

Summary
The growth phase during which probiotic bacteria are harvested and consumed can strongly influence their performance as health‐promoting agents. In this study, global transcriptomic and proteomic changes were studied in the widely used probiotic Lactobacillus rhamnosus GG during growth in industrial‐type whey medium under strictly defined bioreactor conditions. The expression of 636 genes (P ≤ 0.01) and 116 proteins (P < 0.05) changed significantly over time. Of the significantly differentially produced proteins, 61 were associated with alterations at the transcript level. The most remarkable growth phase‐dependent changes occurred during the transition from the exponential to the stationary growth phase and were associated with the shift from glucose fermentation to galactose utilization and the transition from homolactic to mixed acid fermentation. Furthermore, several genes encoding proteins proposed to promote the survival and persistence of L. rhamnosus GG in the host and proteins that directly contribute to human health showed temporal changes in expression. Our results suggest that L. rhamnosus GG has a highly flexible and adaptable metabolism and that the growth stage during which bacterial cells are harvested and consumed should be taken into consideration to gain the maximal benefit from probiotic bacteria.
Introduction
Lactobacillus rhamnosus GG (ATCC 53103) is one of the most intensively studied probiotic strains worldwide. It has most of the characteristics generally desired for a good probiotic bacterium, including the ability to survive passage through and to colonize the human gastrointestinal tract (GIT) (Goldin et al., 1992; Alander et al., 1997; 1999). Adhesion capacity is a prerequisite for colonization, and L. rhamnosus GG has been shown to adhere efficiently to both intestinal mucus and epithelial cells (Jacobsen et al., 1999; Tuomola et al., 2000; Ouwehand et al., 2001). Consumption of L. rhamnosus GG promotes human health by reducing the risk of nosocomial rotavirus‐related diarrhoea in infants (Szajewska et al., 2001) and shortening the duration of acute diarrhoea (Szajewska et al., 2007, recently reviewed by Guarino et al., 2009). In addition to its influence on the GIT, L. rhamnosus GG have other beneficial effects, including reducing upper respiratory tract infections among children in day care (Hatakka et al., 2001; Hojsak et al., 2010), decreasing the risk of developing atopic eczema (Kalliomäki et al., 2001; 2003; 2007), alleviating the symptoms of eczema (Majamaa and Isolauri, 1997; Isolauri et al., 2000) and decreasing the risk of dental caries in children (Näse et al., 2001). So far, the molecular mechanisms identified as being responsible for the health‐promoting effects of lactobacilli mainly relate to immunomodulation, intestinal epithelial barrier protection and microbial balance maintenance (reviewed by Lebeer et al., 2008; 2010). The recently published genome sequence of L. rhamnosus GG (Kankainen et al., 2009) will boost mechanistic and functional genomic research of this well‐known probiotic strain.
In industrial production, dairy strains, including probiotics, are commonly harvested during the late exponential or stationary growth phase to ensure high cell numbers. However, the desired probiotic‐associated factors are not necessarily expressed at the highest levels during the stationary phase of growth. For example, cell surface properties of L. rhamnosus GG have been shown to change during growth, influencing the strain's ability to adhere to epithelial cells (Deepika et al., 2009). Furthermore, consumption of another widely recognized probiotic bacterium, Lactobacillus plantarum, at different growth phases (mid‐exponential or stationary) induced different transcriptional responses in human duodenal mucosa (van Baarlen et al., 2009). Hence, when aiming to optimize the health‐promoting properties of probiotic bacteria in large‐scale industrial cultivations, it is essential to determine the most favorable growth conditions and phase for cell harvesting.
Functional genomics and proteomics analyses are useful tools for identifying the molecular mechanisms behind the health‐promoting features of probiotic lactic acid bacteria. Transcriptomics provides detailed information about global changes in gene expression under particular conditions. Furthermore, microarrays enable reliable measurement of the expression of all genes on the array, regardless of the subcellular location of their corresponding proteins. However, only changes in mRNA level can be measured with this tool, while most biological phenomena result from protein function. In addition, changes occurring at the post‐transcriptional level (due to proteolysis, charged modification, etc.) can be investigated only with proteomic tools. Therefore, it is advisable to use both transcriptomic and proteomic approaches when linking genomic sequences to potential biological functions. Transcript or protein level approaches have been used to investigate global changes in other probiotic lactic acid bacteria during growth transitions (Cohen et al., 2006; Koistinen et al., 2007; Azcarate‐Peril et al., 2009). For example, transcriptomic analysis of Lactobacillus acidophilus cultured in skim milk indicated temporal expression changes in genes involved in the proteolytic system, carbohydrate utilization, adhesion and cellular responses to stress (Azcarate‐Peril et al., 2009). In L. plantarum, protein expression profiles demonstrated differences in several anabolic and stress response pathways during different growth phases (Cohen et al., 2006). A comparison of the proteomes of two L. plantarum strains during growth demonstrated the growth phase‐dependent expression of proteins associated with energy metabolism, such as glycolysis, the phosphoketolase pathway and ribose metabolism (Koistinen et al., 2007).
The global omics‐level analyses conducted on the probiotic L. rhamnosus GG so far include two proteomic studies (Koskenniemi et al., 2009; Sánchez et al., 2009) and a study combining both proteomic and transcriptomic approaches (Koskenniemi et al., 2011). Sánchez and colleagues (2009) tested different extraction methods for surface‐associated proteins, and could identify 18 different possibly surface‐associated proteins of L. rhamnosus GG. Comparison of the proteomes of L. rhamnosus GG grown in an industrial‐type whey medium (the same as used in this study) and in a laboratory medium demonstrated fundamental effects of culture conditions on the protein production of L. rhamnosus GG (Koskenniemi et al., 2009). In a recent study, transcript and protein level responses of L. rhamnosus GG towards bile were shown to include diverse and specific changes in general stress responses as well as alterations in cell envelope‐related functions (Koskenniemi et al., 2011). The present study involves complementary approaches, i.e. DNA microarray analyses combined with two‐dimensional difference gel electrophoresis (2‐D DIGE), to assess global changes in the transcriptome and proteome of probiotic L. rhamnosus GG during growth in industrial‐type whey medium under strictly controlled bioreactor conditions. In particular, this study aimed to determine whether potential probiotic‐associated factors were affected in a growth phase‐dependent fashion during fermentation in whey.
Results and discussion
Expression profile of L. rhamnosus GG during growth in industrial‐type whey medium
In the present study, 2‐D DIGE and whole‐genome DNA microarrays were used to determine how global gene expression in the widely used probiotic, L. rhamnosus GG, changed over time during growth in industrial‐type whey medium under strictly defined bioreactor conditions (Fig. 1). Overall, the transcript levels of 636 genes were significantly changed (≥ 2‐fold difference, P ≤ 0.01) at one or more time points, representing 22% of the complete genome of L. rhamnosus GG (Fig. 2). This is comparable to results from a previous study, where 21% of the ORFs of the L. acidophilus genome were differentially expressed during shift from exponential to stationary growth phase in skim milk (Azcarate‐Peril et al., 2009). Expression profiles of the 636 significantly differentially expressed genes were clustered into 16 groups (Fig. S1, Table S1). A clear difference could be seen between the transcription profiles of the mid‐exponential (4 h) and stationary growth phases (20 and 31 h), although only a few genes were down‐ (five genes) or upregulated (eight genes) during the whole progression of growth (Fig. 2). Interestingly, clusters of orthologous groups (COGs) classification of significantly differentially expressed genes showed that the majority of genes regulated at time points 12, 16, 20 and 31 h, compared with the 4 h time point, could not be included in any COG category (Fig. 3). At the 20 h time point, Carbohydrate transport and metabolism (G), Energy production and conversion (C) and Transcription (K) seemed to be coordinately upregulated when compared with 4 h, while most of the downregulated genes were involved in Amino acid transport and metabolism (E) or were categorized as ‘General function prediction only’ (R). At 31 h, most of the upregulated genes were assigned to the G and C categories, and the majority of downregulated genes belonged to the Translation, ribosomal structure and biogenesis (J) and Nucleotide transport and metabolism (F) categories.
Figure 1.
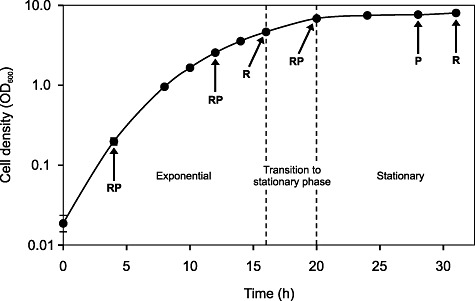
Growth curve of L. rhamnosus GG in whey medium at 37°C at pH 5.8. Cell density values represent the mean of four individual pH‐controlled bioreactor cultivations, and error bars represent the standard deviation. RNA samples were taken at five different time points representing mid‐exponential, late exponential, stationary transition point, early stationary and late stationary phases, while protein samples were collected at four different time points (the same as the RNA sampling phases, excluding the stationary transition point). The time points at which the RNA (R) and protein (P) samples were taken are indicated by arrows.
Figure 2.
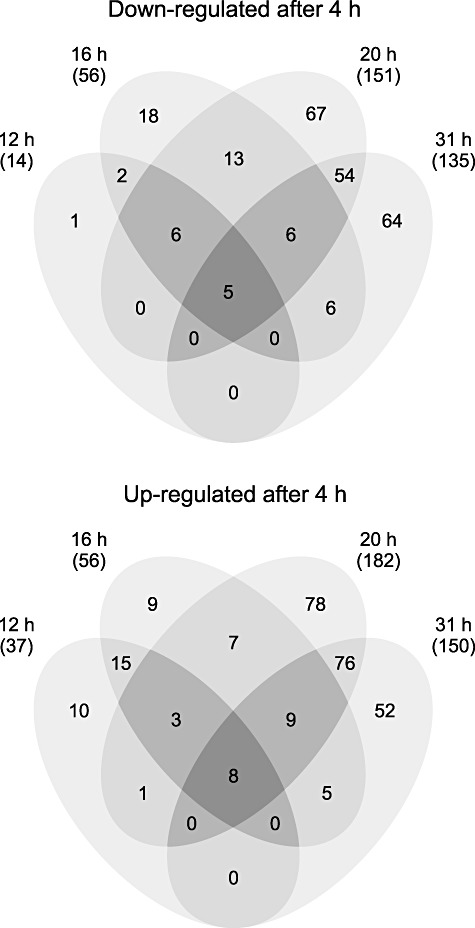
Venn diagrams showing the numbers of significantly differentially transcribed genes (≥ 2‐fold difference, P ≤ 0.01) during growth in whey. Time points at 12 h, 16 h, 20 h and 31 h were compared with the time point at 4 h (4 h, mid‐exponential; 12 h, late exponential; 16 h, stationary transition point; 20 h, early stationary; and 31 h, late stationary phase).
Figure 3.
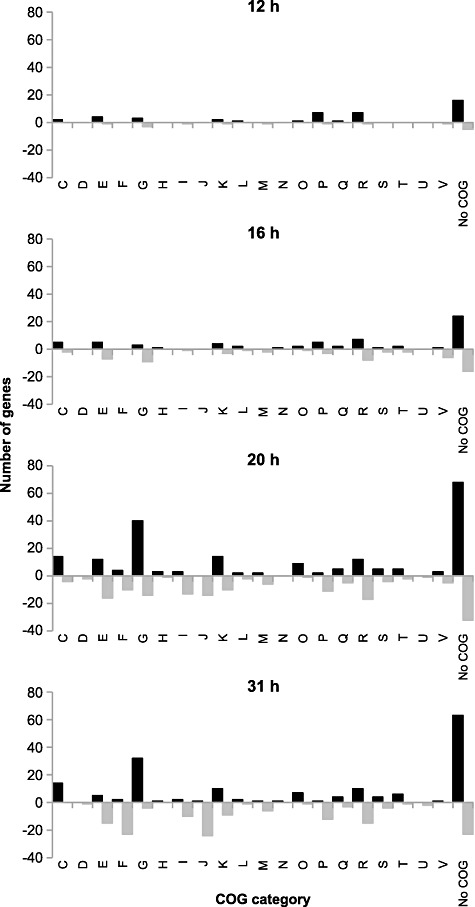
COGs classification of significantly differentially expressed genes at 12, 16, 20 and 31 h compared with 4 h. Black bars indicate upregulated genes, while grey bars represent downregulated genes. COG functional categories are as follows: C, Energy production and conversion; D, Cell cycle control, cell division, chromosome partitioning; E, Amino acid transport and metabolism; F, Nucleotide transport and metabolism; G, Carbohydrate transport and metabolism; H, Coenzyme transport and metabolism; I, Lipid transport and metabolism; J, Translation, ribosomal structure and biogenesis; K, Transcription; L, Replication, recombination and repair; M, Cell wall/membrane/envelope biogenesis; N, Cell motility; O, Post‐translational modification, protein turnover, chaperones; P, Inorganic ion transport and metabolism; Q, Secondary metabolites biosynthesis, transport and catabolism; R, General function prediction only; S, Function unknown; T, Signal transduction mechanisms; U, Intracellular trafficking, secretion and vesicular transport; V, Defence mechanisms.
