

Abstract
Maize represents one of the main cultivar for food and energy and crop yields are influenced by soil physicochemical and climatic conditions. To study how maize plants influence soil microbes we have examined microbial communities that colonize maize plants grown in carbonate-rich soil (pH 8.5) using culture-independent, PCR-based methods. We observed a low proportion of unclassified bacteria in this soil whether it was planted or unplanted. Our results indicate that a higher complexity of the bacterial community is present in bulk soil with microbes from nine phyla, while in the rhizosphere microbes from only six phyla were found. The predominant microbes in bulk soil were bacteria of the phyla Acidobacteria, Bacteroidetes and Proteobacteria, while Gammaproteobacteria of the genera Pseudomonas and Lysobacter were the predominant in the rhizosphere. As Gammaproteobacteria respond chemotactically to exudates and are efficient in the utilization of plants exudate products, microbial communities associated to the rhizosphere seem to be plant-driven. It should be noted that Gammaproteobacteria made available inorganic nutrients to the plants favouring plant growth and then the benefit of the interaction is common.
Introduction
The taxonomical and functional structures of soil microbial communities are influenced by biotic and abiotic factors including the physicochemical characteristics of soil itself, water availability, climate conditions, presence of plants, plant types, and the interactions with other soil prokaryotic and with lower or higher eukaryotic organisms (Pennanen et al., 1999; Oline, 2006; Jones et al., 2009). Plants exert selective pressure on soil microbial populations through modification of the physicochemical characteristics of the surrounding soil and the excretion of exudates consisting of amino acids and organic acids, proteins and other chemicals that act as chemoattractant or repellent molecules (Rambelli, 1973; Espinosa-Urgel et al., 2002; Shaw et al., 2006; Acosta-Martínez et al., 2008; Haichar et al., 2008; Berg and Smalla, 2009; DeAngelis et al., 2009; Lacal et al., 2011). In this study we have focused our attention on the influence of maize, one of the main plant cultivars for animal and human foodstuff. It is known that maize seeds exude a large variety of amino acids, sugars and some weak organic acids that modify the surrounding soil (Vílchez et al., 2000) and that the continuous supply of nutrients via root exudates allows the establishment of a dynamic and nutrient-rich niche in the rhizosphere where the total number of microbes is higher than in bulk soil (Kowalchuk et al., 2002; Nunes da Rocha et al., 2009). Bacteria that colonize the roots and surrounding soil can be pathogens, saprophytes or beneficial plant growth promoters. Among plant growth promoting rhizobacteria (PGPR) are those that solubilize phosphate and nitrogen (Cocking, 2003; Rodríguez et al., 2006; Matilla et al., 2007), and that protect plants against pathogens via the production of antibiotics, antifungal chemicals and insecticides (Preston et al., 2001; Berg et al., 2005).
It is known that only a fraction of soil microbes can be cultured. Because of this limitation a variety of fingerprinting methods, dependent or independent of cloning-sequencing procedures, have been developed (Fierer and Jackson, 2006; Smalla et al., 2007). Microbial phylogenetic diversity can be defined by analysing the gene sequences encoding 16S rRNAs isolated from environmental samples (Giovannoni et al., 1990; DeLong, 1992; Pace, 1997; Huber et al., 2002; Hewson et al., 2003; Rappé and Giovannoni, 2003). The resulting sequences can then be used to generate taxonomic inventories of microbial populations, and the abundance curves from observed frequencies of sequences can be used to predict the number of different microbial taxa in a specific sample (Chao, 1984; Chao et al., 1992; Curtis et al., 2002). Therefore, 16S rRNA analysis is considered an effective tool to compare bacterial community patterns from different samples collected from different environments (Kowalchuk et al., 2002; Smalla et al., 2007; Haichar et al., 2008).
The present study was aimed to examine how maize plants influence the diversity of microbial communities in a typical carbonate-rich Mediterranean soil. Maize is used as a model plant in this study because of its agronomical importance and its use in soils with a wide range of pHs. In this study we have concentrated on a relatively high pH carbonate-rich soil typical of the South Spain (Table 1). We have examined bacterial diversity in the rhizosphere (soil attached to roots) and bulk soil using culture-independent PCR-based methods. Our findings show that plants exerted selective pressure on the microbial communities, causing enrichment of Gammaproteobacteria in the rhizosphere, a group of microbes that are chemotactically attracted by maize exudates that are rich in energy sources.
Table 1.
Physicochemical properties of soils used in this study to grow maize
| Test description | |
|---|---|
| Active lime | 3.70% |
| Carbonates | 13.6% |
| Classification | Type clay loam |
| Assimilable phosphorus | 11 ppm |
| Humic matter | 0.79% |
| Total nitrogen | 0.072% |
| pH | 8.5 |
| Assimilable potassium | 205 ppm |
| Salinity pretest | 0.17 mmhos cm−1 |
| Clay texture | 31.30% |
| Sand texture | 37.02% |
| Silt texture | 31.68% |
Soil assays were performed by the Andalucian Service of soil analysis laboratory using International Standard methods.
Results and discussion
Roots progressing in ‘bulk soil’ introduce labile carbon and nutrients while creating water ways and deposits of antimicrobial compounds and hormones (Brimecombe et al., 2001; Bringhurst et al., 2001; Hawkes et al., 2007) in time (hours or days) (Lubeck et al., 2000). As many soil microbes exhibit limitations to carbon (Paul and Clark, 1996), they could be expected to respond quickly to root-induced changes, by reprogramming their activity (Heijnen et al., 1995; Herman et al., 2006). We have analysed microbial biodiversity in bulk soil, as well as in the more tightly root-adhering soil as is the rhizosphere of maize plants. To this end the different types of soil were collected and total DNA extracted and used for a PCR-based 16S rDNA gene diversity survey of microbial communities (see Experimental procedures). Species richness was represented in rarefaction curves and was measured based on at least 220 sequences and the number of operational taxonomic units (OTUs) using a cut-off of 97% for sequence similarity, a commonly known level for comparative analysis of whole and partial 16S rRNA sequences (Konstantinidis et al., 2006). Rarefaction analysis was used to compare bacterial richness between the rhizosphere soil and bulk soil samples. Figure 1 is a rarefaction curve based on best match for each sequence of 16S rDNA genes and their frequency of recovery. The results show that as the number of sequences in the samples increased, the number of OTUs tended to level (Fig. 1, Fig. S1 for cut-off values different of 97% sequence similarity). The numbers of OTUs for a similar number of sequences were always higher in the bulk soil than in the rhizosphere.
