

SUMMARY
Troyer syndrome is a hereditary spastic paraplegia caused by human spartin (SPG20) gene mutations. We have generated a Drosophila disease model showing that Spartin functions presynaptically with endocytic adaptor Eps15 to regulate synaptic growth and function. Spartin inhibits bone morphogenetic protein (BMP) signaling by promoting endocytic degradation of BMP receptor wishful thinking (Wit). Drosophila fragile X mental retardation protein (dFMRP) and Futsch/MAP1B are downstream effectors of Spartin and BMP signaling in regulating microtubule stability and synaptic growth. Loss of Spartin or elevation of BMP signaling induces age-dependent progressive defects resembling hereditary spastic paraplegias, including motor dysfunction and brain neurodegeneration. Null spartin phenotypes are prevented by administration of the microtubule-destabilizing drug vinblastine. Together, these results demonstrate that Spartin regulates both synaptic development and neuronal survival by controlling microtubule stability via the BMP-dFMRP-Futsch pathway, suggesting that impaired regulation of microtubule stability is a core pathogenic component in Troyer syndrome.
Keywords: hereditary spastic paraplegia, Troyer syndrome, microtubule stability, BMP signaling, NMJ synapse, neuronal survival, Drosophila
INTRODUCTION
Trans-synaptic retrograde signaling from postsynaptic target cells to presynaptic terminals plays critical roles in synapse formation, growth and plasticity, as well as presynaptic neuronal survival (Regehr et al., 2009; Zweifel et al., 2005). In Drosophila, the BMP retrograde signal Glass bottom boat (Gbb) secreted from postsynaptic muscles acts on type II receptor wishful thinking (Wit) and type I receptors thickveins (Tkv) and saxophone (Sax) on presynaptic terminals to regulate proper growth and function of neuromuscular junction (NMJ) synapses (Aberle et al., 2002; Marques et al., 2002; McCabe et al., 2003; Rawson et al., 2003). Upon Gbb binding, two type I and two type II receptors form a tetrameric complex, which phosphorylates the Smad transcription factor Mothers against decapentaplegic (Mad). Phosphorylated Mad (P-Mad) then makes complexes with the co-Smad Medea (Med) to enter the nucleus and regulate transcription.
Drosophila NMJ studies have shown that BMP signaling is tightly regulated at multiple levels. Postsynaptic Gbb secretion is negatively regulated by the Cdc42-Wsp pathway (Nahm et al., 2010a; Nahm et al., 2010b) and presynaptic signaling is attenuated by endocytic proteins regulating internalization and endosomal trafficking of BMP receptors (O’Connor-Giles et al., 2008; Sweeney and Davis, 2002; Wang et al., 2007). Interestingly, regulators of BMP receptor endocytosis and trafficking include Drosophila homologs of human proteins associated with neurodegenerative diseases (Bayat et al., 2011), including hereditary spastic paraplegia 6 (HSP6) (Wang et al., 2007) and multiple sclerosis (MS) (Kim et al., 2010). However, the mechanisms by which elevated BMP signaling causes neuronal degeneration remain largely speculative.
HSPs comprise a heterogeneous group of neurodegenerative diseases characterized by progressive lower limb spasticity and weakness (Blackstone et al., 2011). These neuropathies are due to distal degeneration of corticospinal tract axons. Troyer syndrome is an autosomal recessive, complicated HSP characterized by dysarthria, mental retardation, distal muscle wasting and short stature, in addition to lower extremity spastic weakness (Cross and McKusick, 1967). Troyer syndrome HSP is caused by loss-of-function mutations in the human spartin gene (SPG20) (Manzini et al., 2010; Patel et al., 2002), which encodes a protein consisting of several domains: an N-terminal MIT (contained in microtubule-interacting and trafficking molecules) domain, a central Eps15-interacting domain, and a C-terminal senescence domain (Bakowska et al., 2005; Ciccarelli et al., 2003).