Changes in protein production in L. rhamnosus GG during growth were studied at 4, 12, 20 and 28 h. Using the 2‐D DIGE technique, proteome composition at each time point was compared with the 20 h time point in three separate DIGE experiments (Table S2). The 20 h time point was selected as the baseline because it was assumed to involve the broadest selection of different proteins. In total, 267 protein spots showing a statistically significant difference (≥ 1.5‐fold difference, P < 0.05) in abundance were detected and cut out from the gels, and 201 of these could be identified using MALDI‐TOF mass spectrometry and/or LC‐MS/MS (Fig. S2, Tables 1–3 and Tables S3–S5). In seven cases, two proteins were identified from one spot. Conversely, 30 proteins were differentially abundant and identified in more than one experiment, and 26 proteins were found to occur as different isoforms, i.e. they were identified in two or more separate spots in the same experiment. Overall, the identified protein spots represented 116 distinct gene products. The numbers of differentially abundant protein spots between the time points 4 versus 20 h, 12 versus 20 h and 20 versus 28 h were 132, 46 and 23 respectively. The most remarkable growth phase‐dependent changes in protein abundance occurred in the set of carbohydrate metabolism proteins (COG category G), consistent with the RNA results. The abundance of these proteins was continuously changing during the fermentation process. For most other proteins, the differences were most obvious when comparing the mid‐exponential and early stationary phases (4 h and 20 h). Altered abundances were detected among others in proteins from COG categories F (Nucleotide transport and metabolism), E (Amino acid transport and metabolism) and J (Translation, ribosomal structure and biogenesis).
Table 1.
Proteins with differing abundance between the mid‐exponential (4 h) and early stationary (20 h) growth phases and transcription differences in the corresponding genes.
| Locus ID | Gene name | Function | Proteomic (fold change) | Transcriptomic (Log2R) 20 h/4 h | |
|---|---|---|---|---|---|
| Spot No. | 20 h/4 h | ||||
| Amino acid metabolism | |||||
| LGG_00568 | cysK | Cysteine synthase | 1 | 1.9 | 0.518 |
| LGG_01158 | pepD | Dipeptidase A | 2 | −2 | –0.507 |
| LGG_01201 | sufS | Cysteine desulfurase | 3 | 1.6 | –0.281 |
| LGG_01478 | pepO | Endopeptidase O | 4 | 1.6 | 1.788 |
| LGG_02639 | – | Aminotransferase | 5 | 1.7 | 3.741 |
| LGG_02708 | selA | Pyridoxal phosphate‐dependent enzyme | 6a | 2.5 | 3.039 |
| Carbohydrate metabolism | |||||
| Galactose metabolism | |||||
| LGG_00341 | lacC | Tagatose‐6‐phosphate kinase | 7 | 3.4 | 7.214 |
| LGG_00653 | galK | Galactokinase | 8 | 2.5 | 2.755 |
| LGG_00664 | lacC | Tagatose‐6‐phosphate kinase | 9 | 7.9 | 6.897 |
| LGG_00665 | lacD | Tagatose 1,6‐diphosphate aldolase | 10 | 4.4 | 6.910 |
| LGG_00666 | lacB | Galactose‐6‐phosphate isomerase subunit B | 11,12 | 23.9 | 6.923 |
| LGG_00667 | lacA | Galactose‐6‐phosphate isomerase subunit A | 13,14 | 31.9 | 6.988 |
| LGG_00668 | lacR | Lactose phosphotransferase system repressor | 15 | 15 | 7.055 |
| LGG_01062 | galU | UTP‐glucose‐1‐phosphate uridylyltransferase | 16,17 | 2.1 | 0.872 |
| LGG_02575 | lacD | Tagatose 1,6‐diphosphate aldolase | 18 | 4 | −0.07 |
| Glycolysis/gluconeogenesis | |||||
| LGG_00524 | fba | Fructose‐bisphosphate aldolase | 19 | −1.5 | −1.97 |
| LGG_00921 | pgm | Phosphoglucomutase | 20 | 1.5 | 1.686 |
| LGG_00933 | gapA | Glyceraldehyde‐3‐phosphate dehydrogenase | 21–25 | 2.9 | 0.644 |
| LGG_00933 | gapA | Glyceraldehyde‐3‐phosphate dehydrogenase | 26,27 | −1.8 | 0.644 |
| LGG_00934 | pgk | Phosphoglycerate kinase | 28,29,30a | 3.1 | 0.286 |
| LGG_00935 | tpiA | Triosephosphate isomerase | 31 | 2.2 | 0.147 |
| LGG_00935 | tpiA | Triosephosphate isomerase | 32 | −1.9 | 0.147 |
| LGG_00936 | eno | Enolase | 33 | 2 | 0.672 |
| LGG_01375 | pyk | Pyruvate kinase | 34–36 | 4.2 | −2.004 |
| Others | |||||
| LGG_00052 | eda | 2‐Dehydro‐3‐deoxyphosphogluconate aldolase/4‐hydroxy‐2‐oxoglutarate aldolase | 37 | −1.7 | 0.64 |
| LGG_00321 | deoC | Deoxyribose‐phosphate aldolase | 38 | 1.9 | 0.859 |
| LGG_00342 | srlD | Sorbitol‐6‐phosphate 2‐dehydrogenase | 39,40 | 53.6 | 7.877 |
| LGG_00757 | adhE | Aldehyde‐alcohol dehydrogenase | 41–44 | 18 | 5.86 |
| LGG_01322 | pdhC | Pyruvate dehydrogenase complex E2 component, dihydrolipoyllysine‐residue acetyltransferase | 45 | −1.6 | 0.796 |
| LGG_01360 | fruB | 1‐Phosphofructokinase | 46 | −3 | −1.659 |
| LGG_01421 | pflB | Formate acetyltransferase | 47–49 | 4.7 | 4.288 |
| LGG_01914 | citF | Citrate lyase, alpha subunit, Citrate CoA‐transferase | 50 | 2.8 | −2.673 |
| LGG_02025 | glgD | Glucose‐1‐phosphate adenylyltransferase regulatory subunit | 51 | 1.9 | 3.588 |
| Lipid metabolism | |||||
| LGG_00509 | dhaK | Dihydroxyacetone kinase | 52,53 | 3.4 | 1.109 |
| LGG_02113 | fabZ | (3R)‐hydroxymyristoyl‐[acyl‐carrier‐protein] dehydratase | 54 | −5.7 | −6.321 |
| LGG_02115 | fabF | 3‐Oxoacyl‐[acyl‐carrier‐protein] synthase II | 55,56 | −2.8 | −5.969 |
| LGG_02118 | fabK | Enoyl‐(Acyl‐carrier‐protein) reductase II | 57 | −1.9 | −5.285 |
| Membrane transport | |||||
| LGG_00345 | gatA | PTS system, galactitol‐specific IIA component | 58 | 5.5 | 7.069 |
| LGG_00951 | malE | ABC transporter, sugar transporter periplasmic component | 59a | 1.9 | −0.74 |
| LGG_01940 | oppF | ABC transporter, oligopeptide transporter ATPase component | 60 | 3.2 | 2.51 |
| LGG_01941 | oppD | ABC transporter, oligopeptide transporter ATPase component | 61 | 2.3 | 2.733 |
| LGG_02838 | manA | PTS system, mannose‐specific IIAB component | 62,63 | 1.8 | 1.199 |
| Metabolism of cofactors and vitamins | |||||
| LGG_02764 | entB | Isochorismatase family protein | 64 | 4.7 | 3.538 |
| LGG_02765 | pncB | Nicotinate phosphoribosyltransferase | 65 | 4.7 | 3.707 |
| LGG_01258 | nifS | Aminotransferase class V | 66 | −3.2 | −0.546 |
| Nucleotide metabolism | |||||
| Purine metabolism | |||||
| LGG_00249 | guaB | Inosine‐5′‐monophosphate dehydrogenase | 67 | −2.1 | −1.349 |
| LGG_01803 | purD | Phosphoribosylamine‐glycine ligase | 30a | 1.7 | −1.771 |
| LGG_01805 | purH | Bifunctional purine biosynthesis protein purH | 68,69 | 2.3 | −1.698 |
| LGG_01807 | purM | Phosphoribosylformylglycinamidine cyclo‐ligase | 70a | 2 | −1.574 |
| LGG_01808 | purF | Amidophosphoribosyltransferase | 71,72 | 2.8 | −1.625 |
| LGG_01809 | purL | Phosphoribosylformylglycinamidine synthase II | 73 | 3.8 | −1.653 |
| LGG_01812 | purC | Phosphoribosylaminoimidazole‐succinocarboxamide synthase | 74 | 2.5 | −1.466 |
| LGG_01813 | purK | Phosphoribosylaminoimidazole carboxylase, ATPase subunit | 6a,75,76 | 2.8 | −1.539 |
| LGG_01968 | guaA | GMP synthase | 77 | 1.8 | −1.916 |
| LGG_02466 | adk | Adenylate kinase | 78 | −1.7 | −1.865 |
| Pyrimidine metabolism | |||||
| LGG_01175 | upp | Uracil phosphoribosyltransferase | 79 | −1.6 | −1.009 |
| LGG_01456 | carB | Carbamoyl‐phosphate synthase, large subunit | 80,81 | −2.3 | −3.819 |
| LGG_01458 | pyrC | Dihydroorotase | 82 | −1.9 | −3.269 |
| LGG_01459 | pyrB | Aspartate carbamoyltransferase | 83 | −1.6 | −3.143 |
| LGG_01461 | pyrR | Pyrimidine operon regulatory protein, bifunctional protein pyrR | 84 | −2.2 | −2.752 |
| LGG_02546 | pyrG | CTP synthase | 85 | −2 | −0.964 |
| Others | |||||
| LGG_01474 | nrdE | Ribonucleoside‐diphosphate reductase, alpha subunit | 86,87 | −2.3 | −1.339 |
| Peptidoglycan biosynthesis | |||||
| LGG_00254 | dacA | d‐alanyl‐d‐alanine carboxypeptidase | 59a | 1.9 | 0.149 |
| LGG_00662 | – | Beta‐lactamase class C‐related penicillin‐binding protein | 88 | 7.3 | 6.844 |
| LGG_01282 | murD | UDP‐N‐acetylmuramoylalanine‐d‐glutamate ligase | 89 | −1.7 | −0.545 |
| LGG_01768 | murC | UDP‐N‐acetylmuramate‐l‐alanine ligase | 90 | −1.9 | −1.239 |
| Protein synthesis | |||||
| LGG_01628 | rpsB | SSU/30S ribosomal protein S2P | 70a | 2 | −1.408 |
| LGG_01628 | rpsB | SSU/30S ribosomal protein S2P | 91,92 | −2.1 | −1.408 |
| LGG_01690 | rplU | LSU/50S ribosomal protein L21P | 93 | −2.5 | −0.991 |
| LGG_02493 | fusA | Protein translation elongation factor G (EF‐G) | 94 | −1.7 | −1.288 |
| Stress | |||||
| LGG_01367 | clpB | ATP‐dependent chaperone ClpB | 95 | 1.7 | 0.783 |
| LGG_01604 | dnaK | Chaperone protein dnaK | 96 | −1.5 | −0.027 |
| LGG_02151 | usp | Universal stress protein, UspA family | 97 | 5.6 | −0.364 |
| LGG_02239 | groEL | 60 kDa chaperonin GROEL | 98,99 | 2.3 | 0.058 |
| LGG_02499 | clpC | ATP‐dependent Clp protease ATP‐binding subunit | 100 | 4.5 | 0.25 |
| LGG_02806 | htrA | Serine protease | 101 | −1.6 | −0.454 |
| Transcription | |||||
| LGG_02461 | rpoA | DNA‐directed RNA polymerase, subunit alpha | 102 | −1.8 | −1.536 |
| LGG_02498 | rpoB | DNA‐directed RNA polymerase, beta chain | 103,104 | −2.5 | −0.533 |
| Translation | |||||
| LGG_00848 | leuS | Leucyl‐tRNA synthetase | 105 | −2.4 | −0.541 |
| LGG_01261 | valS | Valyl‐tRNA synthetase | 106 | −2.1 | −0.427 |
| LGG_01786 | argS | Arginyl‐tRNA synthetase | 107 | −1.8 | −0.827 |
| LGG_02332 | gltX | Glutamyl‐tRNA synthetase | 108 | −1.6 | −1.553 |
| LGG_02584 | metG | Methionyl‐tRNA synthetase/protein secretion chaperonin, CsaA | 109,110 | −2 | −0.586 |
| Miscellaneous | |||||
| LGG_00226 | – | Pyridoxine 5′‐phosphate oxidase V related favin‐nucleotide‐binding protein | 111 | −3.4 | −3.048 |
| LGG_00491 | – | NADH peroxidase | 112 | 4.1 | −0.197 |
| LGG_00615 | yeaE | Aldo/keto reductase | 113 | 2.1 | 0.153 |
| LGG_00634 | – | Dyp‐type peroxidase family protein | 114 | 2.5 | 2.248 |
| LGG_00663 | – | Conserved protein | 115 | 4.9 | 6.808 |
| LGG_00740 | gph | Hydrolase, haloacid dehalogenase‐like family | 116 | −1.8 | −0.495 |
| LGG_00744 | dkgA | Aldo/keto reductase | 117 | 1.6 | 0.71 |
| LGG_00899 | secA | Protein translocase subunit secA | 118 | −2.1 | −0.076 |
| LGG_01265 | mreB | Rod shape‐determining protein MreB | 119 | 1.6 | −0.331 |
| LGG_01311 | ykqC | Metallo‐beta‐lactamase superfamily protein | 120 | −2 | 0.493 |
| LGG_01327 | typA | GTP‐binding protein TypA | 121 | −1.8 | −2.590 |
| LGG_01395 | – | Conserved protein | 122 | 3.4 | 3.504 |
| LGG_01433 | – | Nitroreductase | 123 | 2.3 | 1.344 |
| LGG_01465 | fhs | Formate‐tetrahydrofolate ligase | 124 | 1.9 | −1.209 |
| LGG_01468 | – | Nitroreductase family protein | 125,126 | −3.6 | −3.852 |
| LGG_01821 | ptsH | Phosphocarrier protein HPr | 127 | 2.2 | 0.884 |
| LGG_01837 | mvaS | Hydroxymethylglutaryl‐CoA synthase | 128 | −1.5 | 0.071 |
| LGG_02050 | glf | UDP‐galactopyranose mutase | 129 | −1.7 | −1.711 |
| LGG_02098 | – | Conserved protein | 130 | 2.4 | −0.526 |
| LGG_02415 | cueO | Multicopper oxidase | 131 | 3.9 | 1.73 |
| LGG_02630 | yghZ | Aldo/keto reductase (oxidoreductase) | 132 | 2.6 | 1.3 |
Two proteins were identified from these spots. It remains unclear which of these proteins was more abundant.