Fig. 1.
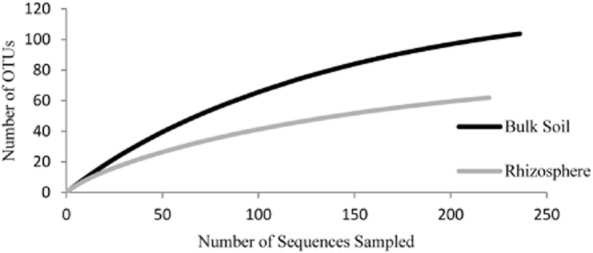
Rarefaction analysis for rhizosphere and bulk soil. Rarefaction curves were constructed using DOTUR software. Rarefaction is shown for OTU with differences that do not exceed 3%.
A series of statistical analyses were performed and several indexes related to biodiversity were calculated to estimate the biodiversity of samples (Table 2). While the Chao 1 index suggested that the maximum OTU value for bulk soil and rhizosphere should be 118 and 78, Good's coverage index gave 0.57 for bulk soil and 0.73 for the rhizosphere (Good, 1953; Zaballos et al., 2006). The Shannon index value was 4.40 for bulk soil and 3.42 for the rhizosphere, while the Simpson's index value was 0.01 for bulk soil and 0.059 for the rhizosphere. These results suggest that the bacterial community present in bulk soil seems more complex than that of the rhizosphere, although we consider that our analysis may underestimate the true richness of it because of the limited number of sequences we obtained, although the Chao 1 value versus the OTU coverage indicates that our analysis had sufficient depth.
Table 2.
Statistical indexes
| Bulk soil | Rhizosphere | |
|---|---|---|
| Good index | 0.57 | 0.73 |
| Shannon index | 4.4 | 3.42 |
| Simpson index | 0.01 | 0.059 |
| Chao 1 | 118 | 78 |
DOTUR software was used to compute the statistical indexes for the bacterial sequences.
Phylogenetic reconstruction showed that the sequences were unevenly scattered through the phylogenetic tree (see Figs S2–S4). In the rhizosphere niche six phyla groups were recovered, whereas nine phyla were recovered from bulk soils (Table 3). 16S rDNA gene sequences in the bulk soil belonged predominantly to three phyla including Acidobacteria (∼39%), Bacteroidetes (∼24%) and Proteobacteria (∼20%). Other typical soil microorganisms included Planctomycetes, Actinobacteria and uncultured members of the TM7 and the OP11 candidate divisions (non-culturable microbes) were found. Similar proportions of these phyla were reported in agricultural and forest soil samples (Roesch et al., 2007; Fulthorpe et al., 2008; Uroz et al., 2010).
Table 3.
Phylogenetic affiliation of bacterial 16S rRNA genesa
| Phylogenetic group | Bulk soil (%) | Rhizosphere (%) |
|---|---|---|
| Candidate division OP11 | 0.4 | 0 |
| Candidate division TM7 | 0.8 | 3.9 |
| Cyanobacteria | 2.9 | 0 |
| Actinobacteria | 2 | 1.3 |
| Acidobacteria | 38.8 | 6.9 |
| Planctomycetes | 2.1 | 0 |
| Deltaproteobacteria | 2.4 | 0 |
| Bacteroidetes | 23.7 | 1.7 |
| Chloroflexi | 1.2 | 0 |
| Alphaproteobacteria | 4.1 | 13.7 |
| Betaproteobacteria | 10.2 | 3 |
| Gammaproteobacteria | 6.5 | 65.2 |
| Firmicutes | 1.7 | |
| Unclassified Bacteria | 4.9 | 2.6 |
Percentage of clones assigned to known and candidate divisions from the 16S rRNA gene libraries from bulk soil and rhizosphere.
In the rhizosphere of Avena fatua DeAngelis and colleagues (2009) reported that a significantly larger number of live cells were detected in the rhizosphere in comparison with bulk soil; their study reported as many as 10-fold more cells detected in the root hairs and the root tip rhizosphere in comparison with bulk soil. In that study the authors used ribosomal RNA-targeted oligonucleotide microarrays (Phylochips) and identified the presence of typical rhizosphere phyla such as Proteobacteria and Firmicutes, as well as other less well-documented rhizosphere colonizers such as Actinobacteria, Verrucomicrobia and Nitrospira. Richness of Bacteroidetes and Actinobacteria decreased in soil close to the root tip in comparison with bulk soil, but then increased in older root areas.
The rhizosphere soil showed a shift in the most frequently represented microbes and an overall reduction in the number of phyla represented (Table 3). Weisskopf and colleagues (2005) also previously reported a decrease in the richness of bacterial communities from the bulk to the rhizosphere soil, when culturable bacteria were analysed.
The most predominant 16S rRNA gene sequences in the rhizosphere were those of Gammaproteobacteria (∼65%) followed by Alphaproteobacteria (∼14%) and Acidobacteria (∼7%) (Table 3). In a recent meta-analysis of 19 libraries of bacterial clones associated to the roots of 14 plant species, over 1200 distinguishable taxa from 35 different taxonomic orders were described (Hawkes et al., 2007). Proteobacteria dominated the rhizosphere in 16 of the 19 studies included, presumably because of their relatively rapid growth rates (Atlas and Bartha, 1998). Our observations that Proteobacteria are frequent in rhizosphere soils are in agreement with studies carried out with microarrays to detect soil bacteria by Sanguin and colleagues (2006). Our data also showed that the proportion of Actinobacteria found in bulk and rhizosphere soil is independent on the presence of plants. This finding is in agreement with the results by Acosta-Martínez and colleagues (2008), who found that levels of Acidobacteria were similar regardless of the type of plantation (grass or wheat) and land management practice.
