

Summary
Pseudomonas putida KT2440 is a non‐pathogenic prototrophic bacterium with high potential for biotechnological applications. Despite all that is known about this strain, the biosynthesis of essential chemicals has not been fully analysed and auxotroph mutants are scarce. We carried out massive mini‐Tn5 random mutagenesis and screened for auxotrophs that require aromatic amino acids. The biosynthesis of aromatic amino acids was analysed in detail including physical and transcriptional organization of genes, complementation assays and feeding experiments to establish pathway intermediates. There is a single pathway from chorismate leading to the biosynthesis of tryptophan, whereas the biosynthesis of phenylalanine and tyrosine is achieved through multiple convergent pathways. Genes for tryptophan biosynthesis are grouped in unlinked regions with the trpBA and trpGDE genes organized as operons and the trpI, trpE and trpF genes organized as single transcriptional units. The pheA and tyrA gene‐encoding multifunctional enzymes for phenylalanine and tyrosine biosynthesis are linked in the chromosome and form an operon with the serC gene involved in serine biosynthesis. The last step in the biosynthesis of these two amino acids requires an amino transferase activity for which multiple tyrB‐like genes are present in the host chromosome.
Pseudomonas putida strain mt‐2 was isolated from garden soil in Japan based on its ability to use 3‐methylbenzoate (3MB) as the sole C‐source (Nakazawa, 2002). The strain contains the TOL plasmid pWW0, which encodes a meta‐cleavage pathway for the degradation of this aromatic carboxylic acid (Worsey and Williams, 1975; Ramos et al., 1997). A variant of the mt‐2 strain is the KT2440 strain, which has lost the TOL plasmid and concomitantly the ability to use 3MB as a C‐source (Table 1, Franklin et al., 1981).
Table 1.
Strains and plasmids used in this study.
| Strain or plasmid | Genotype or relevant characteristic(s) | Reference |
|---|---|---|
| Strains | ||
| P. putida | ||
| KT2440R | Wild‐type, Rifr, derivative of KT2440 | Espinosa‐Urgel and Ramos (2004) |
| M717 | trpI::mini‐Tn5‐Km; Kmr, Rifr | Duque et al. (2007) |
| Aux‐1 | Insertion at 7th codon of trpA | This study |
| Aux‐2 | Insertion at 32th codon of trpC | This study |
| Aux‐3 | Insertion at 184th codon of trpD | This study |
| Aux‐4 | Insertion at 57th codon of trpE | This study |
| Aux‐5 | site‐specific homologous inactivation of trpF | This study |
| Aux‐6 | Insertion at 6th codon of pheA | This study |
| Aux‐7 to 9 | Insertion at 37th, 42nd and 120th codon of pheA | This study |
| Aux‐10 | Insertion at 84th codon of tyrA | This study |
| E. coli | ||
| HB101 pRK600 | Host used for cloning assays | Herrero et al. (1990) |
| CC118λpir pUT‐Km | Source of mini‐Tn5; Kmr | Herrero et al. (1990) |
| Plasmids | ||
| pCHESIΩKm | Kmr | Llamas et al. (2003) |
Kmr and Rifr stand for resistance to kanamycin and rifampicin respectively.
Because of its non‐pathogenic character P. putida KT2440 has been used as a model microorganism to study heterologous gene expression (Marqués et al., 2006), and because of its easy genetic manipulation it has also been used to study the expansion of catabolic pathways and gene evolution (Abril et al., 1989; Contreras et al., 1991; Nüsslein et al., 1992; Duque et al., 1993; Ramos et al., 1994; Timmis, 2002; Hansen et al., 2007), as well as for the development of biosensors to detect pollutants (Werler et al., 2004; Garmendia et al., 2008). It has also been shown that P. putida KT2440 is an excellent root colonizer in a number of agriculturally important plants (Molina et al., 2000; Espinosa‐Urgel and Ramos, 2004; Matilla et al., 2007).
Pseudomonas putida KT2440 can grow on minimal medium and should therefore contain all the necessary genetic information for the biosynthesis of all proteinogenic amino acids. However, no detailed global functional study has ever been undertaken regarding the biosynthesis of amino acids in this strain, and few studies have dealt with this issue in other strains of the genus Pseudomonas (Isaac and Holloway, 1968; Calhoun and Weary, 1969;Buvinger et al., 1981; Cuppels 1986; Essar et al., 1990a,b; Lindow et al., 1993). The number of auxotrophs described for P. putida mt‐2 and its derivatives is restricted to a tryptophan auxotroph of P. putida mt‐2 (Worsey and Williams, 1975). Although few mutants requiring aromatic amino acids have been isolated, information regarding the biochemical biosynthesis of these amino acids has received considerable attention (Gussin, 2004). As in other eubacteria, the biosynthesis of aromatic amino acids in Pseudomonas starts with chorismate as the ultimate intermediate (Fig. 1), which can be transformed into either anthranilate, by the action of anthranilate synthase (TrpE/TrpG), or prephenate by one of the activities associated to the pheA gene product. The biosynthesis of tryptophan from anthranilate follows a one‐way pathway (Fig. 2) (Gussin, 2004), whereas the biosynthesis of tyrosine and phenylalanine is achieved through branched pathways in which one of the most surprising findings is that the pheA gene product participates in multiple steps (Berry, 1996 and Fig. 1). In this branched pathway TyrA is also a multifunctional enzyme and a number of TyrB aminotransferases are involved in the biosynthesis of phenylalanine and tyrosine from phenylpyruvate and 4‐hydroxyphenylpyruvate respectively (Whitaker et al., 1982). Ultimately, l‐tyrosine is also made from l‐phenylalanine in a reaction catalysed by PhhA (Zhao et al., 1994; Arias‐Barrau et al., 2004; Herrera and Ramos, 2007).
Figure 1.
