

Summary
In recent years, the application of approaches for harvesting DNA from the environment, the so‐called, ‘metagenomic approaches’ has proven to be highly successful for the identification, isolation and generation of novel enzymes. Functional screening for the desired catalytic activity is one of the key steps in mining metagenomic libraries, as it does not rely on sequence homology. In this mini‐review, we survey high‐throughput screening tools, originally developed for directed evolution experiments, which can be readily adapted for the screening of large libraries. In particular, we focus on the use of in vitro compartmentalization (IVC) approaches to address potential advantages and problems the merger of culture‐independent and IVC techniques might bring on the mining of enzyme activities in microbial communities.
Introduction
According to the well‐established dilemma of environmental microbiology only a minority of microorganisms is readily culturable (Raes et al., 2007). For this reason, a wide range of approaches collectively described as ‘environmental genomics’ or ‘metagenomics’ have been developed to study such communities without culturing individual organisms. The term ‘metagenomics’ has been used broadly to encompass research ranging from examining environmental DNA in enzyme screenings and drug discovery to randomly sample the genomes (whole‐sequencing) from a small subset of organisms present in an environment (Gabor et al., 2007).
The main task of the sequencing‐based metagenomics, initiated through shotgun sequencing and now becoming feasible through 454 pyro‐sequencing is to reconstruct the metabolism of the organisms comprising the community and to predict their functional roles in the ecosystem: 134 whole metagenomic projects using 454 technology platform are running at the moment (Liolios et al., 2008). Near‐complete genome reconstructions were achieved for the dominant members of communities with a small number of organism types and for a few highly abundant organisms from diverse communities (see examples in Tables 1 and 2). The genome sequence information also serves as cornerstone of enzyme discovery because of the possibility to access, through virtual sequence homology screening, to an immense repertoire of millions of known and unknown proteins predicted by the environmental sequence information (Ferrer et al., 2007). However, the clear disadvantage of this approach for enzyme discovery is its full reliance on the existing genome annotations: the in silico comprehensive functional annotation analysis of proteins remains difficult, error‐prone and machine‐based processes and the options for comparative analyses are limited (Hallin et al., 2008). Better annotation quality and curated functional information would enable improved gene function predictions in newly sequenced organisms and environmental samples, and allow high‐throughput (HT) evaluation of context‐dependent expression and function (Segota et al., 2008).
Table 1.
Analysis of microbial communities through shotgun metagenomic sequencing (only environmental samples are shown).
| Sample | Library size | Host or vector system used | Average insert size (kbp) | Biodiversity | References |
|---|---|---|---|---|---|
| Sargasso Sea | 1 985 561a | Bst XI linearized pBR322 derivative | 2–6 | Samples were dominated by genes from Proteobacteria (primarily subgroups Alpha, Beta and Gamma) with moderate contributions from Firmicutes, Cyanobacteria and species in the CFB phyla (Cytophaga, Flavobacterium and Bacteroides). Poor sequencing coverage enabled the assembly of only two near‐complete genomes. Here, 1.6 G base pair of unique metagenomic DNA sequences were obtained. | Venter et al. (2004) |
| Soil | 1129 (Bacteria)a 527 (Archaea)a 919 (Fungi)a 4577 (Viruses)a | pCR®2.1‐TOPO (Bacterial, Archaeal and Fungal) pSMART (viral) | 0.49 (Viruses) | This is the first study to use sequencing to characterize soil viral communities. Within each of the four microbial groups, data showed minimal taxonomic overlap between sites, suggesting that soil archaea, bacteria, fungi and viruses are globally as well as locally diverse. | Fierer et al. (2007) |
| Acid mine drainage biofilm | 103 462a | pUC18 | 3.2 | Authors report the reconstruction of near‐complete genomes of Leptospirillum group II and Ferroplasma type II and partial recovery of three other genomes. | Tyson et al. (2004) |
| Global Ocean | 7 697 926a | Bst XI linearized pBR322 derivative | 2 | Authors report a metagenomic study of the marine planktonic microbiota in which surface (mostly marine) water samples were analysed as part of the Sorcerer II Global Ocean Sampling expedition. The resulting 7.7 million sequencing reads form 41 samples provide an unprecedented look at the great diversity and heterogeneity in naturally occurring microbial populations. | Rusch et al. (2007) |
| Soil | 1 186 200b | pJN105/pCF430 (small inserts) pBeloBAC11 (big inserts) | 2.7–45 | Authors designed a metagenomic analysis to isolate antibiotic resistance genes from 6 libraries of soil. They identified nine clones expressing resistance to aminoglycoside antibiotics and one expressing tetracycline resistance. | Riesenfeld et al. (2004) |
Number of reads produced or
b. independent clones.
Table 2.