Studies on cultured mammalian cells have shown that Spartin is a multifunctional protein in several subcellular compartments. Spartin interacts with endocytic trafficking protein Eps15 and plays a role in intracellular trafficking of epidermal growth factor receptor (EGFR), transiently localizing to endosomes upon EGF stimulation (Bakowska et al., 2007). Spartin also localizes transiently to midbodies during cell division through interaction with increased sodium tolerance 1 (Ist1), a component of endosomal sorting complexes required for transport (ESCRT)-III complex (Renvoise et al., 2010). Moreover, Spartin associates with lipid droplets (LDs) in cells treated with oleic acid and regulates their turnover, possibly by recruiting ubiquitin E3 ligases of the Nedd4 family (Eastman et al., 2009; Edwards et al., 2009; Hooper et al., 2010). Spartin also associates with mitochondria via interaction with cardiolipin, a phospholipid of the outer membrane, and regulates mitochondrial Ca2+ homeostasis (Joshi and Bakowska, 2011). However, it remains unclear whether these Spartin activities are involved in neuronal function and maintenance in vivo.
The Drosophila genome contains a single spartin gene. To model Troyer syndrome, we have analyzed loss-of-function phenotypes at the larval NMJ synapse as well as in the adult brain. We show that Spartin localizes to NMJ presynaptic membrane and endosomes, and works with Eps15 to regulate synaptic endocytosis and growth. We show that Spartin-mediated endocytosis and endosomal sorting are required for downregulation of the BMP receptor Wit, which initiates a retrograde signal controlling microtubule stability (Ellis et al., 2010; Wang et al., 2007) and synaptic growth (Aberle et al., 2002; Marques et al., 2002; McCabe et al., 2003; Rawson et al., 2003). We show that Spartin/BMP signaling regulates synaptic growth via modulation of dfmr1 expression, which encodes Drosophila fragile X mental retardation protein (dFMRP) (Coffee et al., 2010). dFMRP in turn negatively regulates the expression of the Drosophila MAP1B Futsch (Zhang et al., 2001), which is required for the maintenance of stable microtubules (Bettencourt da Cruz et al., 2005; Roos et al., 2000). Importantly, both spartin mutation and elevated BMP signaling cause progressive neurodegeneration. Rescue with the microtubule-destabilizing drug vinblastine shows that Spartin/BMP signaling-mediated regulation of microtubule stability drives both synaptic growth and neuronal survival. This study establishes a novel synaptic role for Spartin and suggests a specific pathogenic mechanism for Troyer syndrome and other BMP-related neurodegenerative diseases.
RESULTS
Generation of spartin Null Mutant
BLASTP search of the Drosophila genome identified a single homolog of human spartin (CG12001). Sequence alignment showed Drosophila Spartin has an identical domain organization, with 21% identity and 44% similarity (Figure 1A). EP element G8635 was imprecisely excised to generate a 1,957 bp deletion from the EP insertion site into the second exon of spartin, generating the spartin1 allele (Figure 1B). Homozygous spartin1 mutants and transheterozygotes with deficiency Df(3R)110 (hereafter refer to as Df) are viable and fertile. In these animals, spartin RNA expression was abolished (Figure 1C), but adjacent Karyβ3 RNA expression was unaffected, showing that spartin1 represents a specific transcriptional null allele.
Figure 1. Molecular Characterization of Drosophila spartin Gene and Mutants.

(A) Alignment of Drosophila and human Spartin. Identical residues are indicated by white letters on black background. Similar residues are indicated by black letters on gray background. Spartin domain abbreviations: microtubule-interacting and trafficking protein domain (MIT), Eps15-interacting domain (Eps15), senescence domain (SC).
(B) Genomic organization of Drosophila spartin locus, showing exon/intron organization of spartin and neighboring gene Karyβ3, position of P element G8635 (inverted triangle), and spartin1 deletion generated by G8635 excision. Introns indicated by horizontal lines, exons by boxes, coding regions by black boxes, and translation initiation sites by arrows.
(C) RT-PCR analysis of wildtype (WT), spartin1/spartin1, and spartin1/Df(3R)110 (spartin1/Df) third instar larvae using primers specific for spartin, Karyβ3 and rp49.