Table 3.
Proteins with differing abundance between the early stationary (20 h) and late stationary (28 h) growth phases and transcription differences in the corresponding genes.
| Locus ID | Gene name | Function | Proteomic (fold change) | Transcriptomic (Log2R) 31 h/20 h | |
|---|---|---|---|---|---|
| Spot No. | 28 h/20 h | ||||
| Amino acid metabolism | |||||
| LGG_02639 | – | Aminotransferase | 179 | 2.1 | 0.824 |
| Carbohydrate metabolism | |||||
| Galactose metabolism | |||||
| LGG_00341 | lacC | Tagatose‐6‐phosphate kinase | 180 | 2.1 | −1.548 |
| LGG_00664 | lacC | Tagatose‐6‐phosphate kinase | 181 | 2.3 | −2.407 |
| LGG_00665 | lacD | Tagatose 1,6‐diphosphate aldolase | 182 | 2 | −2.313 |
| LGG_00666 | lacB | Galactose‐6‐phosphate isomerase subunit B | 183,184 | 2.6 | −2.027 |
| LGG_00667 | lacA | Galactose‐6‐phosphate isomerase subunit A | 185,186 | 2 | −2.163 |
| LGG_00668 | lacR | Lactose phosphotransferase system repressor | 187 | 2.3 | −2.18 |
| Glycolysis/gluconeogenesis | |||||
| LGG_00921 | pgm | Phosphoglucomutase | 188 | 1.6 | 1.085 |
| LGG_01375 | pyk | Pyruvate kinase | 189 | −1.5 | −0.329 |
| Others | |||||
| LGG_00342 | srlD | Sorbitol‐6‐phosphate 2‐dehydrogenase | 190,191 | 2.4 | −2.053 |
| LGG_00757 | adhE | Aldehyde‐alcohol dehydrogenase | 192,193 | 2.5 | −0.466 |
| LGG_01421 | pflB | Formate acetyltransferase | 194 | 1.5 | 0.176 |
| LGG_01876 | pck | Phosphoenolpyruvate carboxykinase (ATP) | 195 | 1.8 | 0.693 |
| LGG_02693 | xylB | Alcohol dehydrogenase | 196a | 1.5 | 1.601 |
| Membrane transport | |||||
| LGG_02421 | mtsB | ABC transporter, ATPase component | 197a | −1.5 | −0.438 |
| Peptidoglycan biosynthesis | |||||
| LGG_00662 | – | Beta‐lactamase class C‐related penicillin‐binding protein | 198 | 2.6 | −2.265 |
| Nucleotide metabolism | |||||
| Pyrimidine metabolism | |||||
| LGG_01625 | pyrH | Uridylate kinase | 197a | −1.5 | 0.296 |
| Miscellaneous | |||||
| LGG_01016 | ligA | NAD‐dependent DNA ligase | 199 | −1.6 | −0.432 |
| LGG_01061 | mvaK | Phosphomevalonate kinase | 200 | 1.6 | −0.477 |
| LGG_01395 | – | Conserved protein | 201 | 2.5 | 0.926 |
| LGG_02124 | yqhD | Iron‐containing alcohol dehydrogenase | 196a | 1.5 | 0.64 |
Two proteins were identified from these spots. It remains unclear which of these proteins was more abundant.
In conclusion, the microarray analyses coupled with proteomics revealed that the transcription of 636 genes and the production of 116 proteins were altered in L. rhamnosus GG during the progression of growth from the mid‐exponential to the late stationary growth phases. Of these changes, a total of 61 could be confirmed by gene expression analysis at both the transcript and protein level.
Genes and proteins involved in central metabolic pathways are modulated in a growth‐dependent manner
Carbohydrate transport and metabolism. The whey medium used in this study contains glucose and galactose derived from hydrolysed lactose. In L. rhamnosus GG, there are two different pathways for the catabolism of the galactose moiety of lactose. The Leloir pathway (proteins encoded by the galKETRM operon and pgm) converts galactose to glucose‐6‐phosphate, and the tagatose‐6‐phosphate pathway (proteins encoded by the lacCDBAR operon) metabolizes galactose to glyceraldehyde‐3‐phosphate and dihydroxyacetone phosphate (Kankainen et al., 2009). Genes encoding enzymes from both the Leloir pathway and the tagatose‐6‐phosphate pathway showed a significant increase in expression at the mRNA level when the culture shifted from the exponential to the stationary phase of growth (Fig. 4A, Table S1). The same expression pattern was also evident at the protein level, as galactose utilization enzymes were more abundant in the early stationary growth phase (20 h) than in the exponential growth phase (4 or 12 h) (Tables 1 and 2). Furthermore, chemical analyses of the cultures showed that glucose was no longer detectable (detection limit 0.05%) in the early stationary phase of growth while galactose was still found in high amounts (1.0%). Therefore, increased expression of genes involved in galactose utilization at the stationary‐phase transition point was expected because L. rhamnosus GG first metabolizes the readily fermented glucose moiety; only after that does it metabolize the less easily exploited galactose portion of the hydrolysed lactose in the whey medium. Applying a proteomic approach, Cohen and colleagues (2006) also detected upregulation of the Leloir pathway in L. plantarum WCFS1 during stationary growth in the laboratory MRS medium, which possibly contained trace amounts of galactose. Our transcript results also indicated that genes encoding components of the phosphotransferase system (PTS), currently annotated as galactitol‐specific (LGG_00343, LGG_00345–00346), were significantly upregulated upon entry into the stationary growth phase and clustered together with lacCDBAR genes (cluster 2 in Fig. S1). Increased expression of the IIA component (LGG_00345) of that PTS system was also detected at the protein level (Tables 1 and 2). These results suggest that this particular PTS is involved in the transportation of galactose, and revision of its present annotation could be considered. It is noteworthy that current annotations of PTS transporters do not necessarily indicate their true role in the metabolism of particular sugars because the specificities of many lactobacilli PTS transporters are incorrectly annotated (Francl et al., 2010), and current computational methods are unreliable for the prediction of substrate specificity. Therefore, further experiments are needed to characterize the PTS transporter specificities of L. rhamnosus GG; for example, this could be done by studying transcript expression profiles in response to different carbohydrates.
Figure 4.
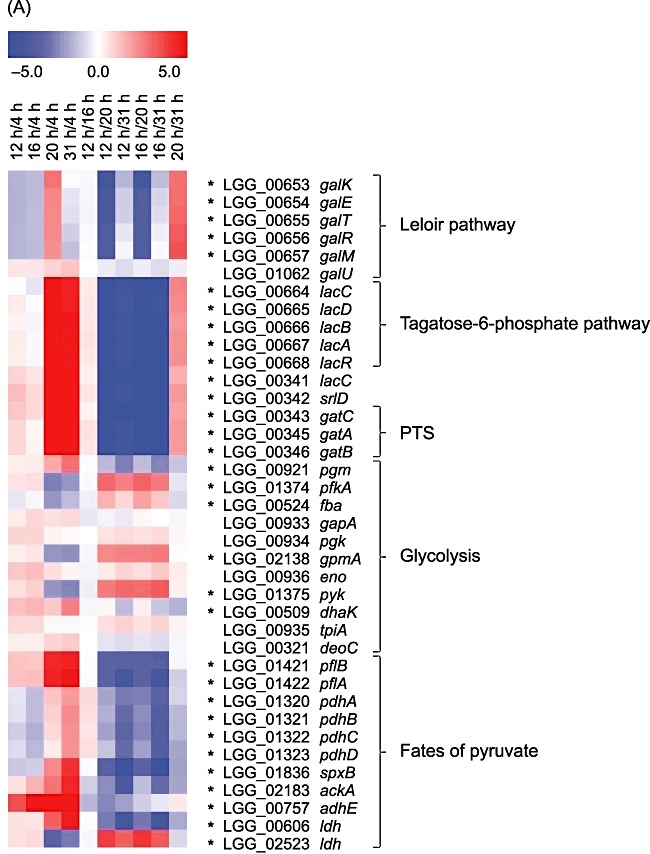
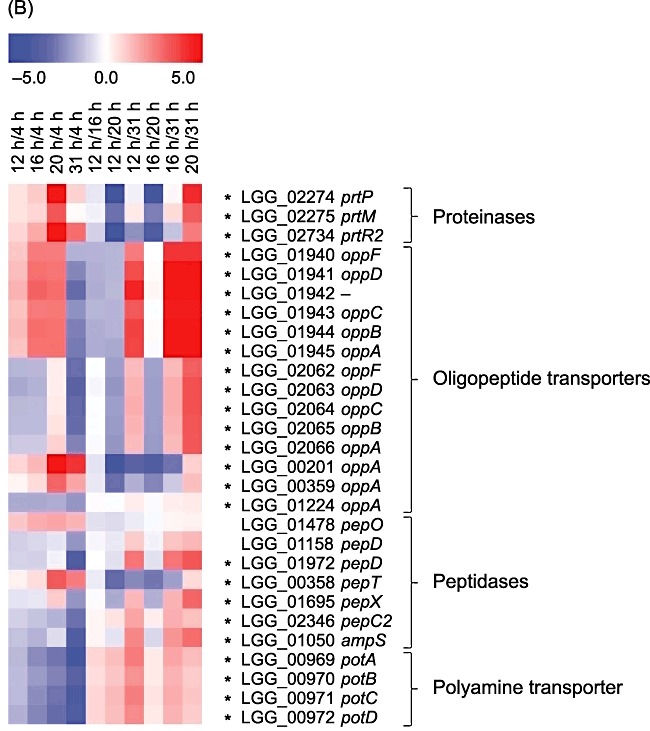

Gene expression patterns of selected genes and operons during growth in whey coding for (A) carbohydrate and pyruvate metabolic proteins, (B) proteinases, peptidases and amino acid transporters and (C) probiotic‐associated factors. Gene expression changes (averages of three biological replicates) between paired time points are represented colorimetrically, with dark red indicating an expression ratio of 5.0 and dark blue indicating an expression ratio of −5.0 on a log2 scale. The first four lanes show results of comparison of time points 12, 16, 20 and 31 h to the time point 4 h corresponding to the mid‐exponential phase. Statistically significant changes (P‐value ≤ 0.01) are marked with asterisks.
Table 2.