Metagenomic populations characterized through the 454 pyrosequencing technology (only environmental samples are shown).
| Sample | 454‐library sizea | Average length of reads | Biodiversity | References |
|---|---|---|---|---|
| Solar saltern | 582 681 | ≈100 bp | 151 genomic fragments were dominated by different halophilic archaea and by Salinibacter. | Krause et al. (2008) |
| Soudan Mine | 334 386 (Red sample) 388 627 (Black sample) | 106 bp (RS) 99.1 bp (BS) | 76 16S rDNA‐fragments dominated by Alpha‐ and Gammaproteobacteria (Red Sample) and 24 16S rDNA‐fragments dominated by Actinobacteria (Black Sample). | Edwards et al. (2006) |
| Coral Porites astreoides | 316 279 | 102 bp | The most prominent bacterial groups were Proteobacteria, Firmicutes, Cyanobacteria and Actinobacteria. | Wegley et al. (2007) |
| North Atlantic Deep Water and Axial Seamount | 118 778 | < 120 bp | Nearly 50% of the population corresponds to divergent Epsilonproteobacteria. | Sogin et al. (2006) |
| Ocean surface waters | 414 323 (DNA) 128.324 (cDNA) | 110 bp (DNA) 114 bp (cDNA) | The genus Prochlorococcus and Alphaproteobacteria (genus Pelagibacter) were the two most highly represented taxonomic groups in both DNA and cDNA libraries. | Frias‐Lopez et al. (2008) |
| Global soil | 314 041 | 96.4 bp | Results indicate that crenarchaeota may be the most abundant ammonia‐oxidizing organisms in soil ecosystems on Earth. | Leininger et al. (2006) |
| Marine virome of four oceanic regions | 1 768 297 | 102 bp | Metagenomic analyses of 184 viral assemblages collected over a decade and representing 68 sites in four major oceanic regions. This work provides evidence that the composition of viral assemblages varies in different geographic regions. | Angly et al. (2006) |
| Northwest Atlantic & Eastern Tropical Pacific Seawater | ≈100 bp | Analysis of 7.7 million sequencing reads (6.3 billion bp) from the microbes collected across a several‐thousand km marine transects. | Yooseph et al. (2007) | |
| Surface and hypersaline marine, freshwater samples | ≈100 bp | Metagenomic analysis of 37 samples. Results showed that most of the 154 662 viral peptide sequences identified were not similar to those in the current database and that only few thousands genes encoding metabolic and cellular functions could be unambiguously identified. | Williamson et al. (2008) |
Number of reads.
For this reason, the selection‐based approach that involves construction of small‐ to large‐insert expression libraries, especially those made in lambda phage, cosmid or copy‐control fosmid vectors, which are further implemented for a direct activity screening (Gabor et al., 2007), is the best option for enzyme discovery. In fact, the exploitation of natural microbial diversity for the identification and isolation of novel enzymes is currently an extremely active field in microbiology (see examples in Beloqui et al., 2008). Further, the powers of metagenomics approaches can be combined with directed evolution methodologies for the generation of a biotechnological platform that will allow for the discovery and engineering of novel enzymes (Fig. 1). Here, a critical step is to screen for genes encoding novel enzymes among millions or billions of unrelated DNA sequences, which highlights the need of high‐throughput screening (HTS) methodologies in order to increase the chance of identifying, isolating and generating novel enzymes. We will review important HTS strategies that enable the screening or selection of extremely large libraries for a variety of enzymatic activities, with an emphasis on the use of in vitro compartmentalization (IVC).
Figure 1.
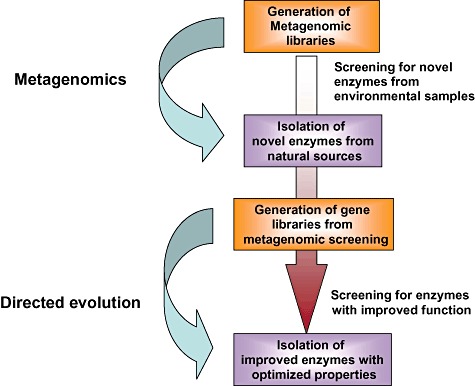
Flow chart illustrating the identification, isolation and further engineering of novel enzymes using metagenomic and directed evolution approaches. Newly identified enzymes from metagenomic libraries can serve as an ideal starting point for the directed evolution of enzymes with improved properties. High‐throughput screening for enzymatic activity can be used, both for screening large metagenomic libraries and subsequently, for directed evolution experiments.