Our overall results are in line with those of Kowalchuk and colleagues (2000), who showed that using culture-independent techniques wild plant species were able to influence the composition of bacterial diversity in the rhizosphere. In their specific study they compared the influence of Cynoglossum officinale (hound's tongue) and Cirsium vulgare (spear thistle) on soil-borne bacterial communities and found differences in the corresponding microbial communities of the rhizosphere.
The ability of plants to alter microbial diversity and distribution in the rhizosphere may be due to their ability to create a microenvironment that is rich in carbohydrates, carboxylic acids and amino acids, and therefore differences in plant exudates may be behind this discrimination (Grayston et al., 1998; Molina et al., 2000; Uroz et al., 2010). In agreement with the notion that the rhizosphere is more nutrient-rich niche than bulk soil, we found that the levels of alkaline phosphatase, β-glucosidase and dehydrogenase activities in bacterial cells recovered from the rhizosphere were statistically higher than the same activities assayed in cells recovered from bulk soil (Table 4); differences were statistically significant in Student's tests (P ≤ 0.05); this increase in activity probably reflected the induction of bacterial catabolic enzymes to nutrients in the exudates, as reported by Vílchez and colleagues (2000), who found a transient increase in proline degradation enzymes in response to maize exudates. Martínez-Iñigo and colleagues (2009) reported that in calcareous soils polluted with heavy metals the microbial enzymatic activity was higher in planted soils than in bare soils at the contamination level of 600 mg of total heavy metals per kilogram of soil. In this soil new bands appeared in the PCR–DGGE profiles of the rhizosphere bacterial community as a response to the exposure to heavy metals, which may indicate that the growth of certain microbes is favoured by the soil/plant interaction. Therefore, soil microorganisms in the rhizosphere show higher levels of activities related to C, N and P cycles, likely representing their induction in response to nutrients. This kind of orchestrated response is known to be under the control of multiple transcriptional regulators (Ishihama, 2010).
Table 4.
Phosphatase, β-glucosidase and dehydrogenase activities in rhizosphere soil and bulk soil
| Phosphatase | β-Glucosidase | Dehydrogenase | |
|---|---|---|---|
| Rhizosphere | 325 ± 40 | 320 ± 50 | 8 ± 1 |
| Bulk soil | 130 ± 15 | 30 ± 2 | 1.5 ± 0.3 |
Enzymatic activities measurements and units are described in Experimental procedures. The results are the average of three independent assays performed by duplicate. Data were analysed using STATGRAPHICS Plus Statistical Software (Statistical Graphics, Princeton, NJ, USA) and Student's t-test was used to compare mean values.
Previous studies have shown various degrees of a ‘rhizosphere effect’ using either culture-dependent (Miller et al., 1989; Germida et al., 1998; Grayston et al., 1998) or culture-independent strategies (Marilley and Aragno, 1999; Miethling et al., 2000; Duineveld et al., 2001; Smalla et al., 2001; Sanguin et al., 2006). The general results of these studies suggest that different plant species differ in the degree and manner in which they influence microbial community structure in the rhizosphere, as was indeed the case when microbial populations of oilseed rape were compared with those of strawberry (Duineveld et al., 2001; Smalla et al., 2001; Berg et al., 2005; Berg and Smalla, 2009). The effect of different plant species on soil microbial communities has been demonstrated for rhizosphere (Grayston et al., 1998; Söderberg et al., 2002; Iovieno et al., 2010) and bulk soil (Myers et al., 2001; Carney and Matson, 2006), both for trees (Saetre, 1998; Myers et al., 2001; Priha et al., 2001; Zak et al., 2003; Grayston and Prescott, 2005; Carney and Matson, 2006) and herbaceous plants (Söderberg et al., 2002; Zak et al., 2003). The influence of plants on the soil microbial community has even been found for different genotypes of the same species (Grayston et al., 1998; Schweitzer et al., 2008).
In this regard we have carried out detailed analyses of the relative distributions of the genera, families, orders and phyla between microbes in the bulk soil and in the maize rhizosphere (Table 3). These analyses are based on partial 16S rRNA sequence analyses and their location in phylogenetic trees based on the RDP programme (see Figs S2–S4). First, among the genera detected in these two niches only nine common family genera or candidate division were found, namely unclassified Sphingomonas, Acidobacteria GP6 and GP7, unclassified Chitinophagaceae, unclassified Rhizobiales, Pseudomonas, TM7, and unclassified Gammaproteobacteria and Lysobacter. Analysis of the eight most abundant genera detected in the bulk soil environment were Acidobacteria GP6, GP4 and GP7, Adheribacter, Hymenobacter, Massilia, and unclassified bacteria, each consisting of at least 5% of the total, with GP6 and GP4 being the most abundant (19% and 14% respectively). In the rhizosphere, Pseudomonas and Lysobacter genera were clearly dominant constituting to 45% of the total microbial abundance, followed by Pseudoaminobacter, unclassified Xanthomonadaceae and Acidobacteria GP7, each in the range of 5–10%.
Analysis of Proteobacteria in bulk soil revealed that Proteobacteria represent ∼23% of total sequences with Betaproteobacteria being the most prevalent (∼45% of total Proteobacteria), followed by gamma (∼27%), alpha (∼18%) and delta (11%). Among the Proteobacteria, Burkholderia was the most common genera followed by Xanthomonas. In the rhizosphere, analysis of the Proteobacteria phylum showed that there were significantly more Gammaproteobacteria (∼75%) than any other Proteobacteria with Pseudomonas spp. and Lysobacter spp. being the dominant genera. This contrasts with studies of the rhizosphere of grape in which there were significantly more Betaproteobacteria in the rhizosphere than in the bulk soil, and significantly more Alphaproteobacteria in the bulk soil than in rhizosphere (Sanguin et al., 2006; Haichar et al., 2008).