Proteins with differing abundance between the late exponential (12 h) and early stationary (20 h) growth phases and transcription differences in the corresponding genes.
| Locus ID | Gene name | Function | Proteomic (fold change) | Transcriptomic (Log2R) 20 h/12 h | |
|---|---|---|---|---|---|
| Spot No. | 20 h/12 h | ||||
| Carbohydrate metabolism | |||||
| Galactose metabolism | |||||
| LGG_00653 | galK | Galactokinase | 133,134 | 4 | 4.176 |
| LGG_00654 | galE | UDP‐glucose 4‐epimerase | 135 | 3.1 | 3.817 |
| LGG_00655 | galT | Galactose‐1‐phosphate uridylyltransferase | 136 | 3.7 | 3.831 |
| LGG_00664 | lacC | Tagatose‐6‐phosphate kinase | 137 | 5.3 | 6.928 |
| LGG_00665 | lacD | Tagatose 1,6‐diphosphate aldolase | 138 | 2.3 | 6.461 |
| LGG_00666 | lacB | Galactose‐6‐phosphate isomerase subunit B | 139,140 | 30.1 | 6.675 |
| LGG_00667 | lacA | Galactose‐6‐phosphate isomerase subunit A | 141 | 26.8 | 6.561 |
| LGG_00668 | lacR | Lactose phosphotransferase system repressor | 142 | 15.9 | 6.699 |
| LGG_02575 | lacD | Tagatose 1,6‐diphosphate aldolase | 143,144 | 2.1 | −1.145 |
| Glycolysis/gluconeogenesis | |||||
| LGG_00921 | pgm | Phosphoglucomutase | 145 | 1.9 | 1.331 |
| LGG_00933 | gapA | Glyceraldehyde‐3‐phosphate dehydrogenase | 146,147 | 1.6 | 0.239 |
| LGG_00933 | gapA | Glyceraldehyde‐3‐phosphate dehydrogenase | 148 | −2.9 | 0.239 |
| LGG_01375 | pyk | Pyruvate kinase | 149,150 | 2 | −2.596 |
| LGG_01375 | pyk | Pyruvate kinase | 151 | −1.8 | −2.596 |
| Others | |||||
| LGG_00342 | srlD | Sorbitol‐6‐phosphate 2‐dehydrogenase | 152,153 | 26.1 | 6.56 |
| LGG_00373 | rbsK | Ribokinase | 154 | 2.1 | 4.074 |
| LGG_00757 | adhE | Aldehyde‐alcohol dehydrogenase | 155–157 | 5.6 | 2.279 |
| LGG_01421 | pflB | Formate acetyltransferase | 158 | 2.2 | 3.134 |
| LGG_02523 | ldh | l‐lactate dehydrogenase | 159a | −1.6 | −3.868 |
| Membrane transport | |||||
| LGG_00345 | gatA | PTS system, galactitol‐specific IIA component | 160 | 13 | 6.286 |
| Metabolism of cofactors and vitamins | |||||
| LGG_02764 | entB | Isochorismatase family protein | 161 | 2.6 | 1.32 |
| LGG_02765 | pncB | Nicotinate phosphoribosyltransferase | 162,163 | 3.3 | 1.623 |
| Nucleotide metabolism | |||||
| Purine metabolism | |||||
| LGG_00249 | guaB | Inosine‐5′‐monophosphate dehydrogenase | 164 | −1.6 | 0.419 |
| LGG_01808 | purF | Amidophosphoribosyltransferase | 165 | 1.7 | −2.035 |
| Peptidoglycan biosynthesis | |||||
| LGG_00662 | – | Beta‐lactamase class C‐related penicillin‐binding protein | 166 | 5.2 | 6.994 |
| Stress | |||||
| LGG_01823 | clpE | ATP‐dependent Clp protease ATP‐binding subunit | 167 | 1.8 | 0.722 |
| LGG_02151 | usp | Universal stress protein, UspA family | 168 | 2.3 | −2.413 |
| LGG_02499 | clpC | ATP‐dependent Clp protease ATP‐binding subunit | 169 | 1.9 | 0.399 |
| Translation | |||||
| LGG_00767 | alaS | Alanyl‐tRNA synthetase | 170 | −1.5 | −0.758 |
| LGG_01019 | gatA | Aspartyl/glutamyl‐tRNA(Asn/Gln) amidotransferase subunit A | 171 | −1.7 | −0.603 |
| Miscellaneous | |||||
| LGG_00226 | – | Pyridoxine 5′‐phosphate oxidase V‐related favin‐nucleotide‐binding protein | 172 | −2.1 | −1.605 |
| LGG_00491 | – | NADH peroxidase | 173 | 1.9 | −2.539 |
| LGG_00634 | – | Dyp‐type peroxidase family protein | 174 | 1.5 | 1.042 |
| LGG_00763 | ytqI | Phosphoesterase, DHH family protein | 159a | −1.6 | −0.088 |
| LGG_01395 | – | Conserved protein | 175 | 2.4 | 3.379 |
| LGG_01465 | fhs | Formate‐tetrahydrofolate ligase | 176 | −1.5 | −1.411 |
| LGG_01468 | – | Nitroreductase family protein | 177 | −1.6 | −1.985 |
| LGG_02630 | yghZ | Aldo/keto reductase (oxidoreductase) | 178 | 1.6 | −0.024 |
Two proteins were identified from this spot. It remains unclear which of these proteins was more abundant.
The abundance of several glycolytic enzymes and the transcription of the corresponding genes changed in a growth phase‐dependent fashion. Genes coding for 6‐phosphofructokinase (pfkA), fructose‐bisphosphate aldolase (fba), phosphoglyceromutase (gpmA) and pyruvate kinase (pyk) were significantly downregulated during the stationary‐phase transition (Fig. 4A, Table S1). At the protein level, Fba was more abundant in the mid‐exponential growth phase than in the early stationary phase, consistent with the mRNA results (Table 1). No clear trend in the abundance changes of glycolytic proteins was seen during growth, however. Many glycolytic proteins (GapA, Pgk, TpiA, Eno, Pyk) were detected in several parallel protein spots (two to seven spots) on 2‐D gels, and in some of these spot sets (GapA, TpiA, Pyk), both a decrease and an increase in abundance were detected (Tables 1 and 2). For instance, five glyceraldehyde‐3‐phosphate dehydrogenase (GapA) isoforms were more abundant during the early stationary growth phase (compared with 4 h) while two other isoforms were more abundant during the mid‐exponential growth phase (compared with 20 h). Post‐translational protein modifications such as phosphorylation (Cozzone, 1998) and methionine formylation (Bandow et al., 2003) may alter the migration of proteins in 2‐D gels, which could also explain the appearance of parallel spots in this case. In Lactococcus lactis, glycolysis has been shown to be regulated at the transcriptional level only to a lesser degree, and post‐translational modifications might be more important for regulation (discussed in Soufi et al., 2008). In Lactococcus, Escherichia coli and Bacillus, several glycolytic proteins have been shown to be phosphorylated (Eymann et al., 2007; Macek et al., 2007; 2008; Soufi et al., 2008), and characterization of the phosphoproteome suggests conservation of this type of modification of glycolytic enzymes in L. rhamnosus GG (J. Koponen, K. Laakso, K. Koskenniemi, M. Kankainen, K. Savijoki, T.A. Nyman, S. Tynkkynen, N. Kalkkinen and P. Varmanen, in preparation). Transcripts of genes coding for GapA, Pgk, TpiA and Eno were only slightly upregulated in the early stationary growth phase compared with the exponential growth phase in our study, suggesting that they are constitutively expressed (Fig. 4A, Tables 1 and 2).
Dihydroxyacetone kinase (DhaK), which catalyses the phosphorylation of dihydroxyacetone, was more abundant in early stationary‐phase cells than in exponential‐phase cells at both the transcript and protein levels (Fig. 4A, Table 1). Dihydroxyacetone phosphate is channelled to glycolysis via the activity of triosephosphate isomerase (TpiA), which catalyses the formation of glyceraldehyde‐3‐phosphate from dihydroxyacetone phosphate. Deoxyribose‐phosphate aldolase (DeoC, LGG_00321), which converts deoxyribose‐5‐phosphate to glyceraldehyde‐3‐phosphate (a glycolytic intermediate), and its concomitant gene were upregulated upon entry into the stationary phase of growth (Fig. 4A, Table 1). One of the most highly differentially expressed proteins and genes was sorbitol‐6‐phosphate 2‐dehydrogenase (SrlD, LGG_00342), which channels sorbitol‐6‐phosphate to glycolysis via the conversion of sorbitol‐6‐phosphate to fructose‐6‐phosphate (Fig. 4A, Tables 1–3). The expression of SrlD increased over time, especially when the culture shifted from the exponential growth to the stationary phase. Transcription of the genes encoding the glucitol/sorbitol‐PTS (LGG_02717–02719) was induced when the culture shifted from exponential growth to stationary growth (Table S1). These results suggest that L. rhamnosus GG starts to use alternative energy sources other than the previously mentioned galactose, namely sorbitol, dihydroxyacetone and deoxyribose‐5‐phosphate, at the beginning of the stationary phase after the exhaustion of glucose. In Lactobacillus casei, the sorbitol operon is repressed by glucose (Alcántara et al., 2008), and this seems to also apply for L. rhamnosus GG because the upregulation of SrlD occurs when glucose has been used up (according to chemical analyses, detection limit 0.05%).
In summary, during the transition to the stationary growth phase, the utilization of glucose as the main carbon and energy source was replaced in L. rhamnosus GG by alternative pathways for carbohydrate metabolism, such as galactose utilization, using the tagatose‐6‐phosphate and the Leloir pathways.
Energy production and conversion (pyruvate metabolism). Under normal glucose fermentation conditions, lactic acid bacteria reduce pyruvate to lactate by a NAD+‐dependent lactate dehydrogenase. In addition, lactic acid bacteria may have alternative means of utilizing pyruvate, including the pyruvate‐formate lyase system, the pyruvate dehydrogenase pathway, the pyruvate oxidase pathway and the diacetyl/acetoin pathway (Axelsson, 2004). Genes encoding pyruvate‐formate lyase (LGG_01421–01422), aldehyde‐alcohol dehydrogenase (LGG_00757), pyruvate dehydrogenase (LGG_01320–01323) and enzymes of the pyruvate oxidase pathway (LGG_01836, pyruvate oxidase and LGG_02183, acetate kinase) were induced at the stationary‐phase transition point (Fig. 4A, Table S1). The PflB subunit of pyruvate‐formate lyase and aldehyde‐alcohol dehydrogenase (AdhE) were also more abundant in stationary‐phase cells than in exponential‐phase cells at the protein level, supporting the transcript results (Table 1–3). However, proteomic analyses indicated that pyruvate dehydrogenase (PdhC) was more abundant in the exponential growth phase than in the stationary growth phase, in contrast with the transcript results (Table 1). A gene encoding lactate dehydrogenase (LGG_00606) clustered together with the pyruvate‐formate lyase and acetate kinase genes, and its expression was significantly increased over time (cluster 4 in Fig. S1). Expression of another gene coding for a lactate dehydrogenase (LGG_02523) was, however, decreased upon entry into the stationary growth phase, and the same tendency was seen at the protein level (12 h versus 20 h) (Table 2). Thus, our results suggest that L. rhamnosus GG changes from homolactic fermentation to a mixed acid fermentation when the culture reaches the stationary phase, and the end‐products formed, in addition to lactate, are formate, acetate and ethanol. This is probably due to the shift detected from glucose to galactose utilization in L. rhamnosus GG at the stationary‐phase transition point. The same trend has been observed in Lactococcus strains, which shift from homolactic to mixed acid fermentation when glucose is limited or during galactose fermentation (reviewed by Neves et al., 2005). In L. plantarum cells, more lactate dehydrogenase was produced in the exponential than in the stationary growth phase (Cohen et al., 2006), supporting our conclusions.
Nucleotide transport and metabolism. The abundance of pyrimidine biosynthetic gene cluster (pyr) transcripts was reduced upon entry into the stationary phase of growth, and the downregulation of the pyrimidine biosynthesis pathway was also detected at the protein level (Table 1 and Table S1). In addition, the expression of several other genes and gene products involved in nucleotide metabolism were decreased over time in whey. This is probably due to protracted growth and, thus, reduced requirements for pyrimidine nucleotides. However, in the purine biosynthesis pathway, no clear downregulation was observed. At the mRNA level, the purine biosynthesis operon (purBCDFHKLM and guaAB) was expressed at lower levels in the stationary growth phase than in the exponential growth phase, while the PurCDFHKLM and GuaA proteins were more abundant at 20 h (early stationary) than at 4 h (mid‐exponential) (Table 1 and Table S1). The whey medium used here does not supply L. rhamnosus GG cells with purines. It has previously been shown that in L. rhamnosus GG cells grown in whey medium, during the stationary phase, the abundance of Pur proteins is elevated compared with cells grown in medium rich in purines (Koskenniemi et al., 2009). Therefore, our results suggest that under the growth conditions described here, purine biosynthesis in L. rhamnosus GG cells remains active during the switch from the exponential to the stationary phase. Furthermore, the identified growth phase‐associated changes in Pur protein abundance were not associated with changes in the corresponding transcript levels, suggesting either the involvement of post‐transcriptional regulation mechanisms or that these proteins have a long half‐life.