Metagenome analysis requires HTS strategies
There are two distinct mining strategies taken in metagenomics to capture the largest amount of the available enzymatic resources in environmental DNA (Fig. 2). First, the application of the sequence‐based approach that involves the design of PCR primers or hybridization probes for the target genes that are derived from conserved regions of already known protein families (Gabor et al., 2007). The use of microarrays to profile libraries offers to this strategy an effective approach for characterizing many clones rapidly in a HT fashion (Sebat et al., 2003). This format is referred to as a metagenome microarray (MGA). In the MGA format, the ‘probe’ and ‘target’ concept is a reversal of those of general cDNA and oligonucleotide microarrays: targets (fosmid clones) are spotted on a slide and a specific gene probe is labelled and used for hybridization. This format of microarray may offer an effective HTS approach for identifying clones from metagenome libraries rapidly without the need of laborious procedures for screening various target genes. As an example, Sebat and colleagues (2003) and Park and colleagues (2008) used microarray platforms to screen microbial genomes and whole community genomes. However, the difficulty and limitation of this approach is related to achieving high hybridization efficiency and that the target genes derived from conserved regions of already known protein families reduce our chances for obtaining fundamentally new proteins (Gabor et al., 2007). On the other hand, based on the few metagenome surveys of microbial and viral communities completed to date, a consensus is emerging that environmental communities are extraordinarily diverse and contain a high proportion (over 60%) of novel sequences with unknown functions that are relatively distant from better known representatives in sequenced genomes (see examples shown in Tables 1 and 2).
Figure 2.
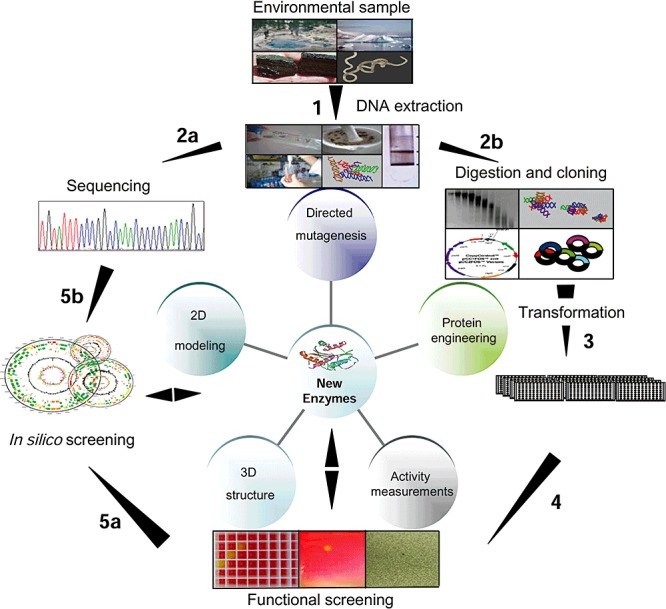
Mining genomes and metagenomes for novel enzymes. A gene library is created from environmental samples (Step 1–3) and used to screen for novel genes (Step 4) cloned into bacteria which can be sequenced (Step 5a). The encoded proteins expressed in appropriate host are then subjected to structure‐function analyses (central panel). Alternatively, large‐scale sequencing of bulk DNA is used for archiving and sequence homology screening purposes to capture the largest amount of the available genetic resources present in environmental samples (Step 5b).
The application of the alternative, activity‐based approach, available at HT scale by using chromogenic or fluorogenic substrates, analyses in a sequence‐independent way dozens of thousands of clones in a single screen (Fig. 2). Certainly, the shortcoming of functional‐screening approach is that even large libraries only provide a small fraction of the environmental diversity and the low frequency of novel enzymes among vast unrelated DNA sequences. The diversity that can be accessed by metagenomic analysis is overwhelming: it was shown that a pristine soil sample contains more than 104 different microbial species and over thousand million open reading frames, many of which encode putative enzymes (Raes et al., 2007). To generate an effective metagenomic library, improved HT‐DNA extraction methods and cloning strategies are required so that the individual genomes in the library provide an acceptable representation of the entire metagenome (Ferrer et al., 2007). The target genes encoding for novel enzymes represent a tiny fraction, in some cases less than 0.01%, of the total nucleic acid sample extracted from environmental sources and therefore the low abundance of the gene in the library may play a critical role in mining programmes (Gabor et al., 2007). For this reason, currently, several enrichment methodologies exist that are either dependent or independent of sequence information, i.e. cultural enrichment methodologies including size‐selective filtration and stable isotope probing, to cite some. However, the main drawbacks of both tools are the danger of enriching fast growing microorganisms that do not utilize the supplied nutrients and the inefficient labelling efficiency and preventing cross‐feeding or recycling of the label within the microbial community (Schloss and Handelsman, 2003) respectively. Another crucial factor in the generation and expression of metagenomic library is the insert size, the gene or operon length and the use of an adequate host organism that is able to express the target gene and other factors in trans to facilitate expression and folding (such as chaperones, cofactors, etc.) (Galvao et al., 2005). Collectively these factors highlight the need for HT cloning and expression independent screenings to increase the chance of isolating novel enzymes. It is therefore conceivable to screen environmental DNA rapidly by applying different IVC‐like strategies using single microdroplets and cell‐free translation systems together with sorting speed screenings (see below). Those may complement the low‐to‐medium colorimetric and genetic traps screening which are not covered here and have been extensively reviewed elsewhere (see Ferrer et al., 2007; Gabor et al., 2007 and references therein).
High‐throughput functional screening or selection assays
In the sections below, we focus on the principles of HTS approaches with the focus on the use of IVC.