Bacterial communities are acknowledged as one of the major components of soil function, playing a key role in niche maintenance. Our study shows an increase in the proportion of Pseudomonas spp. and Lysobacter spp. in the rhizosphere. Pseudomonas spp. are well known root colonizers (Molina et al., 2000) and are able to proliferate by using plant-secreted amino acids such as proline, lysine, phenylalanine, glutamate and others (Vílchez et al., 2000; Espinosa-Urgel and Ramos, 2001; Herrera and Ramos, 2007). In addition, bacteria of this genus exhibit positive chemotaxis towards plant exudates (Espinosa-Urgel et al., 2002), a response in which several chemosensors such as McpS are involved (Lacal et al., 2011). Because of the parallel increase in the proportion of Lysobacter spp. and Pseudomonas spp. in the rhizosphere, we suggest that Lysobacter spp. could be both able to efficiently use the same carbon and nitrogen sources as Pseudomonas spp. and that bacteria of this genera are efficient colonizers of the rhizosphere of plants; however, this will need further in vitro assays with culturable Lysobacter spp. Our results showed that nitrogen-fixing microbes are of low abundance in this soil and do not apparently play a key role in the mobilization of nitrogen between rhizosphere and bulk soil; instead, microbes capable of metabolizing inorganic nitrogen are present, which is consistent with the historical use of inorganic nitrogen sources at this field site.
In short, our results suggest that the predominant bacterial populations in a carbonate-rich soil are influenced by plants and that this effect is most notable in the rhizosphere, defined here as the root surface and adhering soil. In our study we have analysed 16S rDNA gene sequences, and only assessed the detection of numerically predominant bacterial populations with Pseudomonas spp. and Lysobacter spp. as the dominant ones. Our results provide data on how certain bacterial populations become dominant in the rhizosphere through a mechanism that is most likely due to the microenvironment created by the presence of maize exudates and bacterial chemotaxis towards nutrients in the exudates. In general, this main conclusion in a soil with a relatively high pH is in agreement with studies that suggest that soil characteristics may be most important factor determining the dominant bacterial populations in bulk soil (Felske and Akkermans, 1998; Kowalchuk et al., 2000), while the microbial communities found in the rhizosphere are, to a greater extent, plant-driven.
Experimental procedures
Isolation of DNA from soil and rhizosphere samples
Five 1 kg pots were filled with soil collected at the Estación Experimental del Zaidín (Granada), [+37°9′56.50″N, −3°35′31.13″O] 678 m, and each planted with maize seeds. Plants were kept in a greenhouse with 12 h/12 h light–dark cycle, 50% humidity and watered daily. The soil physicochemical parameters were analysed at the ‘Instituto Agroalimentario de Atarfe’ (Table 1). Thirty corn seeds were surface sterilized according to Espinosa-Urgel and colleagues (2000) and sown in pots containing the soil. After 2 weeks maize plants were removed from the soil and the soil which tightly adhered to roots to the plants was separated using glass beads; this soil was the rhizosphere, whereas the soil that did not adhere was taken as the bulk soil. Bulk and rhizosphere soil samples were sieved through a 4 mm mesh (Molina et al., 2000).
Soil samples were processed immediately for DNA extraction. Several methods were used to extract DNA and in terms of quality of DNA we found that the most efficient was that in which total DNA was isolated directly from cells after matrix separation by density gradient centrifugation with Nycodenz (Axis-Shield PoC, Norway), as described by Ferrer and colleagues (2011). DNA was extracted using the GNOME®DNA commercial kit (QBIOgene) and visualized using 0.8% (wt/vol) agarose gel electrophoresis.
Construction of 16S RNA gene clone libraries, DNA sequencing and sequence analysis
For PCR amplification of the 16S rRNA gene serial dilutions of DNA template were used. An approximately 1450 bp amplification product was obtained using universal primers GM3F (5′-AGAGTTTGATCMTGGC-3′) and GM4R (5′-TACCTTGTTACGACTT-3′). Amplification was carried out in 50 μl reaction volume with 2.5 U recombinant Taq DNA polymerase, 25 ng of metagenomic DNA, 250 μM of each of the four deoxynucleotide triphosphates, 1.5 mM MgCl2, 200 nM of each primer and the appropriate buffer supplied by the manufacturer (Roche), according to the PCR protocol described by Uroz and colleagues (2010).
PCR amplicons were purified through 0.8% (wt/vol) agarose gels. DNA was excised using a QIAQUICK Gel Extraction Kit (Qiagen, Germany) and this DNA was ligated into the pGEM-T plasmid vector (Promega, Madison, WI, USA), with subsequent transformation into competent cells of Escherichia coli DH5α. DNA encoding bacterial 16S rRNA were sequenced using the M13 forward and M13 reverse primers. To minimize the effects of random sequencing errors, sequence chromatograms were manually checked to eliminate ambiguities. On average, this stringent trimming procedure reduced the number of sequences by 20% and the average size of the analysed sequences was about 700 bp.
Preliminary phylogenetic analysis of the 16S rRNA clones was performed using the Classifier tool of the Ribosomal Data Project (Cole et al., 2009) (confidence level of 85%). Sequences were checked for possible chimeric origin by using the Ribosomal Database Project's CheckChimera program, which is based on the Pintail algorithm (Ashelford et al., 2005). Then, phylogenetic inference was carried out using the ARB software package (Ludwig et al., 2004). Sequences were automatically aligned using SINA aligner against SILVA SSURef 100 (Pruesse et al., 2007) and LTPs100 (Yarza et al., 2008). The alignments were manually inspected to correct inaccurately misplaced bases. Two independent reference phylogenetic trees were reconstructed to improve resolution at lower taxonomic levels – one comprising only members of the phylum Proteobacteria and a second one containing the remaining bacterial phyla. The phylogeny was reconstructed with the neighbour-joining algorithm using the Jukes-Cantor correction.
ARB-generated 16S sequence alignments were used to create Jukes-Cantor corrected distance matrices. These matrices were used as input for the DOTUR program (see below, Schloss and Handelsman, 2005).
Nucleotide sequence accession numbers
The 16S rRNA gene sequences of the samples analysed in this study were deposited at the GenBank under accession numbers JN366808–JN367265.
Index calculations
The microbial diversity was evaluated using several species-diversity indices (Atlas and Bartha, 1998). The DOTUR software program was used to compute the statistical indexes and to generate rarefaction curves (Heck et al., 1975). For both libraries the coverage was estimated using the Good index (Good, 1953), and the diversity was calculated using the Shannon-Weiner and Simpson's indexes (Magurran, 1998). Sequences were grouped at equal or higher than 97% identity as the standard cut-off. In addition, we determined the non-parametric index Chao as an estimator of species richness (Heltsche and Forrester, 1983; Chao, 1984; Colwell and Coddington, 1994).