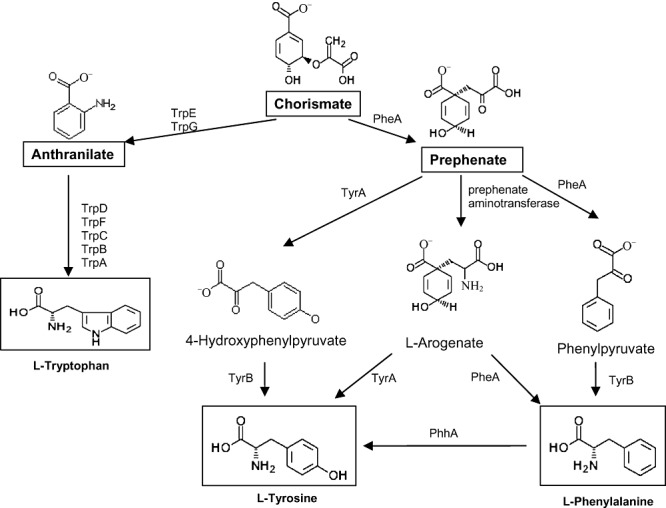
Proposed pathway for the biosynthesis of aromatic amino acids phenylalanine, tyrosine and tryptophan in P. putida KT2440. Chorismate is the ultimate common precursor of all three aromatic acids. The enzymes are TrpE (anthranilate synthase component I), TrpG (anthranilate synthase component II), TrpD (anthranilate phosphoribosyltransferase), TrpF (N‐5′‐phosphoribosyl)anthranilate isomerase, TrpC (indole‐3‐glycerol‐phosphate synthase), TrpA (tryptophan synthase alpha‐subunit), TrpB (tryptophan synthase subunit beta), PheA (chorismate mutase/prephenate dehydratase), PP1770 or TyrA (prephenate dehydrogenase, putative/3‐phosphoshikimate 1‐carboxyvinyltransferase), TyrB‐1 (aromatic‐amino‐acid aminotransferase) and PhhA (phenylalanine‐4‐hydrolase).
Figure 2.
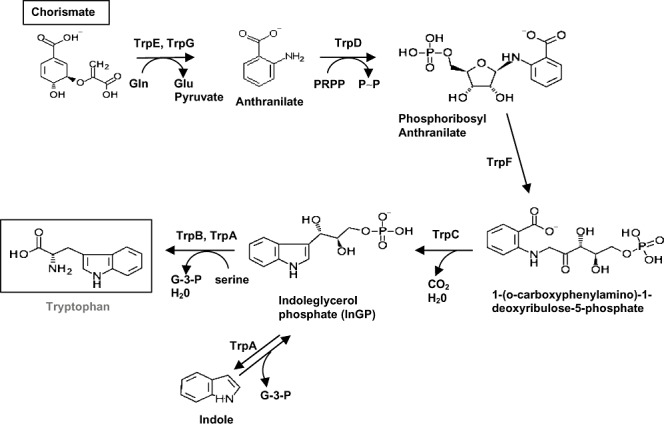
Detailed biosynthetic pathway for tryptophan biosynthesis. Details are as in the legend for Fig. 1.
This study was undertaken to establish a collection of auxotrophs requiring aromatic amino acids in P. putida KT2440, as well as to test their rhizosphere colonization capacity. After massive mutagenesis and screening of almost 150 000 mini‐Tn5 mutants, we isolated a limited number of auxotrophs that require tryptophan, phenylalanine and tyrosine. We characterized these auxotrophs in detail to gather information on the biosynthesis of these aromatic amino acids in this microorganism.
Results and discussion
Isolation of auxotrophs requiring aromatic amino acids for growth
To search for auxotrophs of P. putida we carried out triparental matings to transfer mini‐Tn5‐Km into the KT2440 host chromosome, and KmR transconjugants were selected on M9 minimal medium plates with citrate as a carbon source and supplemented with 0.6 mM of tryptophan, tyrosine or phenylalanine or a mixture of 0.2 mM tyrosine plus 0.4 mM phenylalanine. With the aid of a colony‐picking robot, we screened almost 150 000 clones which resulted in the isolation of four auxotrophs for tryptophan, four auxotrophs for phenylalanine and another whose growth on minimal medium was restored by adding phenylalanine or tyrosine.
Identification of the insertion sites in the four tryptophan auxotrophs. Feeding with pathway intermediates and complementation analyses
The four mutants requiring tryptophan were called Aux‐1 through Aux‐4 (Table 1). The insertion site of mini‐Tn5 in the Aux‐1 mutant was located at the 7th codon after the first ATG of the gene‐encoding PP0082, whereas the insertion site in Aux‐2 and Aux‐3 was located in the gene‐encoding PP0422 (32nd codon after the first ATG) and PP0421 (184th codon after the first ATG). blast analysis indicated that PP0082 exhibited high similarity to the tryptophan synthase alpha‐subunit (trpA) gene, whereas PP0421 encoded anthranilate phosphoryl transferase (trpD). The PP0422 protein exhibited high similarity to the TrpC protein of Escherichia coli– an enzyme with indole‐3‐glycerol phosphate synthase activity. The fourth mutant (Aux‐4) had an insertion in the 57th codon of the open reading frame (ORF) encoding PP0417, which corresponds to anthranilate synthase component I (TrpE) (Fig. 2). A random insertion mutant in PP0084 encoding the TrpI transcriptional regulator was available at the Pseudomonas Stock Centre (Duque et al., 2007); however, the mutant did not exhibit the tryptophan requirement, in agreement with its role as a repressor.
All of the trp genes in the genome of P. putida KT2440 were identified by blast (Essar et al., 1990a,b). Open reading frames were found in two clusters, (i) PP0082 to PP0084 and (ii) PP0417 to PP0422, plus a single monocistronic unit (PP1995) unlinked to any of the two other clusters (Gussin, 2004 and Fig. 3). In the first cluster two ORFs, trpA and trpB, encoded the two subunits of tryptophan synthase. The trpA and trpB overlapped by one nucleotide, suggesting that they formed part of an operon, which was confirmed with reverse transcription (RT)‐PCR assays (Fig. 3B). The trpI (PP0084) gene was divergent with respect to trpBA genes, and is a monocistronic unit.
Figure 3.