Spartin Localizes Presynaptically at the NMJ
We generated a Spartin antibody against amino acids 102–292 (Figure 1A). In Western blots of lysates derived from third instar larvae, the antibody recognized a single 60 kDa band, the expected size of Spartin, which was absent in spartin1/Df (Figure 2A), demonstrating antibody specificity. Spartin was detected broadly in larval brain and ventral nerve cord, colocalizing with synaptic protein Synaptotagmin 1 (Syt) in neuropil regions (Figure 2B). Spartin was not detected in the CNS of spartin1/Df. The protein was similarly enriched at larval NMJ synapses (Figure 2C). Double labeling with the anti-HRP neuronal membrane marker revealed that Spartin was closely associated with the presynaptic membrane, with a small portion localized to bouton central regions, but was not detectable in postsynaptic muscles (Figure 2C). Spartin was not detected in the NMJ of spartin1/Df (Figure 2D). At the presynaptic membrane, Spartin was not colocalized with active zone marker Bruchpilot/NC82, but rather localized in distinctive domains adjacent to active zones (Figure 2E). Within synaptic boutons, Spartin showed a punctate distribution partially overlapping with early endosomal markers, Rab5 and GFP-2xFYVE (Figures 2F and 2G). Similarly in Drosophila S2 cells, HA-Spartin overlapped with GFP-Rab5 at the plasma membrane and in intracellular punctate structures (Figure 2H). Consistent with previous reports (Eastman et al., 2009; Edwards et al., 2009), HA-Spartin was also detected on lipid droplets in S2 cells (Figure 2H, asterisk). Thus, Spartin localizes to distinctive plasma membrane domains and Rab5-positive early endosomes in both NMJ synapses and non-neuronal cells.
Figure 2. Spartin Localizes to Drosophila NMJ Presynaptic Terminal.

(A) Western blot of total extracts prepared from wildtype and spartin1/Df third instar larvae with anti-Spartin and anti-β-actin.
(B) Confocal images of third instar CNS from wildtype and spartin1/Df larvae double labeled with anti-Spartin and anti-Synaptotagmin 1 (Syt). Inset shows anti-Syt staining in spartin1/Df.
(C–H) Single confocal slices of third instar NMJ 6/7 (C–G) and S2 cells (H). (C and D) Wildtype and spartin1/Df labeled with anti-HRP and anti-Spartin antibodies. (E and F) Wildtype double labeled with anti-Spartin and anti-NC82 (E) or anti-Rab5 (F) antibodies. (G) UAS-GFP-2xFYVE driven by C155-GAL4, labeled with anti-GFP and anti-Spartin. Arrowheads show Spartin puncta co-stained for Rab5 or GFP-2xFYVE. (H) S2 cells expressing GFP-Rab5 and HA-Spartin labeled with anti-GFP and anti-HA. Spartin and Rab5 partially colocalize at the plasma membrane and in cytoplasmic punctate structures. Spartin, but not Rab5, also on lipid droplets (asterisk). Scale bars: (B) 100 μm, (G and H) 5 μm.
Spartin Required Presynaptically for Normal NMJ Growth
Given the presynaptic localization of Spartin, we next tested effects of spartin loss on synapse development and function. Compared with control (w1118), homozygous spartin1 and spartin1/Df mutants both displayed a clear overgrowth of the third instar NMJ (Figures 3A and 3B and Table S1). When normalized to muscle surface area, overall bouton number and satellite bouton number were increased by ~40% and ~90%, respectively, in spartin mutants compared with control (p < 0.001 for both) (Figure 3B). Revertant (precise excision of EP element G8635) animals showed normal NMJ morphology (Figure 3B). Synaptic overgrowth in spartin1/Df was completely rescued by presynaptic expression of HA-tagged Drosophila spartin (UAS-HA-spartin) or Myc-tagged human spartin (UAS-Myc-spartin-human) from panneuronal driver C155-GAL4 (Figure 3B). In contrast, expression with muscle driver BG57-GAL4 failed to rescue NMJ phenotypes (Figure 3B). Overexpression of UAS-HA-spartin in the wildtype background using C155-GAL4 significantly reduced overall bouton and satellite bouton numbers compared with control (p < 0.01 and p < 0.001, respectively) (Figure 3B). These gain-of-function phenotypes are the opposite of those observed in spartin nulls, revealing an inverse relationship between presynaptic Spartin level and synaptic growth. NMJ morphology in first instar spartin1/Df larvae (1 hr after hatching) was normal (data not shown), indicating that loss of spartin function does not impair embryonic synapse formation. Thus, Spartin functions in neurons to restrain synaptic growth during postembryonic development.