Determination of soil enzymatic activities
Dehydrogenase activity was determined by the reduction of 2-p-iodo-nitrophenyl-tetrazolium chloride (INT) to iodo-nitrophenyl formazan (INTF) as described by Skujins (1976) and modified by García-Gil and colleagues (2000). Dehydrogenase activity was measured using 1 g of soil, following incubation in the dark with 0.2 ml of 0.4% INT for 20 h at 37°C. The INTF was extracted with a mixture of acetone : tetrachloroethene (1.5:1) by shaking vigorously for 2 min and measuring absorbance at 490 nm in a spectrophotometer. Assays without soil and without INT were carried out simultaneously as controls. Activity is expressed as μg INTF produced g−1 dry soil h−1.
Phosphatase and β-glucosidase activities were determined using disodium p-nitrophenyl phosphate (PNPP, 0.115 M) and p-nitrophenyl-β-D-glucopyranoside (PNG, 0.05 M) as substrates respectively. These assays are based on the production and detection of p-nitrophenol (PNP). Two millilitres of 0.1 M maleate buffer (pH 6.5 for both phosphatase and β-glucosidase activities) and 0.5 ml of substrate were added to a 0.5 g sample and incubated at 37°C for 2 h. The reaction was determined by adding 0.5 M CaCl2 and 2 ml of 0.5 M NaOH and the mixture was centrifuged at 3500 g for 10 min. The amount of PNP was determined using a spectrophotometer at 398 nm (Tabatabai and Bremner, 1969). The same procedure was followed for the controls except that the substrate was added to the soil immediately before stopping the reaction. Activity is expressed as μg PNP production g−1 dry soil h−1.
Acknowledgments
This work was funded by grants from the Ministry of Science and Innovation (Consolider-Ingenio CSD2007-00005; and Explora-Reverse) and FEDER from the Junta de Andalucía (Grupo CVI-191). We thank M.M. Fandila for secretarial assistance and Ben Pakuts for critically reading the manuscript.
Conflict of interest
None declared.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Fig. S1. Rarefaction analysis for (A) bulk soil and (B) rhizosphere samples based on pairwise distance. Rarefaction is shown for OTUs with differences that do not exceed 3%, 5%, or 10%.
Fig. S2. Neighbour-joining tree of proteobacterial SSU rRNA gene sequences from clone libraries established from bulk soil community DNA. Clones sequenced in this work are marked in red.
Fig. S3. Neighbour-joining tree of non-proteobacterial SSU rRNA gene sequences from clone libraries established from bulk soil community DNA. Clones sequenced in this work are marked in red.
Fig. S4. Neighbour-joining tree of proteobacterial SSU rRNA gene sequences from clone libraries established from rhizosphere community DNA. Clones sequenced in this work are marked in red.
References
- Acosta-Martínez V, Dowd S, Sun Y, Allen V. Tag-encoded pyrosequencing analysis of bacterial diversity in a single soil type as affected by management and land use. Soil Biol Biochem. 2008;40:2762–2770. [Google Scholar]
- Ashelford KE, Chuzhanova NA, Fry JC, Jones AJ, Weightman AJ. At least 1 in 20 16S rRNA sequence records currently held in public repositories is estimated to contain substantial anomalies. Appl Environ Microbiol. 2005;71:7724–7736. doi: 10.1128/AEM.71.12.7724-7736.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atlas RM, Bartha R. Microbial Ecology. 4th edn. Redwood City, CA, USA: Benjamin/Cummings Publishing Company; 1998. Fundamentals and applications; pp. 523–530. [Google Scholar]
- Berg G, Smalla K. Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. FEMS Microbiol Ecol. 2009;68:1–13. doi: 10.1111/j.1574-6941.2009.00654.x. [DOI] [PubMed] [Google Scholar]
- Berg G, Zachow C, Lottmann J, Götz M, Costa R, Smalla K. Impact of plant species and site on rhizosphere-associated fungi antagonistic to Verticillium dahliae kleb. Appl Environ Microbiol. 2005;71:4203–4213. doi: 10.1128/AEM.71.8.4203-4213.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brimecombe MJ, DeLeij FA, Lynch JM. The effect of root exudates on rhizosphere microbial populations. In: Pinton R, Varanini Z, Nannipieri P, editors. The Rhizosphere: Biochemistry and Organic Substances at the Soil-Plant Interface. New York, NY, USA: Marcel-Dekker; 2001. pp. 95–140. [Google Scholar]
- Bringhurst RM, Cardon ZG, Gage DJ. Galactosides in the rhizosphere: utilization by Sinorhizobium meliloti and development of a biosensor. Proc Natl Acad Sci USA. 2001;98:4540–4545. doi: 10.1073/pnas.071375898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney KM, Matson PA. The influence of tropical plant diversity and composition on soil microbial communities. Microb Ecol. 2006;52:226–238. doi: 10.1007/s00248-006-9115-z. [DOI] [PubMed] [Google Scholar]
- Chao A. Nonparametric estimation of the number of classes in a population. Scand J Stat. 1984;11:265–270. [Google Scholar]
- Chao A, Ma CA, Yang MCK. Estimating the number of classes via sample coverage. J Am Stat Assoc. 1992;87:210–217. [Google Scholar]
- Cocking EC. Endophytic colonisation of plant roots by nitrogen-fixing bacteria. Plant Soil. 2003;252:169–175. [Google Scholar]
- Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, et al. The ribosomal database project: improvement alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009;37:D141–D145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colwell RK, Coddington JA. Estimating terrestrial biodiversity through extrapolation. Philos Trans R Soc Lond B Biol Sci. 1994;345:101–118. doi: 10.1098/rstb.1994.0091. [DOI] [PubMed] [Google Scholar]