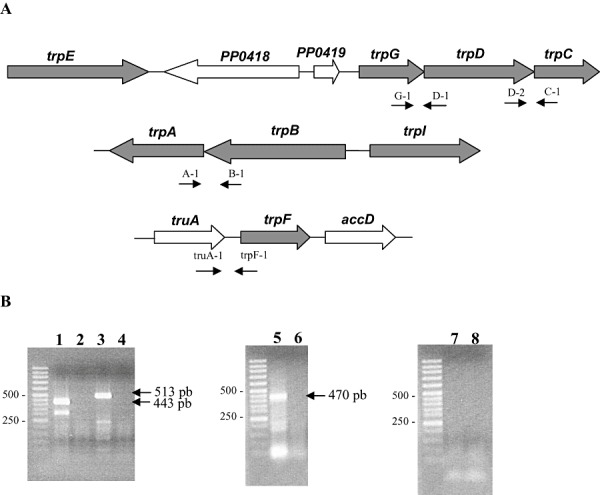
A. Gene organization of the clusters encoding the enzymes for the tryptophan biosynthesis pathway in P. putida KT2440. Genes involved in tryptophan biosynthesis appear in grey. B. Transcriptional organization of the trp genes. Electrophoresis of cDNA amplified with primers G‐1/D‐1 (lane 1); D‐2/C‐1 (lane 3); A‐1/B‐1 (lane 5); truA‐1/trpF‐1 (lane 7). Lanes 2, 4, 6 and 8 corresponded to the negative controls without reverse transcriptase. M refers to molecular weight markers, whose sizes are on the left.
In the second cluster, four genes were related to tryptophan biosynthesis. PP0417 encoded anthranilate synthase subunit I, and it was separated by two genes of unknown function, followed by a cluster of three genes, PP0420 through to PP0422, namely trpG, trpD, trpC. All three genes had the same orientation: the trpD and trpC genes overlapped by six nucleotides and the distance between trpG and trpD was only nine nucleotides, which suggested that they formed an operon. Analysis of this cluster by RT‐PCR revealed two transcriptional units: a single transcript for trpE (PP0417) and an operon made up of the trpG, trpD and trpC genes (Fig. 3B).
The unlinked trpF gene (PP1195) is proposed to encode a phosphoribosyl anthranilate isomerase. The gene is in a cluster of nine genes, all of which are transcribed in the same direction, and no obvious connection with tryptophan biosynthesis was deduced from blast analyses. The preceding ORF, PP1994, was 61 nucleotides away from PP1995, and the ATG of the following ORF (encoding PP1996) was 222 nucleotides away from the PP1995 stop codon. This suggested that these three genes belonged to independent transcriptional units; nonetheless, we tested by RT‐PCR whether the genes were part of the same transcript. Our results were negative, indicating that trpF was most probably a single cistron.
Because no mutants in the trpF gene were found in the original screens, we decided to generate a trpF mutant by site‐directed mutagenesis. To do this we followed the protocol described in Experimental procedures and generated a knockout mutant based on the pCHESI strategy (Llamas et al., 2003). As expected, the trpF‐deficient clones were found to be auxotrophs for tryptophan, although very small colonies were detected on plates without tryptophan supplementation.
To further confirm the pathway shown in Fig. 2 we carried out precursor feeding tests. To this end the mutant cultures were supplemented with commercially available intermediates (chorismate, anthranilate) or products that upon in vivo transformation can yield a pathway intermediate (i.e. indole, which yields indoleglycerol phosphate) (see Fig. 2 and Table 2). The trpA mutant grew on M9 minimal medium if and only if tryptophan (0.6 mM) was added, whereas trpD and trpF mutants grew with tryptophan and indole. However, growth of trpE mutant occurred when anthranilate, indole or tryptophan was added. The pattern of growth of the mutants was as expected from the biochemical pathway shown in Fig. 2.
Table 2.
Growth of parental strain and isogenic trp mutants on mineral medium with different supplements.
| Strain | M9 | M9 + tryptophan | M9 + chorismate | M9 + anthranilate | M9 + indole |
|---|---|---|---|---|---|
| KT2440 | + | + | + | + | + |
| TrpA | − | + | − | − | − |
| TrpD | − | + | − | − | + |
| TrpF | +/− | + | +/− | +/− | + |
| TrpI | + | + | + | + | + |
| TrpE | − | + | − | + | + |
The carbon source in all cases was 16 mM citrate. All of the added nutrients were used at a final concentration of 0.2 mM.
The symbol ‘+’ means that turbidity of the culture after 24 h at 660 nm was equal to or higher than 1 unit.
The symbol ‘−’ means that turbidity of the culture was < 0.1 units at 660 nm.
The symbol ‘+/−’ means that turbidity of the culture was as high as 0.3 units after 24 h at 660 nm.
The tryptophan biosynthetic pathway and genes have been proposed to be highly conserved in proteobacteria (Bae et al., 1989; Essar et al., 1990a; Gussin, 2004). This is clearly observed in that P. putida tryptophan biosynthesis enzymes exhibit a high degree of similarity to the corresponding proteins from other sources and are highly conserved in the genus Pseudomonas (see Fig. S1). However, certain differences were apparent in the physical organization of the genes. For instance, in Acinetobacter calcoaceticus and Burkholderia acidovorans all seven genes coding for the enzymes responsible for tryptophan synthesis map to three chromosomal locations. Two three‐gene clusters, one (trpGDC) specifying the small subunit of anthranilate synthase, phosphoribosyl transferase and indoleglycerol phosphate synthase, and the other (trpFBA) specifying phosphoribosyl anthranilate isomerase and both tryptophan synthase subunits, are not linked to each other or to the trpE gene encoding the large anthranilate synthase subunit (Buvinger et al., 1981). In Vibrio harvey, genetic mapping experiments demonstrated that the trp genes were located in two clusters: trpCDE and trpFBA (Bieger and Crawford, 1983). In P. putida our analysis of physical gene organization revealed clustering of the trpAB genes, which were transcribed divergently from the trpI gene, which encodes a repressor. On the other hand, trpGDC forms an operon whereas trpE and trpF are monocistronic units. The physical organization of the trp genes in P. putida KT2440 − two three‐gene clusters (trpGDC and trpAB, trpI) and two monocistronic trpE and trpF units − is conserved in all Pseudomonas strains whose genomes have been sequenced (Fig. S1).
