasper H.F., Wuhrmann K. Kinetic parameters and relative turnovers of some important catabolic reactions in digesting sludge. Appl Environ Microbiol. 1978b;36:1–7. doi: 10.1128/aem.36.1.1-7.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato S., Kosaka T., Watanabe K. Comparative transcriptome analysis of responses of Methanothermobacter thermautotrophicus to different environmental stimuli. Environ Microbiol. 2008;10:893–905. doi: 10.1111/j.1462-2920.2007.01508.x. [DOI] [PubMed] [Google Scholar]
- Kosaka T., Uchiyama T., Ishii S., Enoki M., Imachi H., Kamagata Y. Reconstruction and regulation of the central catabolic pathway in the thermophilic propionate‐oxidizing syntroph Pelotomaculum thermopropionicum. J Bacteriol. 2006;188:202–210. doi: 10.1128/JB.188.1.202-210.2006. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka T., Kato S., Shimoyama T., Ishii S., Abe T., Watanabe K. The genome of Pelotomaculum thermopropionicum reveals niche‐associated evolution in anaerobic microbiota. Genome Res. 2008;18:442–448. doi: 10.1101/gr.7136508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer D.R., Hansen T.A. Pathway of propionate degradation in Desulfobulbus propionicus. FEMS Microbiol Lett. 1988;49:273–277. [Google Scholar]
- Lettinga G. Anaerobic digestion and wastewater treatment systems. Antonie Leewenhock. 1995;67:3–28. doi: 10.1007/BF00872193. [DOI] [PubMed] [Google Scholar]
- Liu Y., Balkwill D.L., Aldrich H.C., Drake G.R., Boone D.R. Characterization of the anaerobic propionate‐degrading syntrophs Smithella propionica gen. nov., sp. nov. and Syntrophobacter wolinii. Int J Syst Bacteriol. 1999;49:545–556. doi: 10.1099/00207713-49-2-545. [DOI] [PubMed] [Google Scholar]
- Marchler‐Bauer A., Bryant S.H. CD‐Search: protein domain annotations on the fly. Nucleic Acids Res. 2004;32:327–331. doi: 10.1093/nar/gkh454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInerney M.J., Struchtemeyer C.G., Sieber J., Mouttaki H., Stams A.J., Schink B. Physiology, ecology, phylogeny, and genomics of microorganisms capable of syntrophic metabolism. Ann NY Acad Sci. 2008;1125:58–72. doi: 10.1196/annals.1419.005. et al. [DOI] [PubMed] [Google Scholar]
- Pellicer M.T., Badía J., Aguilar J., Baldomà L. glc locus of Escherichia coli: characterization of genes encoding the subunits of glycolate oxidase and the glc regulator protein. J Bacteriol. 1996;178:2051–2059. doi: 10.1128/jb.178.7.2051-2059.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plugge C.M., Dijkema C., Stams A.J.M. Acetyl‐CoA cleavage pathway in a syntrophic propionate oxidizing bacterium growing on fumarate in the absence of methanogens. FEMS Microbiol Lett. 1993;110:71–76. [Google Scholar]
- Plugge C.M., Balk M., Stams A.J.M. Desulfotomaculum thermobenzoicum subsp. thermosyntrophicum subsp. nov., a thermophilic, syntrophic, propionate‐oxidizing, spore‐forming bacterium. Int J Syst Evol Microbiol. 2002;52:391–399. doi: 10.1099/00207713-52-2-391. [DOI] [PubMed] [Google Scholar]
- Rigali S., Derouaux A., Giannotta F., Dusart J. Subdivision of the helix‐turn‐helix GntR family of bacterial regulators in the FadR, HutC, MocR, and YtrA subfamilies. J Biol Chem. 2002;277:12507–12515. doi: 10.1074/jbc.M110968200. [DOI] [PubMed] [Google Scholar]
- Schink B. Energetics of syntrophic cooperation in methanogenic degradation. Microbiol Mol Biol Rev. 1997;61:262–280. doi: 10.1128/mmbr.61.2.262-280.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schink B., Kremer D.R., Hansen T.A. Pathway of propionate formation from ethanol in Pelobacter propionicus. Arch Microbiol. 1987;147:321–327. [Google Scholar]
- Schmidt J.E., Ahring B.K. Effects of hydrogen and formate on the degradation of propionate and butyrate in thermophilic granules from an upflow anaerobic sludge blanket reactor. Appl Environ Microbiol. 1993;59:2546–2551. doi: 10.1128/aem.59.8.2546-2551.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönheit P., Moll J., Thauer R.K. Nickel, cobalt and molybdenum requirement for growth of Methanobacterium thermoautotrophicum. Arch Microbiol. 1979;123:105–107. doi: 10.1007/BF00403508. [DOI] [PubMed] [Google Scholar]
- Sekiguchi Y., Uyeno Y., Sunaga A., Yoshida H., Kamagata Y. Sequence‐specific cleavage of 16S rRNA for rapid and quantitative detection of particular groups of anaerobes in bioreactors. Water Sci Technol. 2005;52:107–113. [PubMed] [Google Scholar]
- Stams A.J.M. Metabolic interactions between anaerobic bacteria in methanogenic environments. Antonie Leeuwenhoek. 1994;66:271–294. doi: 10.1007/BF00871644. [DOI] [PubMed] [Google Scholar]
- Tatusov R.L., Galperin M.Y., Natale D.A., Koonin E.V. The COG database: a tool for genome‐scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov R.L., Natale D.A., Garkavtsev I.V., Tatusova T.A., Shankavaram U.T., Rao B.S. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001;29:22–28. doi: 10.1093/nar/29.1.22. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyeno Y., Sekiguchi Y., Sunaga A., Yoshida H., Kamagata Y. Sequence‐specific cleavage of small‐subunit (SSU) rRNA with oligonucleotides and RNase H: a rapid and simple approach to SSU rRNA‐based quantitative detection of microorganisms. Appl Environ Microbiol. 2004;70:3650–3663. doi: 10.1128/AEM.70.6.3650-3663.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lier J.B., Martin J.L.S., Lettinga G. Effect of temperature on the anaerobic thermophilic conversion of volatile fatty acids by dispersed and granular sludge. Water Res. 1996;30:199–207. [Google Scholar]
- Xu Y., Heath R.J., Li Z., Rock C.O., White S.W. The FadR·DNA complex. Transcriptional control of fatty acid metabolism in Escherichia coli. J Biol Chem. 2001;276:17373–17379. doi: 10.1074/jbc.M100195200. [DOI] [PubMed] [Google Scholar]
- Yang Y.H., Dudoit S., Luu P., Lin D.M., Peng V., Ngai J., Speed T.P. Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic validation. Nucleic Acids Res. 2002;30:e15. doi: 10.1093/nar/30.4.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of genes upregulated under all syntrophic conditions.
Table S2. List of genes downregulated under all syntrophic conditions.
Table S3. Absolute fluorescence intensities in the microarray data for genes for hydrogenases and formate dehydrogenases.
Table S4. Differential expression of genes putatively related to propionate and alcohol metabolism.
Table S5. Differential expression of genes in the putative lactate operon.