The fundamentals of HTS or selection
Screening and selection methodologies should meet the following demands: (i) They should, if possible, directly select for the property of interest –‘you get what you select for’ is an important rule in screening for desired activity. Thus, the substrate should be identical, or as close as possible to the target substrate, and product detection should be under multiple turnover conditions to ensure the selection of effective catalysts. (ii) The assay should be sensitive over the desired dynamic range. The first rounds of screening large libraries demand isolation with high recovery. All variants, including those that exhibit relatively low detectable catalytic activity, should be recovered. The more advanced rounds must be performed at higher stringency so as to ensure isolation of the best variants. A limited dynamic range seems to be a drawback of most selection approaches. (iii) The procedure should be applicable to a HTS format.
Numerous assays enable detection of enzymatic activities in agar colonies or crude cell lysates by the production of a fluorophore or chromophore (see Bornscheuer, 2002; Goddard and Reymond, 2004; Andexer et al., 2006). Assays on agar‐plated colonies typically enable the screening of > 104 variants in a matter of days, but are often limited in sensitivity. Soluble products diffuse away from the colony and hence, only very active variants are detected. Assays based on insoluble products show higher sensitivity, but their scope is rather limited (for example, see Khalameyzer et al., 1999). The range of assays that are applicable for crude cell lysates is obviously much wider, but their throughput is rather restricted. In the absence of sophisticated robotics that are usually unavailable to academic laboratories, only 103–104 variants are typically screened (Geddie et al., 2004). These low‐to‐medium throughput screens have proved effective for the isolation of enzyme variants with improved properties or for the isolation of enzymes from pre‐enriched metagenomic libraries as described in a number of reviews (Beloqui et al., 2008). However, a far more efficient sampling of sequence space is required for the isolation of rare variants from large metagenomic libraries or variants with dramatically altered phenotypes in directed evolution experiments. Here, methods based on the screening of in silico (virtual) libraries with a size up to 1080 variants have been recently applied successfully (Fox et al., 2007).
In vitro compartmentalization
In vitro compartmentalization is based on water‐in‐oil emulsions, where the water phase is dispersed in the oil phase to form microscopic aqueous compartments. Each droplet contains, on average, a single gene, and serves as an artificial cell allowing for transcription, translation and the activity of the resulting proteins, to take place within the compartment. The oil phase remains largely inert and restricts the diffusion of genes and proteins between compartments (Fig. 3). The droplet volume (∼5 femtoliter) enables a single DNA molecule to be transcribed and translated (Griffiths and Tawfik, 2006), as well as the detection of single enzyme molecules (Griffiths and Tawfik, 2003). The high capacity of the system (> 1010 in 1 ml of emulsion), the ease of preparing emulsions and their high stability over a broad range of temperatures, render IVC an attractive system for HTS of enzymes, as well as for many other HT genetic and genomic manipulations (for a recent review, see Griffiths and Tawfik, 2006).
Figure 3.
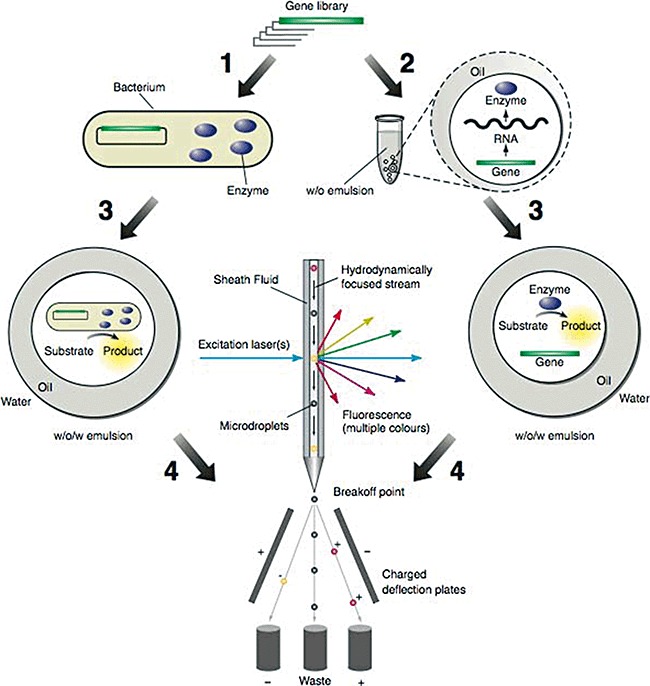
Selections by FACS sorting of double emulsion droplets. A gene library is transformed into bacteria, and the encoded proteins are expressed in the cytoplasm, the periplasm, or on the surface of the cells (Step 1). The bacteria are dispersed to form a water‐in‐oil (w/o) emulsion, with typically one cell per aqueous microdroplet. Alternatively, an in vitro transcription/translation reaction mixture containing a library of genes is dispersed to form a w/o emulsion with typically one gene per aqueous microdroplet. The genes are transcribed and translated within the microdroplets (Step 2). Proteins with enzymatic activity convert the non‐fluorescent substrate into a fluorescent product and the w/o emulsion is converted into a water‐in‐oil‐in‐water (w/o/w) emulsion (Step 3). Fluorescent microdroplets are separated from non‐fluorescent microdroplets using a fluorescence activated cell sorter (FACS) (Step 4). Bacteria or genes from fluorescent microdroplets which encode active enzymes are recovered and the bacteria are propagated or the DNA is amplified using the polymerase chain reaction. These bacteria or genes can then be re‐compartmentalized for further rounds of selection.