In all of the sequenced Pseudomonas strains the trpE gene is separated from the trpGDC operon by one or two genes of unknown function. The trpF gene, which was unlinked to other trp genes, is flanked by truA and accD genes in all Pseudomonas sp. At the amino acid sequence level, the identity of the enzymes of the tryptophan pathway in Pseudomonas sp. was in the range of 71–97% (Table S1).
To provide further unequivocal support for this set of genes being the only one involved in tryptophan biosynthesis, we conjugated each trp auxotroph with E. coli HB101 bearing a library of P. putida genes in the pLAFR1cosmid, as described in Experimental procedures. We rescued complemented clones for all mutants by selecting for growth on M9 minimal medium without tryptophan. Then all complemented clones were subjected to growth tests in the absence of added tryptophan, and we found that all grew on M9 minimal medium at rates similar to those of the parental strain (not shown). PCR analysis revealed that, as expected, each mutant was complemented by the corresponding cosmid bearing the wild‐type version of the mutant genes.
Characterization of mutants in the phenylalanine and tyrosine biosynthetic pathways
In the four mutants in which growth was restored only by phenylalanine, mini‐Tn5 was inserted at different sites within ORF PP1769 (pheA). The insertion in the pheA mutant was located in the 6th, 37th, 42nd and 120th codon (Aux‐6 to 9; Table 1). Growth of the pheA mutants was restored by phenylalanine, but not by tyrosine. As expected PheA‐deficient mutants failed to synthesize prephenate and could not synthesize tyrosine from 4‐hydroxyphenylpyruvate or arogenate, but supplementation with phenylalanine alone guaranteed tyrosine biosynthesis in a reaction mediated by PhhA, in agreement with our earlier observation (Herrera and Ramos, 2007). The fact that tyrosine did not permit the growth of a pheA mutant provides proof that there is no pathway from tyrosine to phenylanine operating in P. putida KT2440.
In the mutant in which growth was restored by phenylalanine or tyrosine, the mini‐Tn5 was inserted at the 84th codon of ORF PP1770 (tyrA). We then tested whether phenylpyruvate or 4‐hydroxyphenylpyruvate could replace the requirement for phenylalanine or tyrosine in all of the above pheA and tyrA mutants. As expected, all clones grew with phenylpyruvate, but 4‐hydroxypyruvate only restored growth of the TyrA‐deficient mutant (Table 3).
Table 3.
Growth of parental strain and isogenic Phe and Tyr mutants on mineral medium with different supplements.
| Strain | M9 | M9 + phenylalanine | M9 + tyrosine | M9 + 4‐hydroxyphenylpyruvate | M9 + phenylpyruvate |
|---|---|---|---|---|---|
| KT2440 | + | + | + | + | + |
| PheA | − | + | − | − | + |
| TyrA | +/− | + | + | + | + |
The carbon source in all cases was 16 mM citrate. All of the added nutrients were used at a final concentration of 0.2 mM.
The symbol ‘+’ means that the turbidity at 660 nm of the culture after 24 h was equal or higher than 1 unit.
The symbol ‘–’ means that turbidity of the culture was < 0.1 units at 660 nm.
The symbol ‘+/−’ means that turbidity of the culture was as high as 0.3 units at 660 nm.
Physical analysis of the ORF PP1769/PP1770 region revealed that ORF PP1769 overlapped with the preceding gene annotated as serC by six nucleotides, and was separated from ORF PP1770 by 61 nucleotides (Fig. 4). As the two ORFs identified as involved in phenylalanine/tyrosine biosynthesis were adjacent, we used RT‐PCR and found that pheA forms an operon with the preceding serC gene and also with PP1770, which encodes the TyrA prephenate dehydrogenase. All three genes are transcribed as part of the same operon. These three genes are also co‐transcribed with cmK and rpsA, located downstream of tyrA, but not with gyrA located upstream of serC (not shown). A supraoperon organization of the serC/pheA/tyrA genes was also found in P. stutzeri (Xie et al., 1999).
Figure 4.
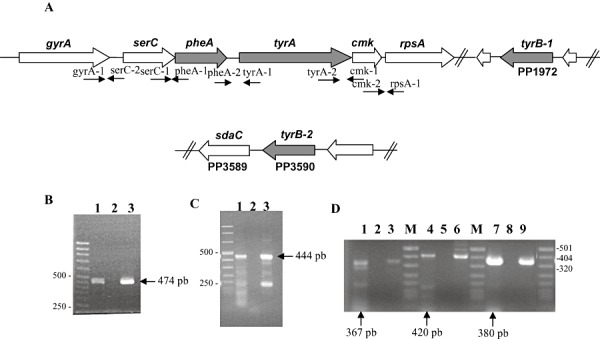
A. Gene organization of the clusters encoding enzymes for the phenylalanine and tyrosine biosynthesis pathway in P. putida KT2440. Genes involved in phenylalanine and tyrosine biosynthesis are in grey. B–D. Transcriptional Organization of the genes involved in phenylalanine and tyrosine biosynthesis. (B) Electrophoresis of cDNA amplified with primers serC‐1/pheA‐1 (lane 1 expected size 474 bp). Lane 2 corresponds to the negative control and lane 3 corresponds to the positive control. (C) Electrophoresis of cDNA amplified with primer pheA‐2/tyrA‐1 (lane 1 expected size 444 bp). Lane 2 corresponds to the negative control and lane 3 corresponds to the positive control. (D) Electrophoresis of cDNA amplified with primers gyrA‐1/serC‐2 (lane 1 expected size 367 bp), tyrA‐2/cmk‐1 (lane 4 expected size 420 bp) and cmk‐2/rpsA‐1 (lane 7 expected size 380 bp). Lanes 2, 5, 8 correspond to the negative controls without reverse transcriptase and lanes 3, 6, 9 correspond to the positive controls. M refers to molecular weight markers.