Lipid transport and metabolism. Fatty acids are constituents of phospholipids and glycolipids found in the cytoplasmic membrane of bacteria. Our results revealed that fatty acid biosynthetic genes (LGG_02110–02122) were strongly repressed at the stationary‐phase transition point (Table S1). Similarly, the abundance of the proteins, FabZ, FabF and FabK, which are involved in the biosynthesis of long‐chain saturated fatty acids, were decreased over time (Table 1). Furthermore, the level of transcripts encoding cyclopropane‐fatty‐acyl‐phospholipid synthase (cfa, LGG_02109), which participates in the cyclopropanation of fatty acids, was reduced upon entry into the stationary phase of growth (Table S1). Fatty acids are energetically the most expensive membrane lipid components to produce, and their production is tightly regulated to match the growth rate of bacterial cells (Zhang and Rock, 2009). Therefore, our observations probably result from the deceleration of the growth rate in the stationary phase. Applying a proteome‐level approach, Cohen and colleagues (2006) observed similar downregulation of several enzymes involved in lipid metabolism in L. plantarum during the shift from the exponential to the stationary phase of growth.
Proteolytic systems, amino acid transport and metabolism
Lactobacillus rhamnosus GG has a limited capacity to synthesize amino acids; therefore, it requires exogenous amino acids and peptides for growth (Kankainen et al., 2009). In this study, several components of the proteolytic system were identified as exhibiting growth phase‐dependent induction at both the transcript and the protein level, including proteinases, which degrade proteins into oligopeptides, peptide transporter systems involved in oligopeptide uptake, and peptidases, which degrade oligopeptides into shorter peptides and amino acids (reviewed by Savijoki et al., 2006). The whey medium used in this study contains free amino acids and oligopeptides derived from hydrolysed casein and whey protein and probably also traces of non‐hydrolysed proteins. During growth in whey medium, L. rhamnosus GG first consumes the accessible free amino acids and, after that, oligopeptides, which require only minimal processing to liberate the free amino acids. Expression of genes encoding cell‐envelope proteinase PrtP, maturation protein PrtM and proteinase PrtR increased during growth of L. rhamnosus GG in whey medium until the early stationary growth phase, suggesting that the non‐hydrolysed or partially hydrolysed proteins in whey medium may be utilized after the exhaustion of other amino acid sources (Fig. 4B, Table S1). In protein gels, no differentially produced proteinases were detected. However, it is well known that high‐molecular‐weight proteins, hydrophobic proteins and proteins with extreme pI values are under‐represented on 2‐D gels possibly explaining the lack of identification of PrtP (206.6 kDa), PrtR (155.8 kDa) and PrtM (pI value 10.3). It has previously been shown that whey‐grown L. rhamnosus GG is unable to hydrolyse intact casein (Kankainen et al., 2009), and the role of the proteinases in the utilization of intact or partially hydrolysed whey proteins remains to be elucidated. Consistent with our findings, in L. acidophilus cultivated in milk, the expression of genes encoding PrtP and PrtM was increased over time (Azcarate‐Peril et al., 2009).
The genome of L. rhamnosus GG holds three complete oligopeptide ABC transporter operons (LGG_01652–01656, LGG_01940–01945 excluding LGG_01942, and LGG_02062–02066) (Kankainen et al., 2009). Each consists of five genes encoding oligopeptide‐binding protein (OppA), two integral membrane proteins (OppB and OppC) and two ATP‐binding proteins (OppD and OppF). Of these operons, LGG_01940–01945 was found to be differentially expressed at both the mRNA and protein levels. Expression of the whole operon at the mRNA level was increased until the early stationary growth phase (Fig. 4B, Table S1), and the ATPase components, OppD and OppF, were more abundant in the early stationary growth phase than in the mid‐exponential growth phase at the protein level, supporting the transcript results (Table 1). LGG_02062–02066 transcripts were more abundant in the mid‐exponential growth phase (4 h) than at the 12, 16 and 31 h time points but less abundant at 4 h than in the early stationary growth phase (20 h) (Fig. 4B, Table S1). Transcription of these genes was therefore transient, with a short peak in the early stationary growth phase. Differential production of the corresponding proteins was not observed possibly because the fold changes were below threshold value of 1.5. In addition to the three oligopeptide‐binding proteins (OppA) encoded within the complete Opp operons, four additional oppA genes are present in the L. rhamnosus GG genome (Kankainen et al., 2009). Of these, three oppA genes were significantly differentially expressed in a growth phase‐dependent manner (Fig. 4B, Table S1). Based on these observations, it could be speculated that different oligopeptide transport systems may have different specificities, and while one Opp transporter system is active in the exponential growth phase, another might be active in the stationary phase of growth. Similar results were obtained in a microarray study of L. acidophilus grown in milk, which showed that different Opp genes were expressed at different growth stages (Azcarate‐Peril et al., 2009).
At the mRNA level, the genes encoding tripeptidase PepT (LGG_00358), proline‐specific aminopeptidase PepX (LGG_01695), dipeptidase PepD (LGG_01972), aminopeptidases PepC2 (LGG_02346) and AmpS (LGG_01050) were significantly differentially expressed at different growth phases (Fig. 4B, Table S1). The gene coding for PepT clustered together with the OppA‐encoding gene (LGG_00359) and was maximally expressed in L. rhamnosus GG at the early stationary phase of the growth curve (cluster 10 in Fig. S1). The transcript encoding PepX clustered together with the ORFs LGG_02062–02066 (the Opp transporter system), and its expression was transiently elevated at 20 h (cluster 6 in Fig. S1). The expression levels of pepD, pepC2 and ampS, as well as genes involved in polyamine transport (potABCD, LGG_00969–00972), were decayed during growth (Fig. 4B, Table S1). Proteomic results indicated that endopeptidase PepO (LGG_01478) was produced at higher levels in the early stationary cultures than in the mid‐exponential cultures while dipeptidase PepD (LGG_01158) was produced more in mid‐exponential cells compared with early stationary cells (Table 1). The same pattern was seen at the mRNA level, although it was not statistically significant (Fig. 4B). These findings suggest that different peptidases are utilized specifically in different growth phases, probably depending on their specificities for the particular peptides available at each stage.
General stress responses
Nutrient limitations activate a variety of bacterial stress responses (reviewed by De Angelis and Gobbetti, 2004). Changes in environmental conditions also rapidly trigger stress‐related proteolytic systems to cope with the accumulation of irreparably damaged proteins (Savijoki et al., 2006). Not surprisingly, the abundance of several stress proteins and the transcription of genes encoding stress responsive proteins was elevated when the cells reached the stationary growth phase. At the mRNA level, transcripts encoding Clp protease subunits, ClpL and ClpE, endopeptidase subunits, HslV (ClpQ) and HslU (ClpY), and heat shock proteins, Hsp1 and Hsp3, were elevated in the stationary phase of growth. However, genes encoding universal stress protein (usp) and 33 kDa chaperonin (hslO) showed their highest expression in the exponential growth phase (Table S1). At the protein level, production of the universal stress protein, the chaperone protein GroEL and the stress‐inducible Clp protease complex subunits ClpC and ClpB was increased over time while the chaperone protein DnaK and the stress‐related serine protease HtrA were more abundant in the mid‐exponential growth phase (4 h) than in the early stationary growth phase (20 h) (Table 1). The corresponding genes, groEL, clpC, clpB, dnaK and htrA, were not significantly differentially expressed. As chaperone proteins are required for de novo protein folding, the expression of chaperones and their corresponding genes in the exponential growth phase, when essential nutrients are still sufficiently available, could be a response to the high protein synthesis rate. Because growth phase‐associated changes in stress proteins were detectable at either the mRNA or protein level, but not at both levels, it is tempting to speculate that post‐transcriptional regulation mechanisms are involved in the adaptation of L. rhamnosus GG to the stationary growth phase conditions. Recently, it has been demonstrated that most of the stress‐related genes showed increasing levels of expression in the stationary growth phase in L. acidophilus (Azcarate‐Peril et al., 2009). Furthermore, in a proteomic study of L. plantarum WCFS1, it has been observed that some stress proteins were highly abundant in the late exponential and early stationary growth phases (ClpL and Hsp3, among others) while some were highly abundant during the exponential growth phase (ClpP, among others) (Cohen et al., 2006). It should be noted that the earlier studies describe bacterial growth phase responses in flask culture conditions where gradual acidification of the cultures causes additional stress. These growth conditions were different from those of the present study where pH‐controlled conditions were used.
Expression of genes mediating potential probiotic‐associated traits changes over time in whey
Lactobacillus rhamnosus GG colonizes the human intestine and adheres efficiently to mucus and epithelial cells (Alander et al., 1997; 1999; Laparra and Sanz, 2009). Cell surface factors, such as cell surface proteins, lipoteichoic acids (LTAs) and exopolysaccharides (EPSs), mediate adherence, biofilm formation and colonization by lactobacilli (reviewed by Lebeer et al., 2008). Recently, MabA (modulator of adhesion and biofilm) encoded by LGG_01865 was shown to contribute to the adhesion of L. rhamnosus GG to the host cells (Vélez et al., 2010). Cell surface‐exposed pilus, encoded by a gene cluster located in a genomic island unique to L. rhamnosus GG, was suggested to play a key role in the adhesion of L. rhamnosus GG to human intestinal mucus (Kankainen et al., 2009). Our transcriptomic results showed that strain‐specific pilus‐encoding genes (spaCBA, LGG_00442–00444) were active in the exponential phase of growth, and their expression was clearly reduced when the culture transitioned to the stationary growth phase (Fig. 4C, Table S1). In contrast, growth phase dependence in the expression pattern of mabA was not observed. However, several other genes encoding proteins predicted to contain an adhesion domain, such as LGG_02337 (inlJ) and LGG_02274 (prtP), were upregulated in L. rhamnosus GG cultures in the stationary growth phase compared with the exponential growth phase (Fig. 4C, Table S1). No changes in the corresponding proteins were observed, which may be due to the hydrophobic nature or the low abundance of these proteins. Furthermore, pilus proteins may occur at heteropolymeric high‐molecular‐weight complexes (Mandlik et al., 2008) that are not detectable using 2‐D DIGE. In L. lactis, cell wall‐anchored PrtP has been shown to have positive effect on cell adhesion to solid surfaces (Habimana et al., 2007); however, its effect on adhesion to human cells or mucus remains to be elucidated. Pili of Gram‐positive pathogens have been extensively investigated and have been shown to promote bacterial adhesion to host cells, play a role in biofilm formation and evoke host immune responses (reviewed by Mandlik et al., 2008). In L. rhamnosus GG, it has also been demonstrated that pili play an important role in adhesion to Caco‐2 epithelial cells and in biofilm formation (Lebeer et al., 2011). In a virulent Streptococcus pneumoniae strain, pilus genes were found to be expressed in a growth phase‐dependent manner, and the regulation of pilus genes was shown to be mediated by a two‐component system (Song et al., 2009). In our study, the expression pattern of genes encoding a two‐component system (LGG_02387–02388, cluster 14 in Fig. S1) was similar to that of the pilus‐encoding genes (cluster 13 in Fig. S1, Table S1), but further studies are needed to establish whether this particular two‐component system plays a role in the regulation of the pilus locus genes in L. rhamnosus GG or whether it is regulating one of the various other functions altered during exponential growth.
The expression of genes involved in d‐alanylation of LTAs (dlt operon, LGG_00777–00780, clusters 13 and 16 in Fig. S1) and most of the genes encoding enzymes required for EPS biosynthesis (LGG_02044–02047 and LGG_02049–02054, clusters 14 and 16 in Fig. S1) was reduced over time in whey (Fig. 4C, Table S1). Consistent with our findings, previous studies have demonstrated that EPS production is growth rate‐dependent (Welman et al., 2003; 2006). In L. rhamnosus GG, EPS plays a negative role in adhesion and biofilm formation, possibly by shielding adhesion factors such as pili (Lebeer et al., 2009). However, EPS appears to play an important role in the protection against host defence mechanisms involving the innate immune system (Lebeer et al., 2010). Because the balance between adhesion properties and resistance against the host immune system seems to be of the utmost importance for the adaptation of L. rhamnosus GG in the intestine, expression of pili and EPS biosynthetic genes are likely to be regulated accordingly. The products of the dlt operon are involved in the d‐alanylation of secondary cell wall polymer LTA. d‐alanyl ester substitutions for LTA add positive charges to otherwise negatively charged LTAs and, in this way, directly affect cell surface charges (Neuhaus and Baddiley, 2003). Lebeer and colleagues (2007) have shown that d‐alanyl‐substituted LTAs serve a modulatory function in the adhesion of L. rhamnosus GG to Caco‐2 cells and in biofilm formation and activate immune responses in the host. Claes and colleagues (2010) recently showed that deletion of dltD resulted in a significant increase in the probiotic efficacy of L. rhamnosus GG in a mouse model of colitis. Therefore, it is suggested that d‐alanylation of LTAs may play a role in the survival and persistence as well as in the probiotic effects of L. rhamnosus GG in the host, and our results indicate that the expression of this probiotic‐associated factor is growth phase‐dependent.