- Curtis TP, Sloan WT, Scannell JC. Estimating prokaryotic diversity and its limits. Proc Natl Acad Sci USA. 2002;99:10494–10499. doi: 10.1073/pnas.142680199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeAngelis KM, Brodie EL, DeSantis TZ, Andersen GL, Lindow SE, Firestone MK. Selective progressive response of soil microbial community to wild oat roots. ISME J. 2009;3:168–178. doi: 10.1038/ismej.2008.103. [DOI] [PubMed] [Google Scholar]
- DeLong EF. Archaea in coastal marine environments. Proc Natl Acad Sci USA. 1992;89:5685–5689. doi: 10.1073/pnas.89.12.5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duineveld BM, Kowalchuk GA, Keizer A, Van Elsas JD, Van Veen JA. Analysis of the bacterial communities in the rhizosphere of chrysanthemum via denaturing gradient gel electrophoresis of PCR amplified 16S ribosomal RNA as well as DNA fragments coding for 16S rRNA. Appl Environ Microbiol. 2001;67:172–178. doi: 10.1128/AEM.67.1.172-178.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa-Urgel M, Ramos JL. Expression of a Pseudomonas putida aminotransferase involved in lysine catabolism is induced in the rhizosphere. Appl Environ Microbiol. 2001;67:5219–5224. doi: 10.1128/AEM.67.11.5219-5224.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa-Urgel M, Salido A, Ramos JL. Genetic analysis of functions involved in adhesion of Pseudomonas putida to seeds. J Bacteriol. 2000;182:2363–2369. doi: 10.1128/jb.182.9.2363-2369.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa-Urgel M, Kolter R, Ramos JL. Root colonization by Pseudomonas putida love at first sight. Microbiology. 2002;148:341–343. doi: 10.1099/00221287-148-2-341. [DOI] [PubMed] [Google Scholar]
- Felske A, Akkermans ADL. Spatial homogeneity of abundant bacterial 16S rRNA molecules in grassland soils. Microb Ecol. 1998;36:31–36. doi: 10.1007/s002489900090. [DOI] [PubMed] [Google Scholar]
- Ferrer M, Guazzaroni ME, Richter M, García-Salamanca A, Yarza P, Suárez-Suárez A, et al. Taxonomic and functional metagenomic profiling of the microbial community in the anoxic sediment of a sub-saline shallow lake (Laguna de Carrizo, Central Spain) Microb Ecol. 2011;62:824–837. doi: 10.1007/s00248-011-9903-y. [DOI] [PubMed] [Google Scholar]
- Fierer N, Jackson RB. The diversity and biogeography of soil bacterial communities. Proc Natl Acad Sci USA. 2006;103:626–631. doi: 10.1073/pnas.0507535103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulthorpe RR, Roesch LF, Riva A, Triplett EW. Distantly sampled soils carry few species in common. ISME J. 2008;2:901–910. doi: 10.1038/ismej.2008.55. [DOI] [PubMed] [Google Scholar]
- García-Gil JC, Plaza C, Soler-Rovira P, Polo A. Long-term effects of municipal solid waste compost application on soil enzyme activities and microbial biomass. Soil Biol Biochem. 2000;32:1907–1913. [Google Scholar]
- Germida JJ, Siciliano SD, De Freitas RJ, Seib AM. Diversity of root-associated bacteria associated with filed-grown canola (Brassica napus L.) and wheat (Triticum aestivum L.) FEMS Microbiol Ecol. 1998;26:43–50. [Google Scholar]
- Giovannoni SJ, Britschgi TB, Moyher CL, Field HG. Genetic diversity in Sargasso Sea bacterioplankton. Nature. 1990;345:60–63. doi: 10.1038/345060a0. [DOI] [PubMed] [Google Scholar]
- Good IJ. The population frequencies of species and the estimation of population parameters. Biometrika. 1953;40:237–264. [Google Scholar]
- Grayston SJ, Prescott CE. Microbial communities in forest floors under four tree species in coastal British Columbia. Soil Biol Biochem. 2005;37:1157–1167. [Google Scholar]
- Grayston SJ, Wang S, Campbell CD, Edwards AC. Selective influence of plant species on microbial diversity in the rhizosphere. Soil Biol Biochem. 1998;30:369–378. [Google Scholar]
- Haichar FZ, Marol C, Berge O, Rangel-Castro JI, Prosser JI, Balesdent J, et al. Plant host habitat and root exudates shape soil bacterial community structure. ISME J. 2008;2:1221–1230. doi: 10.1038/ismej.2008.80. [DOI] [PubMed] [Google Scholar]
- Hawkes CV, DeAngelis KM, Firestone MK. Root interactions with soil microbial communities and processes. In: Cardon Z, Whitbeck J, editors. The Rhizosphere. New York, NY, USA: Elsevier; 2007. pp. 1–31. [Google Scholar]
- Heck KL, Van Belle G, Simberloff D. Explicit calculation of the rarefaction diversity measurement and the determination of sufficient sample size. Ecology. 1975;56:1459–1461. [Google Scholar]
- Heijnen CE, Page S, Van Elsas JD. Metabolic activity of Flavobacterium strain P25 during starvation and after introduction into bulk soil and the rhizosphere of wheat. FEMS Microbiol Ecol. 1995;18:129–138. [Google Scholar]
- Heltsche JF, Forrester NE. Estimating species richness using the jackknife procedure. Biometrics. 1983;39:1–12. [PubMed] [Google Scholar]
- Herman DJ, Johnson KK, Jaeger CH, Schwartz E, Firestone MK. Root influence on nitrogen mineralization and nitrification in Avena barbata rhizosphere soil. Soil Sci Soc Am J. 2006;70:1504–1511. [Google Scholar]
- Herrera MC, Ramos JL. Catabolism of phenylalanine by Pseudomonas putida: the NtrC-family PhhR regulator binds to two sites upstream from the phhA gene and stimulates transcription with σ70. J Mol Biol. 2007;366:1374–1386. doi: 10.1016/j.jmb.2006.12.008. [DOI] [PubMed] [Google Scholar]
- Hewson I, Vargo GA, Fuhrman JA. Bacterial diversity in shallow oligotrophic marine benthos and overlying waters: effects of virus infection containment, and nutrient enrichment. Microb Ecol. 2003;46:322–336. doi: 10.1007/s00248-002-1067-3. [DOI] [PubMed] [Google Scholar]