Figure 3. Presynaptic Spartin Controls NMJ Structure and Function.

(A and B) Mutations in spartin cause overgrowth of the larval NMJ. (A) Confocal images of anti-HRP labeled NMJ 6/7 in wildtype and spartin1/Df. Scale bar, 50 μm. (B) Quantification of total bouton number normalized to muscle surface area and satellite bouton number in the following genotypes: wildtype, precise excision EP line G8635 (revertant), spartin1/spartin1, spartin1/Df, C155-GAL4/+; Df/UAS-HA-spartin,spartin1 (Spartin rescue-pre), C155-GAL4/+; Df/UAS-HA-spartinΔEps15,spartin1 (SpartinΔEps15 rescue-pre), C155-GAL4/+; Df/UAS-Myc-spartin-human,spartin1 (Human Spartin rescue-pre), BG57-GAL4,Df/UAS-HAspartin, spartin1 (Spartin rescue-post), C155-GAL4/+, and C155-GAL4/+; UAS-HA-spartin/+.
(C–F) Synaptic transmission and vesicle fusion frequency are depressed in spartin mutants. (C) Representative evoked EJCs from wandering third instar NMJ (1.0 mM Ca2+) for wildtype, spartin1/spartin1, spartin1/Df, C155-GAL4/+; Df/UAS-HA-spartin,spartin1 (Spartin rescue-pre), and BG57-GAL4,Df/UAS-HA-spartin,spartin1 (Spartin rescue-post). Each family of traces shows ten consecutive evoked EJC responses in a 0.2-Hz stimulus train. (D) Mean amplitude of evoked EJCs. (E) Representative traces of spontaneous mEJCs. Each record shows a typical 3-sec sample from a 2-min recording session. (F) Mean frequency of spontaneous mEJCs. The number of NMJs examined for each genotype is indicated inside the bars. All comparisons are with wildtype unless indicated (*p < 0.001; **p < 0.01; ***p < 0.05; n.s., not significant). Error bars are SEM for all figures.
For a more detailed examination of satellite boutons, we assayed spartin1/Df NMJs using an array of synaptic markers. The synaptic vesicle marker CSP and the active zone marker NC82 were present in each satellite bouton (Figures S1A and S1B). In addition, all satellite boutons were surrounded by DLG-positive subsynaptic reticulum (SSR) (Figure S1C) and apposed to postsynaptic glutamate receptor clusters (GluRIIC) (Figure S1D). BRP-positive active zones were correctly juxtaposed to GluRIIC domains (Figure S1E). Importantly, transmission EM revealed the presence of clear synaptic vesicles, mitochondria and electron-dense active zone T-bars in satellite boutons (Figure S1F). Thus, satellite boutons in spartin mutants display the predicted molecular and structural features of a functional synaptic bouton.