In addition to adhesion factors such as pili and InlJ, contact between L. rhamnosus GG and host cells can also be mediated by secreted, small soluble proteins. For instance, secreted, low‐molecular‐weight proteins from L. rhamnosus GG have been shown to promote in vitro intestinal epithelial cell homeostasis through certain signalling pathways (Yan and Polk, 2002; Tao et al., 2006; Yan et al., 2007; Seth et al., 2008). Expression of a gene coding for the NLP/P60 family secreted protein, p40 (LGG_00031), was reduced during growth (Fig. 4C, Table S1). In contrast, the genes LGG_00504 and LGG_00661, coding for secreted proteins of low molecular mass (< 10 kDa), were strongly induced in stationary‐phase cells (Fig. 4C, Table S1). Interestingly, a gene located next to one of these small‐sized soluble proteins (LGG_00503) and annotated as a myosin‐cross‐reactive antigen orthologue was also highly expressed in the stationary growth phase compared with the exponential growth phase. None of these responses was detected at the protein level, probably because cell surface‐exposed and secreted proteins are usually hydrophobic and are precipitated within the first dimension once they have migrated to their isoelectric point.
Concluding remarks
Consumption of probiotic bacteria harvested at different phases of growth has been shown to cause profoundly different mucosal responses in human (van Baarlen et al., 2009). Accordingly, we assume that the expression status of the genes during the industrial cultivation of a probiotic reflects its activity in the GIT. We investigated how gene expression, at both the transcript and protein levels, in the probiotic bacterium, L. rhamnosus GG, changed over time during growth in industrial‐type whey medium under controlled bioreactor conditions. In particular, we aimed to determine whether any traits that are associated with the probiotic activity of L. rhamnosus GG were affected during the fermentation of whey. Expression of 636 genes, many implicated in the probiotic‐linked actions of L. rhamnosus GG, and 116 proteins were modulated in a growth‐dependent manner and half of the changes in protein abundance were associated with changes in transcript levels. Most of the observed growth phase‐dependent changes at both the mRNA and protein level appeared during the shift from the exponential growth phase to the stationary growth phase. In particular, genes and proteins involved in the galactose utilization pathways were among the highest induced upon entry into stationary phase. Many transcripts and proteins involved in central metabolic pathways such as carbohydrate, nucleotide, lipid and pyruvate metabolism were differentially expressed during growth in whey (Fig. 5). Differential expression of distinct carbohydrate utilization and energy production pathways and several distinct PTS transporters observed in response to changes in nutritional environment (e.g. exhaustion of the preferred carbon source in the medium) indicate that L. rhamnosus GG has a flexible and adaptable metabolism. This adaptability might provide a competitive advantage for L. rhamnosus GG in the host's GIT. Furthermore, expression of many genes encoding adhesion factors and secreted proteins with low molecular masses were increased over time during the growth of L. rhamnosus GG in whey medium (Fig. 5), with the highest expression in the stationary phase. The expression patterns of genes encoding mucus‐binding pili and secreted protein p40 differed from this general pattern (Fig. 5), having the highest expression values in the exponential phase. Like nutritional versatility, these adhesion and probiotic‐associated factors provide a competitive advantage for L. rhamnosus GG by promoting its survival and persistence in the GIT. At present, the number of probiotic‐associated factors of L. rhamnosus GG that has been identified is still limited which makes it impossible to draw conclusions about the optimal harvesting phase for this probiotic bacterium. However, as soon as the key mediators of probiotic traits have been identified, the results presented here together with future studies involving growth conditions mimicking those in the GIT will be instrumental for these purposes.
Figure 5.
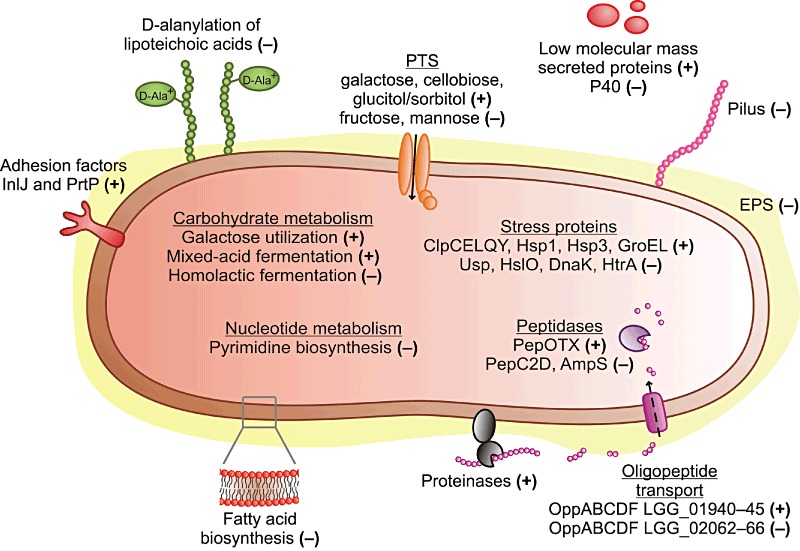
A model for physiological changes in L. rhamnosus GG during the progression of growth. Increased and decreased expression is indicated by plus symbol ‘+’ and minus symbol ‘−’ respectively.
Experimental procedures
Growth conditions and chemical analyses
Lactobacillus rhamnosus GG (ATCC 53103) was maintained in a laboratory culture collection as a glycerol stock at −70°C and propagated at 37°C in MRS medium (Labema). Appropriate dilutions of a culture grown for 17 h were plated on MRS agar and incubated anaerobically at 37°C for 3 days. Single colonies from these plates were resuspended in MRS medium. One individual colony represented one biological replicate. Cells were passaged through two sequential subcultures in MRS medium at 37°C for 12 h and one subculture in industrial‐type whey medium containing 5% hydrolysed whey, 0.6% casein hydrolysate and 0.0015% MnSO4·H2O at 37°C for 17 h. Four Biostat Q fermentation vessels (B. Braun Biotech International) containing 750 ml of whey medium were inoculated at 2% (v/v) with four individual cultures of L. rhamnosus GG. The cultures were grown at 37°C and stirred constantly (150 r.p.m. min−1). The pH was maintained at 5.8 by automatic titration with 5% (v/v) ammonia. To monitor growth, samples were taken at regular intervals from each vessel, and their optical density at 600 nm was measured. The amount of glucose, galactose and lactose in whey medium during fermentation was determined by enzymatic methods based on ISO 5765‐1: 2002 (IDF 79‐1: 2002) and ISO 5765‐2: 2002 (IDF 79‐2: 2002) standards. Samples for RNA extraction were collected at the mid‐exponential (4 h), late exponential (12 h), stationary transition point (16 h), early stationary (20 h) and late stationary (31 h) phases (Fig. 1). Protein samples were collected at the mid‐exponential (4 h), late exponential (12 h), early stationary (20 h) and late stationary (28 h) phases (Fig. 1). Three parallel RNA samples and four parallel protein samples from each sampling point were included in the analyses.
Transcriptomic methods
Experimental design, RNA methods, cDNA synthesis, labeling and hybridization. mRNA samples from three independent biological replicates, i.e. from three fermentations, for each time point were hybridized, each sample independently, to microarrays according to an anova design where all possible direct pair‐wise comparisons within biological replicates were conducted. A total of 30 hybridizations were performed. Dye‐swaps were carried out over biological replicate sets.
Depending on the growth stage, 1–4 ml of culture was added to 2–8 ml of RNAprotect Bacteria reagent (Qiagen), and the samples were processed according to the manufacturer's instructions. Cell pellets were stored at −70°C for later RNA extraction. RNA was extracted and purified as described previously (Koskenniemi et al., 2011).
For each sample, 5 µg of RNA was reverse transcribed to cDNA using the SuperScript Indirect cDNA Labelling System (Invitrogen) and fluorescently labelled with Cy3 or Cy5 mono‐reactive dyes (Amersham Biosciences) according to previously published protocols (Koskenniemi et al., 2011). The labelling efficiency was quantified using a NanoDrop ND‐1000 spectrophotometer (NanoDrop Technologies), and labelled cDNA samples were hybridized to microarrays according to Agilent's procedure, ‘Two‐Color Microarray‐Based Gene Expression Analysis’ (http://www.agilent.com).
Scanning, image analysis and data analyses. Microarrays were scanned at 5 µm resolution with a GenePix 4200 AL scanner (Axon Instruments). The fluorescence signal intensities of each feature were measured and addressed to genomic ORFs using GenePix® Pro 6.0 software (Axon Instruments/Molecular Devices Corp.) with default parameters. The computational alignments were further improved manually, and features that could not be verified were flagged.
This study was a part of a larger experiment, comprising a total of 66 two‐color microarrays, which were analysed as an entity. After pre‐processing, samples involved in this particular study were separated and analysed independently from the remaining data sets. All analyses were performed with the Bioconductor package for statistical analysis using the R programming language (Gentleman et al., 2004). The data were pre‐processed with limma (Smyth and Speed, 2003). The foreground and background median intensity estimates were corrected for the background using the normexp‐function (offset set to 50) (Ritchie et al., 2007), normalized within arrays using loess (100 iterations, suspicious spots and probe sequences matching multiple hits or borders of the coding regions were down weight to zero, and intergenic probe sequences were down weight to 0.1) (Smyth and Speed, 2003) and normalized between arrays using quantile normalization (Yang and Thorne, 2003). Gene expression ratios were obtained by taking the average over the log2‐transformed expression ratios of features describing the same gene and matching a single genetic coding locus. Expression ratios between conditions were calculated using linear models implemented in limma (Smyth, 2004). Gene expression ratios were calculated for 2798 genes out of the total 2944 genes predicted to be encoded by the genome (95% coverage). After pre‐processing, samples involved in this study were separated from the remaining samples, and the statistical significance of the expression ratio of a gene between two conditions was analysed using the paired t‐test method implemented in CyberT (the Bayesian prior estimate of within‐treatment variance was set to five, and the window size was set to 101) (Baldi and Long, 2001). P‐values were adjusted for multiple hypothesis correction using the Bonferroni method and the number of performed t‐tests in total (61 556). Significantly differentially expressed genes with at least a twofold change in expression ratio between paired time points were clustered in MeV version 4.3 (Saeed et al., 2006) by using the K‐means clustering algorithm and a Euclidean distance metric. Data for the classification of COGs were obtained from Kankainen and colleagues (2009). The statistical significance of groups was assessed using the hypergeometric distribution, and the results were corrected with the Bonferroni method. Microarray platform and data details are available at the Gene Expression Omnibus (GEO platform GPL10580 and series GSE28903).
Proteomic methods
Protein extraction and CyDye labeling. Cells were harvested from 1–5 ml of samples by centrifuging at +4°C and washed twice with ice‐cold 50 mM Tris‐HCl pH 8 (Sigma‐Aldrich). Bacterial cells were broken by bead beating (Koskenniemi et al., 2009). The protein samples were processed using a Clean‐up kit (GE Healthcare) and dissolved in 25–50 µl of a buffer containing 7 M urea (Sigma‐Aldrich), 2 M thiourea (Sigma‐Aldrich), 4% 3‐[(3‐Cholamidopropyl)dimethylam‐monio]‐1‐propanesulfonate (Sigma‐Aldrich) and 30 mM Tris (Bio‐Rad). Protein concentrations in the samples were determined using a 2‐D Quant Kit (GE Healthcare).
Protein samples were adjusted to pH 8.5 by adding 2 M Tris. The samples were then labelled using Cy2, Cy3 and Cy5 dyes (CyDye DIGE Fluor minimal dyes; GE Healthcare), according to the Ettan™ 2‐D DIGE protocol and Koskenniemi and colleagues (2009). The experiment was divided into three parts; each time point (4 h, mid‐exponential; 12 h, late exponential; 28 h, late stationary growth state) was separately compared with the early stationary‐phase (20 h) time point. Labelling was performed reciprocally so that both the early stationary‐phase samples and the other time point samples (4 h, 12 h or 28 h) were labelled with Cy3 and Cy5 to account for any preferential protein labelling by the CyDyes (labelling chart as Table S2). Cy2 was used for the internal pooled standard, consisting of equal amounts of each sample.