While IVC provides a facile mean for co‐compartmentalizing genes and the proteins they encode, the selection of an enzymatic activity requires a link between the desired reaction product and the gene. One possible selection format is to have the substrate, and subsequently the product, of the desired enzymatic activity physically linked to the gene. Enzyme‐encoding genes can then be isolated by virtue of their attachment to the product while other genes that encode an inactive protein carry the unmodified substrate. The simplest applications of this strategy come with the selection of DNA‐modifying enzymes, where the gene and substrate comprise the same molecule. Indeed, IVC was first applied for the selection of DNA‐methyltransferases (MTases) (Tawfik and Griffiths, 1998). Other applications include the selection of restriction endonucleases (Doi et al., 2004) and DNA polymerases (Ghadessy et al., 2001; 2004).
The first application of IVC beyond DNA‐modifying enzymes was demonstrated by a selection of bacterial phosphotriesterase variants (Griffiths and Tawfik, 2003). The selection strategy used was based on two emulsification steps. In the first step, microbeads, each displaying a single gene and multiple copies of the encoded protein variant, were formed by translating genes immobilized to microbeads in emulsion droplets and capturing the resulting protein via an affinity tag. The microbeads were isolated and re‐emulsified in the presence of a modified phosphotriester substrate. In the second step, the product and any unreacted substrate were subsequently coupled to the beads. Product‐coated beads, displaying active enzymes and the genes that encode them, were detected using fluorescently labelled anti‐product antibodies and selected by fluorescence‐activated cell sorting (FACS). Selection from a library of > 107 different variants led to the isolation of an enzyme variant with a very high kcat value (> 105 s−1).
Some IVC selection modes take advantage of the fact that this system is solely used in vitro, allowing for selection of substrates, products and reaction conditions that are incompatible with in vivo systems. However, cell‐free translation must be performed under defined pH, buffer, ionic strength and metal ion composition. In the selection for phosphotriesterase using IVC described above (Griffiths and Tawfik, 2003), translation was completely separated from catalytic selection by using two sequential emulsification steps, allowing selection for catalysis under conditions that are incompatible with translation. However, it is also possible to use a single emulsification step, and to modify the contents of the droplets without breaking the emulsion once translation is completed (Bernath et al., 2005).
IVC in double emulsions
The need to link the product to the enzyme‐coding gene complicates and restricts the scope of selection, especially for non‐DNA modifying enzymes. Recently, an alternative strategy was developed, based on compartmentalizing and sorting single genes, together with the fluorescent product molecules generated by their encoded enzymes. The technology makes use of double, water‐in‐oil‐in‐water (w/o/w) emulsions that are amenable to sorting by FACS (Fig. 3). FACS technology, originally developed by Diversa, enables the identification of biological activity within a single cell by incorporating a laser with multiple wavelength capabilities with the ability to screen up to 50 000 clones per second, or over 1 billion clones per day. This, therefore, circumvents the need to tailor the selection for each substrate and reaction, and allows the use of a wide variety of existing fluorogenic substrates. The making and sorting of w/o/w emulsion droplets does not disrupt the content of the aqueous droplets of the primary w/o emulsions. Further, sorting by FACS of w/o/w emulsion droplets containing a fluorescent marker and parallel gene enrichment have been demonstrated (Bernath et al., 2004).
The w/o/w emulsions were also applied for the directed evolution of two different enzymatic systems. New variants of serum paraoxonase (PON1) with thiolactonase activity (Aharoni et al., 2005a) and new enzyme variants with β‐galactosidase activity were selected from libraries of > 107 mutants (Mastrobattista et al., 2005). The β‐galactosidase variants were translated in vitro, as in previously described IVC selections (Tawfik and Griffiths, 1998). In the case of PON1, intact Escherichia coli cells in which the library variants were expressed, emulsified and FACS sorted, thereby demonstrating the applicability of double emulsions for single‐cell phenotyping and directed enzyme evolution (Aharoni et al., 2005b). The same strategy has been recently applied for the selection of lactonase activity (Amitai et al., 2007) using an oxo‐lactone substrate, the enzymatic hydrolysis of which generates a thiol that was subsequently detected with a fluorogenic probe (Khersonsky and Tawfik, 2006). Detailed protocols for the preparation and sorting of double emulsions are available (Miller et al., 2006).