- Huber JA, Butterfield DA, Baross JA. Temporal changes in archaeal diversity and chemistry in a mid-ocean ridge subseafloor habitat. Appl Environ Microbiol. 2002;68:1585–1594. doi: 10.1128/AEM.68.4.1585-1594.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iovieno P, Alfani A, Bááth E. Soil microbial community structure and biomass as effected by Pinus pinea plantation in two Mediterranean areas. Appl Soil Ecol. 2010;45:56–63. [Google Scholar]
- Ishihama A. Prokaryotic genome regulation: multifactor promoters, multitarget regulators and hierarchic networks. FEMS Microbiol Rev. 2010;34:628–645. doi: 10.1111/j.1574-6976.2010.00227.x. [DOI] [PubMed] [Google Scholar]
- Jones RT, Robertson MS, Lauber CL, Hamady M, Knight R, Fiere N. A comprehensive survey of soil acidobacterial diversity using pyrosequencing and clone library analyses. ISME J. 2009;3:442–453. doi: 10.1038/ismej.2008.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinidis KT, Ramette A, Tiedje JM. Toward a more robust assessment of intraspecies diversity, using fewer genetic markers. Appl Environ Microbiol. 2006;72:7286–7293. doi: 10.1128/AEM.01398-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalchuk GA, Stienstra AW, Heilig GHJ, Stephen JR, Woldendorp JW. Changes in the community structure of ammonia-oxidizing bacteria during secondary succession of calcareous grasslands. Environ Microbiol. 2000;2:99–110. doi: 10.1046/j.1462-2920.2000.00080.x. [DOI] [PubMed] [Google Scholar]
- Kowalchuk GA, Buma DS, de Boer W, Klinkhamer PGL, van Veen JA. Effects of above-ground plant species composition and diversity on the diversity of soil-borne microorganisms. Antonie Van Leeuwenhoek. 2002;81:509–520. doi: 10.1023/a:1020565523615. [DOI] [PubMed] [Google Scholar]
- Lacal J, Muñoz-Martínez F, Reyes-Darias JA, Duque E, Matilla M, Segura A, et al. Bacterial chemotaxis towards aromatic hydrocarbons in Pseudomonas. Environ Microbiol. 2011;13:1733–1744. doi: 10.1111/j.1462-2920.2011.02493.x. [DOI] [PubMed] [Google Scholar]
- Lubeck PS, Hansen M, Sorensen J. Simultaneous detection of the establishment of seed-inoculated Pseudomonas fluorescens strain DR54 and native soil bacteria on sugar beet root surfaces using fluorescence antibody and in situ hybridization techniques. FEMS Microbiol Ecol. 2000;33:11–19. doi: 10.1111/j.1574-6941.2000.tb00721.x. [DOI] [PubMed] [Google Scholar]
- Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar BA, et al. ARB: a software environment for sequence data. Nucleic Acids Res. 2004;32:1363–1371. doi: 10.1093/nar/gkh293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magurran AE. Population differentiation without speciation. Philos Trans R Soc Lond B Biol Sci. 1998;353:275–286. [Google Scholar]
- Marilley L, Aragno M. Phylogenetic diversity of bacterial communities differing in degree of proximity of Lolium perenne and Trifolium repens roots. Appl Soil Ecol. 1999;13:127–136. [Google Scholar]
- Martínez-Iñigo MJ, Pérez-Sanz A, Ortiz I, Alonso J, Alarcon R, García P, Lobo MC. Bulk soil and rhizosphere bacterial community PCR-DGGE profiles and β-galactosidase activity as indicators of biological quality in soils contaminated by heavy metals and cultivated with Silene vulgaris (Moench) Garcke. Chemosphere. 2009;75:1376–1381. doi: 10.1016/j.chemosphere.2009.03.014. [DOI] [PubMed] [Google Scholar]
- Matilla MA, Espinosa-Urgel M, Rodríguez-Herva JJ, Ramos JL, Ramos-González MI. Genomic analysis reveals the major driving forces of bacterial life in the rhizospheer. Genome Biol. 2007;8:R179. doi: 10.1186/gb-2007-8-9-r179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miethling R, Wieland G, Backhaus H, Tebbe CC. Variation of microbial rhizosphere communities in response to crop species, soil origin, and inoculation with Sinorhizobium meliloti L33. Microb Ecol. 2000;41:43–56. doi: 10.1007/s002480000021. [DOI] [PubMed] [Google Scholar]
- Miller HJ, Henken G, Veen JA. Variation and composition of bacterial populations in the rhizosphere of maize, wheat, and grass cultivars. Can J Microbiol. 1989;35:656–660. [Google Scholar]
- Molina L, Ramos C, Duque E, Ronchel MC, García JM, Wyke L, Ramos JL. Survival of Pseudomonas putida KT2440 in soil and in the rhizosphere of plants under greenhouse and environmental conditions. Soil Biol Biochem. 2000;32:315–321. [Google Scholar]
- Myers RT, Zak DR, White DC, Peacock A. Landscape-level patterns of microbial community composition and substrate use in upland forest ecosystems. Soil Sci Soc Am J. 2001;65:359–367. [Google Scholar]
- Nunes da Rocha U, van Overbeek L, van Elsas JD. Exploration of hitherto-uncultured bacteria from the rhizosphere. FEMS Microbiol Ecol. 2009;69:313–328. doi: 10.1111/j.1574-6941.2009.00702.x. [DOI] [PubMed] [Google Scholar]
- Oline DK. Phylogenetic comparisons of bacterial communities from serpentine and nonserpentine soils. Appl Environ Microbiol. 2006;72:6965–6971. doi: 10.1128/AEM.00690-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace NR. A molecular view of microbial diversity and the biosphere. Science. 1997;276:734–740. doi: 10.1126/science.276.5313.734. [DOI] [PubMed] [Google Scholar]
- Paul EA, Clark FE. Soil Microbiology and Biochemistry. 2nd edn. San Diego, CA, USA: Academic Press; 1996. [Google Scholar]
- Pennanen T, Liski J, Bååth E, Kitunen VV, Uotila J, Westman CJ, et al. Structure of the microbial communities in coniferous forest soils in relation to site fertility and stand development stage. Microb Ecol. 1999;38:168–179. doi: 10.1007/s002489900161. [DOI] [PubMed] [Google Scholar]