The third protein involved in the biosynthesis of tyrosine and phenylalanine is TyrB (Fig. 2). In the original annotation of the P. putida KT2440 genome, two tyrB genes were identified, namely, tyrB1 (ORF PP1972) and tyrB2 (ORF PP3590), whose gene products exhibit aminotransferase activity that produce l‐phenylalanine from phenylpyruvate and l‐tyrosine from 4‐hydroxyphenylpyruvate. Single tyrB2 and tyrB1 knockout mutants and a double knock‐out mutant of this strain did not exhibit phenylalanine or tyrosine auxotrophy, although they grew a bit more slowly than the parental strain on M9 minimal medium. This is similar to the situation in P. aeruginosa, a microorganism that exhibits at least five different enzymes with aromatic amino transferase activity (Whitaker et al., 1982). In all sequenced Pseudomonas sp. genomes the tyrB genes appear unlinked to the pheA, tyrA operon (Fig. S2). In γ‐proteobacteria, on the other hand, the pheA and tyrA genes are often unlinked.
Aromatic amino acid auxotrophs of Pseudomonas putida KT2440 survived in the absence of these amino acids and exhibited good colonization ability
In P. syringae epiphytic colonization of bean leaves was compromised in auxotroph mutants (Lindow et al., 1993). As P. putida KT2440 was isolated from garden soil (Nakazawa, 2002), we studied colonization of the root system by parental and mutant strains in the rhizosphere of corn plants inoculated individually and in competition with the parental strain. The trp, pheA and tyrA mutant strains established in the root system at a level of about 8 ± 2 × 106 cfu g−1 soil, which is slightly below the number of cfu g−1 soil (7.5 ± 0.5 × 107) recovered for the parental strain. This is in agreement with the findings that root exudates contain all three aromatic compounds to promote growth. However, this finding contrasts with our report that an Δasd P. putida mutant, unable to synthesize several amino acids and diaminopimelic acid, was unable to grow in the rhizosphere of corn plants (Ronchel and Ramos, 2001).
In Bacillus subtilis starvation of tryptophan mutants has been described to lead to a rapid loss of viability (Barlati and Majerfeld, 1970). To test whether this was the case in P. putida, all tryptophan mutants and the pheA and tyrA mutants were grown in M9 minimal medium with glucose and the required amino acid. Cells were then washed and suspended in M9 minimal medium without adding amino acids and kept at 30°C. Then the number of viable cells was monitored with time. No loss in viability was observed during 72 h.
In summary, our findings identify the main requirements for the biosynthesis of aromatic amino acids in Pseudomonas putida KT2440. These biosynthetic pathways seem to be conserved in other Pseudomonas species such as P. aeruginosa, P. fluorescens, P. entomophila and P. syringae, as deduced from conserved gene organization and high sequence similarity of the proteins involved in all metabolic steps.
Experimental procedures
Bacterial strains, plasmids and culture conditions used in this study
We used P. putida KT2440, a derivative of P. putida mt‐2 (Franklin et al., 1981). All strains were grown at 30°C and 200 r.p.m. in 100 ml conical flasks with 20 ml M9 minimal medium supplemented with goodies solution (Abril et al., 1989) and with amino acids (0.6 mM) when required. Glucose (16 mM) or citrate (25 mM) were used as carbon sources.
Escherichia coli CC118[λpir] (pUT‐Km) was used as the source of mini‐Tn5, and E. coli HB101 (pRK600) was used as a helper strain to mobilize the pUT‐Km plasmid into P. putida. Antibiotics were added when necessary to the culture medium at the following concentrations (µg ml−1): ampicillin (Ap), 100; kanamycin (Km), 50; and rifampicin (Rif), 20.
Mutagenesis
Transposon mutagenesis with a mini‐Tn5 (Km) was performed according to the following protocol. Triparental matings of the recipient (P. putida KT2440R), donor (E. coli CC118[λpir] with pUT‐Km) and helper (E. coli HB101 with pRK600) were cultured overnight in Luria–Bertani (LB) with the appropriate antibiotics. After incubation of the recipient at 40°C for 15 min to temporarily inactivate its restriction systems, 0.7 ml of the recipient was mixed with 0.2 ml of the donor cells and 0.1 ml of the helper cells. Cells were collected by centrifugation, suspended in 50 µl fresh LB, spotted on a 0.45 µm filter and plated on the surface of an LB plate. After 6 h of incubation at 30°C, cells were re‐suspended in 1 ml LB, and serial dilutions were plated on selective minimal medium, which was M9 with citrate, Km and Rif, supplemented with 0.6 mM of aromatic amino acids. Around 150 000 clones were tested for amino acid requirements using a Pick‐up robot.
To construct mutant strains bearing an inactivated chromosomal version of the trpF gene, we generated the corresponding knockout using plasmid pCHESIΩKm that is based on pUC18 and bears the origin of transfer oriT of RP4 and the Ω‐Km interposon of plasmid pHP45ΩKm cloned as a HindIII fragment (Llamas et al., 2003). To generate the desired mutation, an internal fragment of about 500 bp of the trpF gene was amplified by PCR with primers containing the XbaI and KpnI (TrpF‐XbaI: 5′‐AGTCTAGATTACCCGCATCGAAG ACG‐3′ TrpF‐2: 5′‐GGCTTTACCTGGGCAATGGC‐3′). The amplified fragment was subsequently cloned between the XbaI and KpnI sities of pCHESIΩKm. The recombinant plasmid was introduced into P. putida KT2440 by electroporation, and a few transformants bearing a co‐integrate of the plasmid in the host chromosome was selected on M9 minimal medium with citrate as a carbon source and Km. The correct insertion of the mutant allele was confirmed by colony‐screening PCR using a primer based on the Km marker gene and another primer that was annealed to the sequence complementary to the cloned gene fragment. The correctness of the construction was confirmed by Southern blotting using the target gene as a probe (not shown).
DNA techniques
Preparation of plasmid and chromosomal DNA, digestion with restriction enzymes (Roche and New England BioLabs), ligation, electrophoresis and Southern blotting were done using standard methods (Sambrook et al., 1989; Ausubel et al., 1991). Plasmid sequencing was done with universal or reverse pUC19/M13 oligonucleotides as primers. The transposon insertion point was identified by random PCR amplification with Taq polymerase (Amersham Pharmacia), using TNEXT2 (5′‐CTTTATTGATTCCATTTTTCACT‐3′) and a mixture of random primers followed by direct sequencing with primers TNEXT2 and TNINT (5′‐AGGCGATTTCAGCGAA GCAC‐3′) (Duque et al., 2007). Sequencing was done on an ABIPRISM 310 automated sequencer. Sequences were analysed with Omiga 2.0 software (Oxford Molecular) and compared with data from the P. putida KT2440 genome, obtained from The Institute for Genomic Research (http://www.tigr.org), and with the GenBank database using blast programs (Altschul et al., 1997).