Perspectives of HT‐IVC screens for metagenome mining
Recent progress has revealed that the capture of genetic resources of complex microbial communities in metagenome libraries allows the discovery of a richness of new genomic and metabolic diversity that had not previously been imagined. Activity‐based screening of such libraries has demonstrated that this new diversity is not simply variations on known sequence themes, but rather the existence of entirely new sequence classes and novel functionalities (Ferrer et al., 2007). IVC‐analysis of bulk environmental DNA may serve as a powerful tool for selecting and dynamically analysing those new sequences through the ability to check biochemical parameters in millions of single cells or microdroplets each of them containing individual ‘metagenes’. The application of IVC‐FACS technologies would enable the screening of billions of bp for enzymatic activity in few days, a processing speed which is impossible to achieve using conventional HT screening approaches.
The only example in the literature so far for using the power of the FACS for sorting metagenomic libraries, is the substrate‐dependent gene‐induction assays (SIGEX). Using this methodology metagenome fragments are ligated into an operon‐trap vector (e.g. p18GFP), and the cells are then separated and analysed by HT‐FACS to select for GFP‐expressing cells (Uchiyama and Watanabe, 2008). The SIGEX approach enriches for functional genes but do no select directly for enzymatic activity and is restricted to cloning and the usage of transcription–translation machinery of E. coli. These drawbacks can be partially circumvented by IVC cell‐free system, which allows the direct screening for enzymatic activity and the advantage of controlling the reaction conditions and introducing novel chemical groups to enzymes (new metals, cofactors and unnatural amino acids). Yet the full recovery of ‘metaenzymes’ by IVC‐FACS is limited by the reliance and low availability on cell‐free systems comprising the essential components for transcription and translation (Griffiths and Tawfik, 2006).
Conclusions
Exploiting the vast environmental genetic diversity by metagenomic approaches is a promising strategy for the identification and isolation of novel enzymes. Identification of such enzymes will enhance our knowledge of the structure, function and evolution of enzymes and will allow definition of many new enzyme families. These enzymes could be highly valuable for industrial applications, especially in the development of new biocatalytic processes. The application of HTS approaches for selecting metagenomic libraries, and especially IVC assays, will enable us to identify novel enzymes for a wider scope of biotransformations. These newly identified enzymes can serve as the ideal starting point for the directed evolution of novel enzymes with improved properties. We believe that in the near future, a new biotechnological platform will emerge, combining the fields of metagenomics and directed evolution for the generation of novel biocatalysts.
Acknowledgments
This research was supported by the Spanish MEC BIO2006‐11738 and CSD2007‐00005 projects. A.B. thanks the Spanish MEC for a FPU fellowship. A.A. acknowledges funding support from the Israel Science Foundation (ISF) Morasha program.
References
- Aharoni A., Amitai G., Bernath K., Magdassi S., Tawfik D.S. High‐throughput screening of enzyme libraries: thiolactonases evolved by fluorescence‐activated sorting of single cells in emulsion compartments. Chem Biol. 2005a;12:1281–1289. doi: 10.1016/j.chembiol.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Aharoni A., Griffiths A.D., Tawfik D.S. High‐throughput screens and selections of enzyme encoding genes. Curr Opin Chem Biol. 2005b;9:210–216. doi: 10.1016/j.cbpa.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Amitai G., Gupta R.D., Tawfik D.S. Latent evolutionary potentials under the neutral mutational drift of an enzyme. HFSP J. 2007;1:67–78. doi: 10.2976/1.2739115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andexer J., Guterl J.K., Pohl M., Eggert T. A high‐throughput screening assay for hydroxynitrile lyase activity. Chem Commun (Camb) 2006;40:4201–4203. doi: 10.1039/b607863j. [DOI] [PubMed] [Google Scholar]
- Angly F.E., Felts B., Breitbart M., Salamon P., Edwards R.A., Carlson C. The marine viromes of four oceanic regions. PLoS Biol. 2006;4:e368. doi: 10.1371/journal.pbio.0040368. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beloqui A., De María P.D., Golyshin P.N., Ferrer M. Recent trends in industrial microbiology. Curr Opin Microbiol. 2008;11:240–248. doi: 10.1016/j.mib.2008.04.005. [DOI] [PubMed] [Google Scholar]