2‐DE and DeCyder analyses. The labelled proteins were separated by isoelectric focusing using immobilized pH gradient strips (24 cm, pH 3–10 non‐linear, Bio‐Rad) and a Protean IEF Cell (Bio‐Rad) for 80 000 Vh (Koskenniemi et al., 2009). The strips were then equilibrated and loaded onto 12% acrylamide gels, and the gels were subjected to electrophoresis in an Ettan™ DALTsix Electrophoresis Unit (GE Healthcare) (Koskenniemi et al., 2009). The gels were imaged using an FLA‐5100 laser scanner (Fujifilm), and the images were cropped to identical sizes by removing areas extraneous to the proteins spots with ImageQuant™ TL 7.0 software (GE Healthcare). After scanning, the gels were fixed in 30% ethanol and 0.5% acetic acid for a minimum of 60 min and then silver stained (O'Connell and Stults, 1997). Image and statistical analyses for the cropped 2‐D DIGE gels were performed using DeCyder™ 2D 7.0 software (GE Healthcare) (Koskenniemi et al., 2009). Approximately 700–800 separate protein spots were detected on each gel. Protein spots were picked and identified if they demonstrated at least a 1.5‐fold difference in average spot volume ratio (average ratio ≥ 1.5 or ≤ −1.5) between different time points in at least three out of four separate biological replicates. Statistical significance was calculated using the Student's t‐test; a P‐value value less than 0.05 was considered significant.
Protein identification by mass spectrometry. MS‐compatible silver staining (O'Connell and Stults, 1997) was performed to visualize the protein spots for identification. Protein spots of interest were digested in‐gel with trypsin and the peptides recovered as previously described (Koskenniemi et al., 2009). The resulting peptides were analysed by peptide‐mass fingerprinting (PMF) or by fragment ion analysis with LC‐ESI‐MS/MS as described in detail previously (Koskenniemi et al., 2011).
The PMF spectra were processed with FlexAnalysis version 3.0 (Bruker Daltonik). The PMF and LC‐ESI‐MS/MS data were searched with the local Mascot version 2.2 (Matrix Science) against the in‐house database of the published ORF set of L. rhamnosus GG, which contains 2944 protein entries (Kankainen et al., 2009), using the Biotools 3.0 (Bruker Daltonik) and ProteinPilot 2.0.1 (Applied Biosystem) interfaces respectively. The search criteria for both Mascot searches were as follows: trypsin digestion with one missed cleavage allowed, carbamidomethyl modification of cysteine as a fixed modification and oxidation of methionine as a variable modification. For the PMF spectra the maximum peptide mass tolerance was ±50 ppm. The protonated molecule ion ‘MH+’ and ‘monoisotopic’ were defined for the peak mass data input. For the LC‐ESI‐MS/MS spectra, both the maximum precursor ion mass tolerance and the MS/MS fragment ion mass tolerance were 0.2 Da, and a peptide charge state of +1, +2 or +3 was used. A successful identification was reported when a significant match (P ≤ 0.05) was obtained. In addition, to consider the LC‐ESI‐MS/MS identification reliable, a minimum of two peptides with an ions score of at least 40 was required.
Acknowledgments
Elina Ahola‐Iivarinen, Hanna Jefremoff, Saija Laakso and Eeva‐Marja Turkki are acknowledged for their technical assistance. This work was supported financially by the Academy of Finland (Grants 210740 and 117746), the Finnish Funding Agency for Technology and Innovations (Grant 201/08) and by an ABS Graduate School scholarship to K.K.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
K‐means clustering of transcriptomic expression profiles. Significant changes in gene expression are represented as log2 intensity ratio values between paired time points. Genes were grouped into 16 clusters using a Euclidean distance metric, and the analysis included 636 genes intotal. The purple expression profile in each cluster represents the cluster median.
Representative 2‐D DIGE images of proteins extracted from different growth phases: (A) 4 h and 20 h, (B) 12 h and 20 h and (C) 20 h and 28 h. The total amount of protein used for CyDye labelling was 105 µg. The numbered protein spots were cut from silver‐stained 2‐D DIGE gels and subjected to MS or MS/MS identification. Protein spots that were more abundant at time point 20 h are shown in red and those more abundant at time points 4 h, 12 h or 28 h are shown in green. The protein spots with no difference in abundance between the treatments are yellow.
Log2 ratio values of significantly (P ≤ 0.01) differentially expressed genes with at least a twofold change in the expression ratio between paired time points. Clusters refer to Fig. S1.
Set‐up of the DIGE experiments. Letters A?D refer to biological replicate samples. Gels 1?4, 5?8 and 9?12 each form a separate DIGE experiment.
Table S3. Proteins with differing abundance between the mid‐exponential (4 h) and early stationary (20 h) growth phases.
Table S4. Proteins with differing abundance between the late exponential (12 h) and early stationary (20 h) growth phases.
Table S5. Proteins with differing abundance between the early stationary (20 h) and late stationary (28 h) growth phases.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alander M., Korpela R., Saxelin M., Vilpponen‐Salmela T., Mattila‐Sandholm T., von Wright A. Recovery of Lactobacillus rhamnosus GG from human colonic biopsies. Lett Appl Microbiol. 1997;24:361–364. doi: 10.1046/j.1472-765x.1997.00140.x. [DOI] [PubMed] [Google Scholar]
- Alander M., Satokari R., Korpela R., Saxelin M., Vilpponen‐Salmela T., Mattila‐Sandholm T., von Wright A. Persistence of colonization of human colonic mucosa by a probiotic strain, Lactobacillus rhamnosus GG, after oral consumption. Appl Environ Microbiol. 1999;65:351–354. doi: 10.1128/aem.65.1.351-354.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcántara C., Sarmiento‐Rubiano L.A., Monedero V., Deutscher J., Pérez‐Martínez G., Yebra M.J. Regulation of Lactobacillus casei sorbitol utilization genes requires DNA‐binding transcriptional activator GutR and the conserved protein GutM. Appl Environ Microbiol. 2008;74:5731–5740. doi: 10.1128/AEM.00230-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelsson L. Lactic acid bacteria: classification and physiology. In: Salminen S., von Wright A., Ouwehand A., editors. Marcel Dekker; 2004. pp. 1–66. [Google Scholar]
- Azcarate‐Peril M.A., Tallon R., Klaenhammer T.R. Temporal gene expression and probiotic attributes of Lactobacillus acidophilus during growth in milk. J Dairy Sci. 2009;92:870–886. doi: 10.3168/jds.2008-1457. [DOI] [PubMed] [Google Scholar]
- van Baarlen P., Troost F.J., van Hemert S., van der Meer C., de Vos W.M., de Groot P.J. Differential NF‐κB pathways induction by Lactobacillus plantarum in the duodenum of healthy humans correlating with immune tolerance. Proc Natl Acad Sci USA. 2009;106:2371–2376. doi: 10.1073/pnas.0809919106. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldi P., Long A.D. A Bayesian framework for the analysis of microarray expression data: regularized t‐test and statistical inferences of gene changes. Bioinformatics. 2001;17:509–519. doi: 10.1093/bioinformatics/17.6.509. [DOI] [PubMed] [Google Scholar]
- Bandow J.E., Becher D., Büttner K., Hochgräfe F., Freiberg C., Brötz H., Hecker M. The role of peptide deformylase in protein biosynthesis: a proteomic study. Proteomics. 2003;3:299–306. doi: 10.1002/pmic.200390043. [DOI] [PubMed] [Google Scholar]
- Claes I.J.J., Lebeer S., Shen C., Verhoeven T.L.A., Dilissen E., De Hertogh G. Impact of lipoteichoic acid modification on the performance of the probiotic Lactobacillus rhamnosus GG in experimental cholitis. Clin Exp Immunol. 2010;162:306–314. doi: 10.1111/j.1365-2249.2010.04228.x. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen D.P.A., Renes J., Bouwman F.G., Zoetendal E.G., Mariman E., de Vos W.M., Vaughan E.E. Proteomic analysis of log to stationary growth phase Lactobacillus plantarum cells and a 2‐DE database. Proteomics. 2006;6:6485–6493. doi: 10.1002/pmic.200600361. [DOI] [PubMed] [Google Scholar]
- Cozzone A.J. Post‐translational modification of proteins by reversible phosphorylation in prokaryotes. Biochimie. 1998;80:43–48. doi: 10.1016/s0300-9084(98)80055-2. [DOI] [PubMed] [Google Scholar]
- De Angelis M., Gobbetti M. Environmental stress responses in Lactobacillus: a review. Proteomics. 2004;4:106–122. doi: 10.1002/pmic.200300497. [DOI] [PubMed] [Google Scholar]
- Deepika G., Green R.J., Frazier R.A., Charalampopoulos D. Effect of growth time on the surface and adhesion properties of Lactobacillus rhamnosus GG. J Appl Microbiol. 2009;107:1230–1240. doi: 10.1111/j.1365-2672.2009.04306.x. [DOI] [PubMed] [Google Scholar]
- Eymann C., Becher D., Bernhardt J., Gronau K., Klutzny A., Hecker M. Dynamics of protein phosphorylation on Ser/Thr/Tyr in Bacillus subtilis. Proteomics. 2007;7:3509–3526. doi: 10.1002/pmic.200700232. [DOI] [PubMed] [Google Scholar]
- Francl A.L., Thongaram T., Miller M.J. The PTS transporters of Lactobacillus gasseri ATCC 33323. BMC Microbiol. 2010;10:77. doi: 10.1186/1471-2180-10-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman R.C., Carey V.J., Bates D.M., Bolstad B., Dettling M., Dudoit S. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin B.R., Gorbach S.L., Saxelin M., Barakat S., Gualtieri L., Salminen S. Survival of Lactobacillus species (strain GG) in human gastrointestinal tract. Dig Dis Sci. 1992;37:121–128. doi: 10.1007/BF01308354. [DOI] [PubMed] [Google Scholar]
- Guarino A., Lo Vecchio A., Canani R.B. Probiotics as prevention and treatment for diarrhea. Curr Opin Gastroenterol. 2009;25:18–23. doi: 10.1097/MOG.0b013e32831b4455. [DOI] [PubMed] [Google Scholar]
- Habimana O., Le Goff C., Juillard V., Bellon‐Fontaine M.‐N., Buist G., Kulakauskas S., Briandet R. Positive role of cell wall anchored proteinase PrtP in adhesion of lactococci. BMC Microbiol. 2007;7:36. doi: 10.1186/1471-2180-7-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakka K., Savilahti E., Pönkä A., Meurman J.H., Poussa T., Näse L. Effect of long term consumption of probiotic milk on infections in children attending day care centres: double blind, randomised trial. BMJ. 2001;322:1–5. doi: 10.1136/bmj.322.7298.1327. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hojsak I., Snovak N., Abdović S., Szajewska H., Mišak Z., Kolaček S. Lactobacillus GG in the prevention of gastrointestinal and respiratory tract infections in children who attend day care centers: a randomized, double‐blind, placebo‐controlled trial. Clin Nutr. 2010;29:312–316. doi: 10.1016/j.clnu.2009.09.008. [DOI] [PubMed] [Google Scholar]
- Isolauri E., Arvola T., Sütas Y., Moilanen E., Salminen S. Probiotics in the management of atopic eczema. Clin Exp Allergy. 2000;30:1604–1610. doi: 10.1046/j.1365-2222.2000.00943.x. [DOI] [PubMed] [Google Scholar]
- Jacobsen C.N., Rosenfeldt Nielsen V., Hayford A.E., Møller P.L., Michaelsen K.F., Pærregaard A. Screening of probiotic activities of forty‐seven strains of Lactobacillus spp. by in vitro techniques and evaluation of the colonization ability of five selected strains in humans. Appl Environ Microbiol. 1999;65:4949–4956. doi: 10.1128/aem.65.11.4949-4956.1999. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalliomäki M., Salminen S., Arvilommi H., Kero P., Koskinen P., Isolauri E. Probiotics in primary prevention of atopic disease: a randomised placebo‐controlled trial. Lancet. 2001;357:1076–1079. doi: 10.1016/S0140-6736(00)04259-8. [DOI] [PubMed] [Google Scholar]
- Kalliomäki M., Salminen S., Poussa T., Arvilommi H., Isolauri E. Probiotics and prevention of atopic disease: 4‐year follow‐up of a randomised placebo‐controlled trial. Lancet. 2003;361:1869–1871. doi: 10.1016/S0140-6736(03)13490-3. [DOI] [PubMed] [Google Scholar]
- Kalliomäki M., Salminen S., Poussa T., Isolauri E. Probiotics during the first 7 years of life: a cumulative risk reduction of eczema in a randomized, placebo‐controlled trial. J Allergy Clin Immunol. 2007;119:1019–1021. doi: 10.1016/j.jaci.2006.12.608. [DOI] [PubMed] [Google Scholar]