Southern hybridization and DNA labelling
DNA fragments were separated in agarose gels and transferred onto nylon membranes by capillary blotting as previously described (Sambrook et al., 1989). Specific probes for hybridization were recovered from agarose gels with an agarose gel DNA extraction kit (Reference No. 1696505; Roche). All probes were labelled with digoxigenin by Klenow random primer extension according to the recommended procedure (Ausubel et al., 1991). Blotted filters were pre‐hybridized, hybridized, washed and immunologically developed according to the supplier's instructions. High‐stringency conditions [50% (v/v) formamide at 42°C] were used.
Complementation assays
A P. putida KT2440 gene library in the pLAFR1 cosmid (M.I. Ramos‐González, unpubl. data) was used for complementation studies. Cosmids were transferred by conjugation with the filter‐mating technique (de Lorenzo and Timmis, 1994) to P. putida auxotroph mutants. Filters with a mixture of donor [E. coli HB101 (pLAFR‐cosmid)], recipient (P. putida auxotroph) and helper [E. coli HB101(pRK600)] strains at a ratio of 1:5:1 were incubated for 4 h at room temperature on LB plates. The cells were suspended in 1 ml M9 minimal medium, and 100 µl was plated on selective minimal medium (M9 minimal medium with 10 mM benzoic acid, 10 µg of Rif per ml and 10 µg of Tc per ml). The transconjugants obtained were tested for their ability to grow on M9 minimal medium without further supplementation, and the cosmid was analysed by PCR, Southern hybridization or both to confirm the nature of the complementing genes.
RNA preparation and RT‐PCR
Bacterial cells were grown on M9 minimal medium with citrate and total RNA from P. putida KT2440 was isolated according to the RNEasy protocol (Qiagen, GmbH). RT‐PCR assays were done with the Titan One Tube RT‐PCR System. We used the pairs of primers A‐1 (5′‐CAGCTGCTACTAGTCC GGTA‐3′) and B‐1 (5′‐TACCAGCACACGGACAGCCC‐3′) to determine contiguity of mRNA of trpA and trpB, and we used primers G1 (5′‐GCCCACCAGTCAATCGGGCA‐3′), D1 (5′‐TGGCGCATGACCTCGCGCAT‐3′), D2 (5′‐ACCCTTTGC CGCCCGCTGGC‐3′) and C1 (5′‐TGCGCTCGGCCACTT CCTGA‐3′) to determine the contiguity of mRNA that contained the trpG, trpD and trpC genes. We used primers TruA‐1 (5′‐CAGTACCTGCTCGGTACCCA‐3′) and TrpF‐1 (5′‐CGAAGCCGATGGCATCGGCC‐3′) to test the contiguity of the truA and trpF genes. To test the contiguity of gyrA, serC, pheA, tyrA, cmK and rpsA we used the following set of primers: gyrA‐1: (5′‐GAGCGTATCCAGGAGCCGTC‐3′) and serC‐2: (5′‐AGTCCAGCATCTCGGCCTGT‐3′), serC‐1: (5′‐CTGACAACGGCTCGATGTAC‐3′) and pheA‐1: (5′‐TCGCT GATCAGCTCGAGAAT‐3′), pheA‐2: (5′‐ATGAGTTGCTGG TGCCGTTC‐3′) and tyrA‐1: (5′‐ATCAGGCCTAGACCGAC CAC‐3′), tyrA‐2: (5′‐GCATCATCATCGACGGTGGC‐3′) and cmk‐1: (5′‐GCGATGAACTGCACATCCAG‐3′), cmk‐2: (5′‐CGAGTCTGCTGGATGAGATT‐3′) and rpsA‐1: (5′‐GATACC GGTGATGATCGCAC‐3′).
Surface sterilization, germination of seeds and root colonization assay
Corn seeds were surface‐sterilized by rinsing with sterile deionized water, washing for 10 min with 70% (v/v) ethanol, then for 15 min with 20% (v/v) bleach, followed by thorough rinsing with sterile deionized water. Surface‐sterilized seeds were pre‐germinated on MS medium (Murashige and Skoog, 1962) containing 0.2% (w/v) phytagel (Sigma P8169, St Louis, MO, USA) and 0.5% (w/v) glucose, which was used instead of sucrose to monitor contamination of the seeds, at 30°C in the dark for 48 h. For root colonization assays, seeds were inoculated with approximately 5 × 106 cfu ml−1 from LB medium overnight culture suspended in M9 salt (Sambrook et al., 1989). After incubation without shaking for 30 min at 30°C, seeds were washed and planted in 50 ml Sterilin tubes containing 40 g sterile washed silica sand and 10% (v/w) plant nutrient solution supplemented with Fe‐EDTA and MS micronutrients as described above. The final inoculum size was 105 cfu. Inoculated plants were maintained in a controlled chamber at 24°C and 55–65% humidity with a daily light period of 16 h. After 6 days plants were collected, the shoots were discarded and the roots were placed in 50 ml Sterilin tubes containing 15 ml M8 salts (Abril et al., 1989) and 4 g glass beads (3 mm diameter). Tubes were vortexed for 2 min and left to stand for 15 s, then cells from bacterial suspensions were collected by centrifugation for 8 min at 6700 g (4°C) in tubes pre‐cooled in liquid nitrogen. Pellets were immediately frozen in liquid nitrogen and stored at −80°C.
Competitive root colonization assays
Surface sterilization, germination of seeds and bacterial inoculation were performed as described in the previous section, except that seedlings were inoculated with a mixture of KT2440RTn7‐Sm used as a control and the strain with the specified gene mutation. Inoculum size differences between wild‐type and mutant strains were less than 2%. At the indicated times, bacterial cells were recovered from the rhizosphere as specified above. LB agar supplied with rifampin and streptomycin (or Km) was used to select KT2440RTn7‐Sm or the mutant strains respectively.