- Bernath K., Hai M., Mastrobattista E., Griffiths A.D., Magdassi S., Tawfik D.S. In vitro compartmentalization by double emulsions: sorting and gene enrichment by fluorescence activated cell sorting. Anal Biochem. 2004;325:151–157. doi: 10.1016/j.ab.2003.10.005. [DOI] [PubMed] [Google Scholar]
- Bernath K., Magdassi S., Tawfik D.S. Directed evolution of protein inhibitors of DNA‐nucleases by in vitro compartmentalization (IVC) and nano‐droplet delivery. J Mol Biol. 2005;345:1015–1026. doi: 10.1016/j.jmb.2004.11.017. [DOI] [PubMed] [Google Scholar]
- Bornscheuer U.T. Methods to increase enantioselectivity of lipases and esterases. Curr Opin Biotechnol. 2002;13:543–547. doi: 10.1016/s0958-1669(02)00350-6. [DOI] [PubMed] [Google Scholar]
- Doi N., Kumadaki S., Oishi Y., Matsumura N., Yanagawa H. In vitro selection of restriction endonucleases by in vitro compartmentalization. Nucleic Acids Res. 2004;32:e95. doi: 10.1093/nar/gnh096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards R.A., Rodriguez‐Brito B., Wegley L., Haynes M., Breitbart M., Peterson D.M. Using pyrosequencing to shed light on deep mine microbial ecology. BMC Genomics. 2006;7:57. doi: 10.1186/1471-2164-7-57. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer M., Golyshina O., Beloqui A., Golyshin P.N. Mining enzymes from extreme environments. Curr Opin Microbiol. 2007;10:207–214. doi: 10.1016/j.mib.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Fierer N., Breitbart M., Nulton J., Salamon P., Lozupone C., Jones R. Metagenomic and small‐subunit rRNA analyses reveal the genetic diversity of bacteria, archaea, fungi, and viruses in soil. Appl Environ Microbiol. 2007;73:7059–7066. doi: 10.1128/AEM.00358-07. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox R.J., Davis S.C., Mundorff E.C., Newman L.M., Gavrilovic V., Ma S.K. Improving catalytic function by ProSAR‐driven enzyme evolution. Nature Biotechnol. 2007;25:338–344. doi: 10.1038/nbt1286. et al. [DOI] [PubMed] [Google Scholar]
- Frias‐Lopez J., Shi Y., Tyson G.W., Coleman M.L., Schuster S.C., Chisholm S.W., Delong E.F. Microbial community gene expression in ocean surface waters. Proc Natl Acad Sci USA. 2008;10:3805–3810. doi: 10.1073/pnas.0708897105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabor E., Liebeton K., Niehaus F., Eck J., Lorenz P. Updating the metagenomics toolbox. Biotechnol J. 2007;2:201–206. doi: 10.1002/biot.200600250. [DOI] [PubMed] [Google Scholar]
- Galvao T.C., Mohn W.W., De Lorenzo V. Exploring the microbial biodegradation and biotransformation gene pool. Trends Biotechnol. 2005;23:497–506. doi: 10.1016/j.tibtech.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Geddie M.L., Rowe L.A., Alexander O.B., Matsumura I. High throughput microplate screens for directed protein evolution. Methods Enzymol. 2004;388:134–145. doi: 10.1016/S0076-6879(04)88012-1. [DOI] [PubMed] [Google Scholar]
- Ghadessy F.J., Ong J.L., Holliger P. Directed evolution of polymerase function by compartmentalized self‐replication. Proc Natl Acad Sci USA. 2001;98:4552–4557. doi: 10.1073/pnas.071052198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghadessy F.J., Ramsay N., Boudsocq F., Loakes D., Brown A., Iwai S. Generic expansion of the substrate spectrum of a DNA polymerase by directed evolution. Nat Biotechnol. 2004;22:755–759. doi: 10.1038/nbt974. et al. [DOI] [PubMed] [Google Scholar]
- Goddard J.P., Reymond J.L. Enzyme assays for high‐throughput screening. Curr Opin Biotechnol. 2004;15:314–322. doi: 10.1016/j.copbio.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Griffiths A.D., Tawfik D.S. Directed evolution of an extremely fast phosphotriesterase by in vitro compartmentalization. EMBO J. 2003;22:24–35. doi: 10.1093/emboj/cdg014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths A.D., Tawfik D.S. Miniaturising the laboratory in emulsion droplets. Trends Biotechnol. 2006;24:395–402. doi: 10.1016/j.tibtech.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Hallin P.F., Binnewies T.T., Ussery D.W. The genome BLASTatlas‐a GeneWiz extension for visualization of whole‐genome homology. Mol Biosyst. 2008;5:363–371. doi: 10.1039/b717118h. [DOI] [PubMed] [Google Scholar]
- Khalameyzer V., Fischer I., Bornscheuer U.T., Altenbuchner J. Screening, nucleotide sequence, and biochemical characterization of an esterase from Pseudomonas fluorescens with high activity towards lactones. Appl Environ Microbiol. 1999;65:477–482. doi: 10.1128/aem.65.2.477-482.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khersonsky O., Tawfik D.S. Chromogenic and fluorogenic assays for the lactonase activity of serum paraoxonases. Chembiochem. 2006;7:49–53. doi: 10.1002/cbic.200500334. [DOI] [PubMed] [Google Scholar]