- Kankainen M., Paulin L., Tynkkynen S., von Ossowski I., Reunanen J., Partanen P. Comparative genomic analysis of Lactobacillus rhamnosus GG reveals pili containing a human‐mucus binding protein. Proc Natl Acad Sci USA. 2009;106:17193–17198. doi: 10.1073/pnas.0908876106. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koistinen K.M., Plumed‐Ferrer C., Lehesranta S.J., Kärenlampi S.O., von Wright A. Comparison of growth‐phase‐dependent cytosolic proteomes of two Lactobacillus plantarum strains used in food and feed fermentations. FEMS Microbiol Lett. 2007;273:12–21. doi: 10.1111/j.1574-6968.2007.00775.x. [DOI] [PubMed] [Google Scholar]
- Koskenniemi K., Koponen J., Kankainen M., Savijoki K., Tynkkynen S., de Vos W.M. Proteome analysis of Lactobacillus rhamnosus GG using 2‐D DIGE and mass spectrometry shows differential protein production in laboratory and industrial‐type growth media. J Proteome Res. 2009;8:4993–5007. doi: 10.1021/pr9003823. et al. [DOI] [PubMed] [Google Scholar]
- Koskenniemi K., Laakso K., Koponen J., Kankainen M., Greco D., Auvinen P. Proteomics and transcriptomics characterization of bile stress response in probiotic Lactobacillus rhamnosus GG. Mol Cell Proteomics. 2011;10:M110.002741. doi: 10.1074/mcp.M110.002741. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laparra J.M., Sanz Y. Comparison of in vitro models to study bacterial adhesion to the intestinal epithelium. Lett Appl Microbiol. 2009;49:695–701. doi: 10.1111/j.1472-765X.2009.02729.x. [DOI] [PubMed] [Google Scholar]
- Lebeer S., Verhoeven T.L.A., Perea Vélez M., Vanderleyden J., De Keersmaecker S.C.J. Impact of environmental and genetic factors on biofilm formation by the probiotic strain Lactobacillus rhamnosus GG. Appl Environ Microbiol. 2007;73:6768–6775. doi: 10.1128/AEM.01393-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebeer S., Vanderleyden J., De Keersmaecker S.C.J. Genes and molecules of lactobacilli supporting probiotic action. Microbiol Mol Biol Rev. 2008;72:728–764. doi: 10.1128/MMBR.00017-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebeer S., Verhoeven T.L.A., Francius G., Schoofs G., Lambrichts I., Dufrêne Y. Identification of a gene cluster for the biosynthesis of a long, galactose‐rich exopolysaccharide in Lactobacillus rhamnosus GG and functional analysis of the priming glycosyltransferase. Appl Environ Microbiol. 2009;75:3554–3563. doi: 10.1128/AEM.02919-08. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebeer S., Vanderleyden J., De Keersmaecker S.C.J. Host interactions of probiotic bacterial surface molecules: comparison with commensals and pathogens. Nat Rev Microbiol. 2010;8:171–184. doi: 10.1038/nrmicro2297. [DOI] [PubMed] [Google Scholar]
- Lebeer S., Claes I.J.J., Verhoeven T.L.A., Vanderleyden J., De Keersmaecker S.C.J. Exopolysaccharides of Lactobacillus rhamnosus GG form a protective shield against innate immune factors in the intestine. Microb Biotechnol. 2011;4:368–374. doi: 10.1111/j.1751-7915.2010.00199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macek B., Mijakovic I., Olsen J.V., Gnad F., Kumar C., Jensen P.R., Mann M. The serine/threonine/tyrosine phosphoproteome of the model bacterium Bacillus subtilis. Mol Cell Proteomics. 2007;6:697–707. doi: 10.1074/mcp.M600464-MCP200. [DOI] [PubMed] [Google Scholar]
- Macek B., Gnad F., Soufi B., Kumar C., Olsen J.V., Mijakovic I., Mann M. Phosphoproteome analysis of E. coli reveals evolutionary conservation of bacterial Ser/Thr/Tyr phosphorylation. Mol Cell Proteomics. 2008;7:299–307. doi: 10.1074/mcp.M700311-MCP200. [DOI] [PubMed] [Google Scholar]
- Majamaa H., Isolauri E. Probiotics: a novel approach in the management of food allergy. J Allergy Clin Immunol. 1997;99:179–185. doi: 10.1016/s0091-6749(97)70093-9. [DOI] [PubMed] [Google Scholar]
- Mandlik A., Swierczynski A., Das A., Ton‐That H. Pili in Gram‐positive bacteria: assembly, involvement in colonization and biofilm development. Trends Microbiol. 2008;16:33–40. doi: 10.1016/j.tim.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Näse L., Hatakka K., Savilahti E., Saxelin M., Pönkä A., Poussa T. Effect of long‐term consumption of a probiotic bacterium, Lactobacillus rhamnosus GG, in milk on dental caries and caries risk in children. Caries Res. 2001;35:412–420. doi: 10.1159/000047484. et al. [DOI] [PubMed] [Google Scholar]
- Neuhaus F.C., Baddiley J. A continuum of anionic charge: structures and functions of d‐alanyl‐teichoic acids in gram‐positive bacteria. Microbiol Mol Biol Rev. 2003;67:686–723. doi: 10.1128/MMBR.67.4.686-723.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves A.R., Wietske A.P., Kok J., Kuipers O.P., Santos H. Overview on sugar metabolism and its control in Lactococcus lactis– the input from in vivo NMR. FEMS Microbiol Rev. 2005;29:531–554. doi: 10.1016/j.femsre.2005.04.005. [DOI] [PubMed] [Google Scholar]
- O'Connell K.L., Stults J.T. Identification of mouse liver proteins on two‐dimensional electrophoresis gels by matrix‐assisted laser desorption/ionization mass spectrometry of in situ enzymatic digests. Electrophoresis. 1997;18:349–359. doi: 10.1002/elps.1150180309. [DOI] [PubMed] [Google Scholar]
- Ouwehand A.C., Tuomola E.M., Tölkkö S., Salminen S. Assessment of adhesion properties of novel probiotic strains to human intestinal mucus. Int J Food Microbiol. 2001;64:119–126. doi: 10.1016/s0168-1605(00)00440-2. [DOI] [PubMed] [Google Scholar]
- Ritchie M.E., Silver J., Oshlack A., Holmes M., Diyagama D., Holloway A., Smyth G.K. A comparison of background correction methods for two‐colour microarrays. Bioinformatics. 2007;23:2700–2707. doi: 10.1093/bioinformatics/btm412. [DOI] [PubMed] [Google Scholar]
- Saeed A.I., Bhagabati N.K., Braisted J.C., Liang W., Sharov V., Howe E.A. TM4 microarray software suite. Methods Enzymol. 2006;411:134–193. doi: 10.1016/S0076-6879(06)11009-5. et al. [DOI] [PubMed] [Google Scholar]
- Sánchez B., Bressollier P., Chaignepain S., Schmitter J.‐M., Urdaci M.C. Identification of surface‐associated proteins in the probiotic bacterium Lactobacillus rhamnosus GG. Int Dairy J. 2009;19:85–88. [Google Scholar]
- Savijoki K., Ingmer H., Varmanen P. Proteolytic systems of lactic acid bacteria. Appl Microbiol Biotechnol. 2006;71:394–406. doi: 10.1007/s00253-006-0427-1. [DOI] [PubMed] [Google Scholar]
- Seth A., Yan F., Polk D.B., Rao R.K. Probiotics ameliorate the hydrogen peroxide‐induced epithelial barrier disruption by a PKC‐ and MAP kinase‐dependent mechanism. Am J Physiol Gastrointest Liver Physiol. 2008;294:G1060–G1069. doi: 10.1152/ajpgi.00202.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article 3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- Smyth G.K., Speed T. Normalization of cDNA microarray data. Methods. 2003;31:265–273. doi: 10.1016/s1046-2023(03)00155-5. [DOI] [PubMed] [Google Scholar]
- Song X.‐M., Connor W., Hokamp K., Babiuk L.A., Potter A.A. The growth phase‐dependent regulation of the pilus locus genes by two‐component system TCS08 in Streptococcus pneumoniae. Microb Pathog. 2009;46:28–35. doi: 10.1016/j.micpath.2008.10.006. [DOI] [PubMed] [Google Scholar]
- Soufi B., Gnad F., Jensen P.R., Petranovic D., Mann M., Mijakovic I., Macek B. The Ser/Thr/Tyr phosphoproteome of Lactococcus lactis IL1403 reveals multiply phosphorylated proteins. Proteomics. 2008;8:3486–3493. doi: 10.1002/pmic.200800069. [DOI] [PubMed] [Google Scholar]
- Szajewska H., Kotowska M., Mrukowicz J.Z., Armańska M., Mikolajczyk W. Efficacy of Lactobacillus GG in prevention of nosocomial diarrhea in infants. J Pediatr. 2001;138:361–365. doi: 10.1067/mpd.2001.111321. [DOI] [PubMed] [Google Scholar]
- Szajewska H., Skórka A., Ruszczyński M., Gieruszczak‐Białek D. Meta‐analysis: Lactobacillus rhamnosus GG for treating acute diarrhoea in children. Aliment Pharmacol Ther. 2007;25:871–881. doi: 10.1111/j.1365-2036.2007.03282.x. [DOI] [PubMed] [Google Scholar]
- Tao Y., Drabik K.A., Waypa T.S., Musch M.W., Alverdy J.C., Schneewind O. Soluble factors from Lactobacillus GG activate MAPKs and induce cytoprotective heat shock proteins in intestinal epithelial cells. Am J Physiol Cell Physiol. 2006;290:C1018–C1030. doi: 10.1152/ajpcell.00131.2005. et al. [DOI] [PubMed] [Google Scholar]
- Tuomola E.M., Ouwehand A.C., Salminen S.J. Chemical, physical and enzymatic pre‐treatments of probiotic lactobacilli alter their adhesion to human intestinal mucus glycoproteins. Int J Food Microbiol. 2000;60:75–81. doi: 10.1016/s0168-1605(00)00319-6. [DOI] [PubMed] [Google Scholar]
- Vélez M.P., Petrova M.I., Lebeer S., Verhoeven T.L., Claes I., Lambrichts I. Characterization of MabA, a modulator of Lactobacillus rhamnosus GG adhesion and biofilm formation. FEMS Immunol Med Microbiol. 2010;59:386–398. doi: 10.1111/j.1574-695X.2010.00680.x. et al. [DOI] [PubMed] [Google Scholar]
- Welman A., Maddox I., Archer R. Exopolysaccharide and extracellular metabolite production by Lactobacillus delbrueckii subsp. bulgaricus, grown on lactose in continuous culture. Biotechnol Lett. 2003;25:1515–1520. doi: 10.1023/a:1025432003915. [DOI] [PubMed] [Google Scholar]
- Welman A.D., Maddox I.S., Archer R.H. Metabolism associated with raised metabolic flux to sugar nucleotide precursors of exopolysaccharides in Lactobacillus delbrueckii subsp. bulgaricus. J Ind Microbiol Biotechnol. 2006;33:391–400. doi: 10.1007/s10295-005-0075-y. [DOI] [PubMed] [Google Scholar]
- Yan F., Polk D.B. Probiotic bacterium prevents cytokine‐induced apoptosis in intestinal epithelial cells. J Biol Chem. 2002;277:50959–50965. doi: 10.1074/jbc.M207050200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan F., Cao H., Cover T.L., Whitehead R., Washington M.K., Polk D.B. Soluble proteins produced by probiotic bacteria regulate intestinal epithelial cell survival and growth. Gastroenterology. 2007;132:562–575. doi: 10.1053/j.gastro.2006.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.H., Thorne N.P. Normalization for two‐color cDNA microarray data. In: Goldstein D.R., editor. Institute of Mathematical Statistics; 2003. pp. 403–418. [Google Scholar]
- Zhang Y.‐M., Rock C.O. Transcriptional regulation in bacterial membrane lipid synthesis. J Lipid Res. 2009;50:S115–S119. doi: 10.1194/jlr.R800046-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
K‐means clustering of transcriptomic expression profiles. Significant changes in gene expression are represented as log2 intensity ratio values between paired time points. Genes were grouped into 16 clusters using a Euclidean distance metric, and the analysis included 636 genes intotal. The purple expression profile in each cluster represents the cluster median.
Representative 2‐D DIGE images of proteins extracted from different growth phases: (A) 4 h and 20 h, (B) 12 h and 20 h and (C) 20 h and 28 h. The total amount of protein used for CyDye labelling was 105 µg. The numbered protein spots were cut from silver‐stained 2‐D DIGE gels and subjected to MS or MS/MS identification. Protein spots that were more abundant at time point 20 h are shown in red and those more abundant at time points 4 h, 12 h or 28 h are shown in green. The protein spots with no difference in abundance between the treatments are yellow.
Log2 ratio values of significantly (P ≤ 0.01) differentially expressed genes with at least a twofold change in the expression ratio between paired time points. Clusters refer to Fig. S1.
Set‐up of the DIGE experiments. Letters A?D refer to biological replicate samples. Gels 1?4, 5?8 and 9?12 each form a separate DIGE experiment.
Table S3. Proteins with differing abundance between the mid‐exponential (4 h) and early stationary (20 h) growth phases.
Table S4. Proteins with differing abundance between the late exponential (12 h) and early stationary (20 h) growth phases.
Table S5. Proteins with differing abundance between the early stationary (20 h) and late stationary (28 h) growth phases.