Acknowledgments
This work was supported by SYSMO project GEN2006‐27750‐C5‐5‐E/SYS. We thank M.M. Fandila and C. Lorente for secretarial assistance, and K. Shashok for improving the use of English in the manuscript.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Organization of trp genes in P. aeruginosa PAO1, P. entomophila L48, P. fluorescens Pf0-1 and P. syringae.
Organization of pheA/tyrA genes in several Pseudomonas.
Conservation of tyrosine and phenylalanine biosynthesis genes among Pseudomonas species.
References
- Abril M.A., Michán C., Timmis K.N., Ramos J.L. Regulator and enzyme specificities of the TOL plasmid‐encoded upper pathway for degradation of aromatic hydrocarbons and expansion of the substrate range of the pathway. J Bacteriol. 1989;171:6782–6790. doi: 10.1128/jb.171.12.6782-6790.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S.F., Madden T.L., Schäffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. Gapped BLAST and PSI‐BLAST: a new generation of protein database search programs. Nucl Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias‐Barrau E., Olivera E.R., Luengo J.M., Fernández C., Galán B., García J.L. The homogentisate pathway: a central catabolic pathway involved in the degradation of L‐phenylalanine, L‐tyrosine, and 3‐hydroxyphenylacetate in Pseudomonas putida. J Bacteriol. 2004;186:5062–5077. doi: 10.1128/JB.186.15.5062-5077.2004. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausubel F.M., Brent R., Kingston R.F., Moore D.D., Seidman J.G., Smith J.A., Struhl K. John Wiley & Sons; 1991. [Google Scholar]
- Bae Y.M., Holmgrem E., Crawford I.P. Rhizobium melliloti authranilate syntase gene: cloning, sequence, and expression in Escherichia coli. J Bacteriol. 1989;171:3471–3478. doi: 10.1128/jb.171.6.3471-3478.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlati S., Majerfeld I. Partial characterization of the factor responsible for trypophanless death in Bacillus subtilis. J Bacteriol. 1970;101:355–360. doi: 10.1128/jb.101.2.355-360.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry A. Improving production of aromatic compounds in Escherichia coli by metabolic. Tibtech. 1996;14:250–256. doi: 10.1016/0167-7799(96)10033-0. engineering. [DOI] [PubMed] [Google Scholar]
- Bieger C.D., Crawford I.P. Tryptophan biosynthesis in the marine luminous bacterium Vibrio harveyi. J Bacteriol. 1983;153:884–894. doi: 10.1128/jb.153.2.884-894.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buvinger W.E., Stone L.C., Heath H.E. Biochemical genetics of tryptophan synthesis in Pseudomonas acidovorans. J Bacteriol. 1981;147:62–68. doi: 10.1128/jb.147.1.62-68.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun D.H., Weary T. Transductional analysis of Pseudomonas aeruginosa methioninless auxotrophs. J Bacteriol. 1969;97:210–216. doi: 10.1128/jb.97.1.210-216.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras A., Molin S., Ramos J.L. Conditional‐suicide containment system for bacteria which mineralize aromatics. Appl Environ Microbiol. 1991;57:1504–1508. doi: 10.1128/aem.57.5.1504-1508.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuppels D.A. Generation and characterization of Tn5 insertion mutations in Pseudomonas syringae pv. tomato. Appl Environ Microbiol. 1986;51:323–327. doi: 10.1128/aem.51.2.323-327.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duque E., Haïdour A., Godoy F., Ramos J.L. Construction of a Pseudomonas hybrid strain that mineralizes 2,4,6‐trinitrotoluene. J Bacteriol. 1993;175:2278–2283. doi: 10.1128/jb.175.8.2278-2283.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duque E., Molina‐Henares A.J., De La Torre J., Molina‐Henares M.A., Del Castillo T., Lam J., Ramos J.L. Towards a genome‐wide mutant library of Pseudomonas putida strain KT2440. In: Ramos J.L., Filloux A., editors. V. Springer; 2007. pp. 227–255. [Google Scholar]
- Espinosa‐Urgel M., Ramos J.L. Cell density‐dependent gene contributes to efficient seed colonization by Pseudomonas putida KT2440. Appl Environ Microbiol. 2004;70:5190–5198. doi: 10.1128/AEM.70.9.5190-5198.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essar D.W., Eberly L., Han C.Y., Crawford I.P. DNA sequences and characterization of four early genes of the tryptophan pathway in Pseudomonas aeruginosa. J Bacteriol. 1990a;172:853–866. doi: 10.1128/jb.172.2.853-866.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essar D.W., Eberly L., Crawford I.P. Evolutionary differences in chromosomal locations of four early genes of the tryptophan pathway in fluorescent pseudomonads: DNA sequences and characterization of Pseudomonas putida trpE and trpGDC. J Bacteriol. 1990b;172:867–883. doi: 10.1128/jb.172.2.867-883.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin F.C.H., Bagdasarian M., Bagdasarian M.M., Timmis K.N. Molecular and functional analysis of the TOL plasmid pWW0 from Pseudomomas putida and cloning of genes for the entire regulated aromatic ring meta‐cleavage pathway. Proc Natl Acad Sci USA. 1981;78:7458–7462. doi: 10.1073/pnas.78.12.7458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garmendia J., De Las Heras A., Calcagno G.T., De Lorenzo V. Tracing explosives in soil with transcriptional regulators of Pseudomonas putida evolved for responding to nitrotoluenes. Microbial Biotechnol. 2008;1:236–426. doi: 10.1111/j.1751-7915.2008.00027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gussin G.N. Activation of transcription initiation and regulation of tryptophan biosíntesis in fluorescent pseudomonads. In: Ramos J.L., editor. Vol. 3. Kluwer Academic/Plenum Publishers; 2004. pp. 293–322. [Google Scholar]
- Hansen S.K., Rainey P.B., Haagensen J.A., Molin S. Evolution of species interactions in a biofilm community. Nature. 2007;445:533–536. doi: 10.1038/nature05514. [DOI] [PubMed] [Google Scholar]