- Preston GM, Bertrand N, Rainey PB. Type III secretion in plant growth promoting Pseudomonas fluorescens SBW25. Mol Microbiol. 2001;41:999–1014. doi: 10.1046/j.1365-2958.2001.02560.x. [DOI] [PubMed] [Google Scholar]
- Priha O, Grayston SJ, Hiukka R, Pennanen T, Smolander A. Microbial community structure and characteristics of the organic matter in soils under Pinus sylvestrisPicea abies and Betula pendula at two forest sites. Biol Fertil Soils. 2001;33:17–24. [Google Scholar]
- Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glöckner FO. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35:7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambelli A. The rhizosphere of mycorrhizae. In: Marks GC, Kozlowski TT, editors. Ectomycorrhizae, Their Ecology and Physiology. New York, NY, USA: Academic Press; 1973. pp. 299–349. [Google Scholar]
- Rappé MS, Giovannoni SJ. The uncultured microbial majority. Annu Rev Microbiol. 2003;57:369–394. doi: 10.1146/annurev.micro.57.030502.090759. [DOI] [PubMed] [Google Scholar]
- Rodríguez H, Fraga R, González T, Bahan Y. Genetics of phosphate solubilization and its potential applications for improving plant growth-promoting bacteria. Plant Soil. 2006;287:15–21. [Google Scholar]
- Roesch LF, Fulthorpe RR, Riva A, Casella G, Hadwin AK, Kent AD, et al. Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J. 2007;1:283–290. doi: 10.1038/ismej.2007.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saetre P. Decomposition, microbial community structure and earthworm effects along a birch-spruce soil gradient. Ecology. 1998;79:834–846. [Google Scholar]
- Sanguin H, Remenant B, Dechesne A, Thioulouse J, Vogel TM, Nesme X, et al. Potential of a 16S rRNA-based taxonomic microarray for analyzing the rhizosphere effects of maize on Agrobacterium spp. and bacterial communities. Appl Environ Microbiol. 2006;72:4302–4312. doi: 10.1128/AEM.02686-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Handelsman J. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol. 2005;71:1501–1506. doi: 10.1128/AEM.71.3.1501-1506.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer JA, Bailey JK, Fischer DG, LeRoy CJ, Lonsdorf EV, Whitham TG, Hart SC. Plant–soil–microorganism interactions: heritable relationship between plant genotype and associated soil microorganisms. Ecology. 2008;89:773–781. doi: 10.1890/07-0337.1. [DOI] [PubMed] [Google Scholar]
- Shaw LJ, Morris P, Hooker JE. Perception and modification of plant flavonoid signals by rhizosphere microorganisms. Environ Microbiol. 2006;8:1867–1880. doi: 10.1111/j.1462-2920.2006.01141.x. [DOI] [PubMed] [Google Scholar]
- Skujins J. Extracellular enzymes in soil. Crit Rev Microbiol. 1976;4:383–421. doi: 10.3109/10408417609102304. [DOI] [PubMed] [Google Scholar]
- Smalla K, Wieland G, Buchner A, Zock A, Parzy J, Kaiser S, et al. Bulk and rhizosphere soil bacterial communities studied by denaturing gradient gel electrophoresis: plant-dependent enrichment and seasonal shifts revealed. Appl Environ Microbiol. 2001;67:4742–4751. doi: 10.1128/AEM.67.10.4742-4751.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalla K, Oros-Sichler M, Milling A, Heuer H, Baumgarte S, Becker R, et al. Bacterial diversity of soils assessed by DGGE, T-RFLP and SSCP fingerprints of PCR-amplified 16S rRNA gene fragments: do the different methods provide similar results? J Microbiol Methods. 2007;69:470–479. doi: 10.1016/j.mimet.2007.02.014. [DOI] [PubMed] [Google Scholar]
- Söderberg KH, Olsson PA, Baath E. Structure and activity of the bacterial community in the rhizosphere of different plant species and the effect of arbuscular mycorrhizal colonisation. FEMS Microbiol Ecol. 2002;40:223–231. doi: 10.1111/j.1574-6941.2002.tb00955.x. [DOI] [PubMed] [Google Scholar]
- Tabatabai MA, Bremner JM. Use of p-nitrophenylphosphate for assay of soil phosphatase activity. Soil Biol Biochem. 1969;1:301–307. [Google Scholar]
- Uroz S, Buée M, Murat C, Frey-Klett P, Martin F. Pyrosequencing reveals a contrasted bacterial diversity between oak rhizosphere and surrounding soil. Environ Microbiol Rep. 2010;2:281–288. doi: 10.1111/j.1758-2229.2009.00117.x. [DOI] [PubMed] [Google Scholar]
- Vílchez S, Manzanera M, Ramos JL. Control of expression of divergent Pseudomonas putida put promoters for proline catabolism. Appl Environ Microbiol. 2000;66:5221–5225. doi: 10.1128/aem.66.12.5221-5225.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisskopf L, Fromin N, Tomasi N, Aragno M, Martinoia E. Secretion activity of white lupin's cluster roots influences bacterial abundance, function and community structure. Plant Soil. 2005;268:181–194. [Google Scholar]
- Yarza P, Richter M, Peplies J, Euzéby J, Amann R, Schleifer K-H, et al. The All-Species Living Tree Project: a 16S rRNA-based phylogenetic tree of all sequenced type strains. Syst Appl Microbiol. 2008;31:241–250. doi: 10.1016/j.syapm.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Zaballos M, López-López A, Ovreas L, Galán-Bartual S, D'Auria G, Alba-Casado J, et al. Comparison of prokaryotic diversity at offshore oceanic locations reveals a different microbiota in the Mediterranean Sea. FEMS Microbiol Ecol. 2006;56:389–405. doi: 10.1111/j.1574-6941.2006.00060.x. [DOI] [PubMed] [Google Scholar]
- Zak DR, Holmes WE, White DC, Peacock AD, Tilman D. Plant diversity, soil microbial communities, and ecosystem function: are there any links? Ecology. 2003;84:2042–2050. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.