- Krause L., Diaz N.N., Goesmann A., Kelley S., Nattkemper T.W., Rohwer F., Edwards R.A., Stoye J. Phylogenetic classification of short environmental DNA fragments. Nucleic Acids Res. 2008;36:2230–2239. doi: 10.1093/nar/gkn038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leininger S., Urich T., Schloter M., Schwark L., Qi J., Nicol G.W. Archaea predominant among ammonia‐oxidizing prokaryotes in soils. Nature. 2006;442:806–809. doi: 10.1038/nature04983. et al. [DOI] [PubMed] [Google Scholar]
- Liolios K., Mavromatis K., Tavernarakis N., Kyrpides N.C. The Genomes On Line Database (GOLD) in 2007: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res. 2008;36:D475–D479. doi: 10.1093/nar/gkm884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastrobattista E., Taly V., Chanudet E., Treacy P., Kelly B.T., Griffiths A.D. High‐throughput screening of enzyme libraries: in vitro evolution of a β‐galactosidase by fluorescence‐activated sorting of double emulsions. Chem Biol. 2005;12:1291–1300. doi: 10.1016/j.chembiol.2005.09.016. [DOI] [PubMed] [Google Scholar]
- Miller O.J., Bernath K., Agresti J.J., Amitai G., Kelly B.T., Mastrobattista E. Directed evolution by in vitro compartmentalization. Nat Methods. 2006;3:561–570. doi: 10.1038/nmeth897. et al. [DOI] [PubMed] [Google Scholar]
- Park S.J., Kang C.H., Chae J.C., Rhee S.K. Metagenome microarray for screaning of fosmid clones containing specific genes. FEMS Microbiol Lett. 2008;284:28–34. doi: 10.1111/j.1574-6968.2008.01180.x. [DOI] [PubMed] [Google Scholar]
- Raes J., Foerstner K.U., Bork P. Get the most out of your metagenome: computational analysis of environmental sequence data. Curr Opin Microbiol. 2007;10:490–498. doi: 10.1016/j.mib.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Riesenfeld C.S., Goodman R.M., Handelsman J. Uncultured soil bacteria are a reservoir of new antibiotic resistance genes. Environ Microbiol. 2004;6:981–989. doi: 10.1111/j.1462-2920.2004.00664.x. [DOI] [PubMed] [Google Scholar]
- Rusch D.B., Halpern A.L., Sutton G., Heidelberg K.B., Williamson S., Yooseph S. The Sorcerer II Global Ocean Sampling Expedition: Northwest. Atlantic through Eastern Tropical Pacific. PLoS Biol. 2007;5:e77. doi: 10.1371/journal.pbio.0050077. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss P.D., Handelsman J. Biotechnological prospects from metagenomics. Curr Opin Biotechnol. 2003;14:303–310. doi: 10.1016/s0958-1669(03)00067-3. [DOI] [PubMed] [Google Scholar]
- Sebat J.L., Colwell F.S., Crawford R.L. Metagenomic profiling: microarray analysis of an environmental genomic library. Appl Environ Microbiol. 2003;8:4927–4934. doi: 10.1128/AEM.69.8.4927-4934.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segota I., Bartonicek N., Vlahovicek K. MADNet: microarray database network web server. Nucleic Acids Res. 2008;36:w332–w335. doi: 10.1093/nar/gkn289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogin M.L., Morrison H.G., Huber J.A., Mark Welch D., Huse S.M., Neal P.R. Microbial diversity in the deep sea and the underexplored ‘rare biosphere. Proc Natl Acad Sci USA. 2006;103:12115–12120. doi: 10.1073/pnas.0605127103. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawfik D.S., Griffiths A.D. Man‐made cell‐like compartments for molecular evolution. Nat Biotechnol. 1998;16:652–656. doi: 10.1038/nbt0798-652. [DOI] [PubMed] [Google Scholar]
- Tyson G.W., Chapman J., Hugenholtz P., Allen E.E., Ram R.J., Richardson P.M. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature. 2004;428:37–43. doi: 10.1038/nature02340. et al. [DOI] [PubMed] [Google Scholar]
- Uchiyama T., Watanabe K. Substrate‐induced gene expression (SIGEX) screening of metagenome libraries. Nat Protoc. 2008;3:1202–1212. doi: 10.1038/nprot.2008.96. [DOI] [PubMed] [Google Scholar]
- Venter J.C., Remington K., Heidelberg J.F., Halpern A.L., Rusch D., Eisen J.A. Environmental genome shotgun sequencing of the Sargasso Sea. Science. 2004;304:66–74. doi: 10.1126/science.1093857. , and et al. [DOI] [PubMed] [Google Scholar]
- Wegley L., Edwards R., Rodriguez‐Brito B., Liu H., Rohwer F. Metagenomic analysis of the microbial community associated with the coral Porites astreoides. Environ Microbiol. 2007;9:2707–2719. doi: 10.1111/j.1462-2920.2007.01383.x. [DOI] [PubMed] [Google Scholar]
- Williamson S.J., Rusch D.B., Yooseph S., Halpern A.L., Heidelberg K.B., Glass J.I. The Sorcerer II Global Ocean Sampling Expedition: metagenomic characterization of viruses within aquatic microbial samples. PLoS ONE. 2008;1:e1456. doi: 10.1371/journal.pone.0001456. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yooseph S., Sutton G., Rusch D.B., Halpern A.L., Williamson S.J., Remington K. The Sorcerer II Global Ocean Sampling expedition: expanding the universe of protein families. PLoS Biol. 2007;5:e16. doi: 10.1371/journal.pbio.0050016. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]