- Herrera M.C., Ramos J.L. Catabolism of phenylalanine by Pseudomonas putida: the NtrC‐family PhhR regulator binds to two sites upstream from the phhA gene and stimulates transcription with σ‐70. J Mol Biol. 2007;366:1374–1386. doi: 10.1016/j.jmb.2006.12.008. [DOI] [PubMed] [Google Scholar]
- Herrero M., De Lorenzo V., Timmis K.N. Transposon vectors containing non‐antibiotic resistance selection markers for cloning and stable chromosomal insertion of foreign genes in gram‐negative bacteria. J Bacteriol. 1990;172:6557–6567. doi: 10.1128/jb.172.11.6557-6567.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac J.H., Holloway B.W. Control of pyrimidine biosynthesis in Pseudomonas aeruginosa. J Bacteriol. 1968;96:1732–1741. doi: 10.1128/jb.96.5.1732-1741.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindow S.E., Andersen G., Beattie G.A. Characteristics of insertional mutants of Pseudomonas syringae with reduced epiphytic fitness. Appl Environ Microbiol. 1993;59:1593–1601. doi: 10.1128/aem.59.5.1593-1601.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llamas M.A., Rodríguez‐Herva J.J., Hancock R.E., Bitter W., Tommassen J., Ramos J.L. Role of Pseudomonas putida tol‐oprL gene products in uptake of solutes through the cytoplasmic membrane. J Bacteriol. 2003;185:4707–4716. doi: 10.1128/JB.185.16.4707-4716.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lorenzo V., Timmis K.N. Analysis and construction of stable phenotypes in Gram‐negative bacteria with Tn5‐ and Tn10‐derived minitransposons. Methods Enzymol. 1994;235:386–405. doi: 10.1016/0076-6879(94)35157-0. [DOI] [PubMed] [Google Scholar]
- Marqués S., Aranda‐Olmedo I., Ramos J.L. Controlling bacterial physiology for optimal expression of gene reporter constructs. Curr Opin Biotech. 2006;17:50–56. doi: 10.1016/j.copbio.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Matilla M., Espinosa‐Urgel M., Rodríguez‐Herva J.J., Ramos J.L., Ramos‐González M.I. Genomic analysis reveals the major driving forces of bacterial life in the rhizosphere. Genome Biol. 2007;8:R179‐1–R179‐13. doi: 10.1186/gb-2007-8-9-r179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina L., Ramos C., Duque E., Ronchel M.C., Garcia J.M., Wyke L., Ramos J.L. Survival of Pseudomonas putida KT2440 in soil and in the rhizosphere of plants under greenhouse and environmental conditions. Soility Biol Biochem. 2000;32:315–321. [Google Scholar]
- Murashige T., Skoog F. A revised medium for rapid growth and bioassays with tobacco tissue culture. Physiol Plant. 1962;15:473–497. [Google Scholar]
- Nakazawa T. Travels of a Pseudomonas, from Japan around the world. Environ Microbiol. 2002;4:782–786. doi: 10.1046/j.1462-2920.2002.00310.x. [DOI] [PubMed] [Google Scholar]
- Nüsslein D., Maris D., Timmis K.N., Dwyer D.F. Expression and transfer of engineered catabolic pathways harbored by Pseudomonas spp. introduced into activated sludge microcosms. Appl Environ Microbiol. 1992;58:3380–3386. doi: 10.1128/aem.58.10.3380-3386.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos J.L., Duque E., Rodríguez‐Herva J.J., Godoy P., Haïdour A., Reyes F., Fernández‐Barrero A. Mechanisms for solvent tolerance in bacteria. J Biol Chem. 1997;272:3887–3890. doi: 10.1074/jbc.272.7.3887. [DOI] [PubMed] [Google Scholar]
- Ramos J.L., Díaz E., Dowling D., De Lorenzo V., Molin S., O' Gara F. The behavior of bacteria designed for biodegradation. Bio/Technology. 1994;12:1349–1358. doi: 10.1038/nbt1294-1349. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronchel M.C., Ramos J.L. Dual system to reinforce biological containment of recombinant bacteria designed for rhizoremediation. Appl Environ Microbiol. 2001;67:2649–2656. doi: 10.1128/AEM.67.6.2649-2656.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J., Fritsch E.F., Maniatis T. 2nd. Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Timmis K.N. Pseudomonas putida: a cosmopolitan opportunist par excellence. Environ Microbiol. 2002;4:779–781. doi: 10.1046/j.1462-2920.2002.00365.x. [DOI] [PubMed] [Google Scholar]
- Werler C., Jasper M.C.M., Van Der Meer J.R. Measurement of biologically available naphthalene in gas and aqueous phases by use of a Pseudomonas putida biosensor. Appl Environ Microbiol. 2004;70:43–51. doi: 10.1128/AEM.70.1.43-51.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker R.J., Gaines C.G., Jensen R.A. A multispecific quintet of aromatic aminotransferases that overlap different biochemical pathways in Pseudomonas aeruginosa. J Biol Chem. 1982;22:13550–13556. [PubMed] [Google Scholar]
- Worsey M.J., Williams P.A. Metabolism for toluene and xylenes by Pseudomonas (putida(arvilla) mt‐2: evidence for a new function of the TOL plasmid. J Bacteriol. 1975;124:7–13. doi: 10.1128/jb.124.1.7-13.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie G., Bonner C.A., Jensen R.A. A probable mixed‐function supraoperon in Pseudomonas exhibits gene organization features of both intergenomic conservation and DNA shuffling. J Mol Evol. 1999;49:108–121. doi: 10.1007/pl00006523. [DOI] [PubMed] [Google Scholar]
- Zhao G., Xia T., Song J., Jensen R.A. Pseudomonas aeruginosa possesses homologues of mammalian phenylalanine hydroxylase and 4 alpha‐carbinolamine dehydratase/DCoH as part of a three‐component gene cluster. Proc Natl Acad Sci USA. 1994;91:1366–1370. doi: 10.1073/pnas.91.4.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Organization of trp genes in P. aeruginosa PAO1, P. entomophila L48, P. fluorescens Pf0-1 and P. syringae.
Organization of pheA/tyrA genes in several Pseudomonas.
Conservation of tyrosine and phenylalanine biosynthesis genes among Pseudomonas species.