Spartin Required Presynaptically for NMJ Neurotransmission
We next examined functional properties of spartin null NMJs by measuring synaptic currents using two-electrode voltage-clamp (TEVC) recording. The mean amplitude of evoked excitatory junction currents (EJCs) was clearly and consistently decreased in spartin mutants compared with control (wildtype: 345 ± 11.1 nA; spartin1/spartin1: 204 ± 9.3 nA, p < 0.001; spartin1/Df: 230 ± 6.3 nA, p < 0.001) (Figures 3C and 3D). This defect was strongly rescued by presynaptic expression of HA-Spartin in the spartin1/Df null background (C155-GAL4/+; Df/UAS-HA-spartin,spartin1: 290 ± 9.5 nA, p = 0.09 from wildtype), while postsynaptic expression had no effect (BG57-GAL4,Df/UAS-HA-spartin, spartin1: 186 ± 9.7 nA, p < 0.001) (Figure 3D). Evoked EJC amplitudes were not significantly different between wildtype and revertant larvae (353 ± 14.14 nA, p = 1.0 from wildtype). Presynaptic overexpression of HA-Spartin in the wildtype background had no effect on synaptic transmission (Figures 3D and 3F). Spontaneous miniature EJCs (mEJCs) were next examined to further differentiate presynaptic versus postsynaptic mechanisms. Compared with controls, mEJC amplitudes were not significantly changed in spartin mutants (wild type, 0.78 ± 0.03 nA; spartin1/spartin1, 0.87 ± 0.05 nA, p = 0.33; spartin1/Df, 0.78 ± 0.05 nA, p = 1.0) (Figures 3E and 3F), further confirming that Spartin is not working postsynaptically. The distribution of mEJC amplitudes was indistinguishable between control and spartin mutants (Kolmogorov-Smirnov test, p = 0.95) (Figure S1G). Average mEJC frequency displayed a downward trend in spartin mutants compared with control (wildtype: 3.4 ± 0.18 Hz; spartin1/spartin1: 2.2 ± 0.21 Hz, p < 0.05; spartin1/Df, 2.78 ± 0.18 Hz, p = 0.81) (Figure 3F).
Spartin Interacts with Eps15 to Regulate Synaptic Growth and Endocytosis
Human Spartin interacts with the endocytic and trafficking protein Eps15 in vitro (Bakowska et al., 2005). We therefore tested physical interactions between Drosophila Spartin and Eps15. First, pull-down experiments with an Eps15 GST fusion lacking the first 582 amino acids (GST-Eps15ΔN) revealed that full-length HA-Spartin interacted specifically with GST-Eps15ΔN, but not with GST alone (Figure S2A). In contrast, a mutant form of HA-Spartin lacking the putative Eps15-binding domain (HA-SpartinΔEps15) failed to interact with GST-Eps15ΔN, demonstrating that the predicted Eps15-binding domain in Drosophila Spartin mediates the interaction with Eps15. Second, interactions between HA-Spartin and Myc-Eps15 were confirmed in S2 cells by co-immunoprecipitation with anti-Myc or anti-HA antibody (Figure S2B). Finally, Spartin and Eps15 closely colocalize at the NMJ (Figure S2C).
At the Drosophila NMJ, eps15 null mutants exhibit a synaptic overgrowth phenotype similar to spartin (Koh et al., 2007), suggesting a functional relationship. To test this possibility, we examined transheterozygous interaction between spartin and eps15. Single heterozygotes lacking one copy of spartin and eps15 alone did not show any growth defects. However, both overall bouton number and satellite bouton number were significantly increased in spartin-eps15 transheterozygotes (p < 0.001) (Figure S2D and Table S2), suggesting that they function in the same pathway.
Eps15 plays an essential role in synaptic vesicle endocytosis (Koh et al., 2007). To test whether Spartin is also involved in this process, we performed FM1-43FX styryl dye uptake experiments, depolarizing NMJ synapses with 90 mM K+. During a 1 min labeling period, dye uptake was decreased by ~50% in spartin1/Df compared with wildtype or revertant animals (p < 0.001) (Figures 4A and 4B). The defect in FM1-43FX uptake was fully rescued by presynaptic expression of wildtype HA-Spartin (Figure 4B). Transheterozygous interaction between spartin and eps15 was also observed during the process of FM1-43FX uptake (Figure 4B), further supporting function in the same pathway. Moreover, neuronal expression of HA-SpartinΔEps15 in spartin1/Df failed to rescue defects in synaptic growth and synaptic vesicle endocytosis (Figures 3B and 4B), showing that Spartin-Eps15 interaction is essential for regulating both synaptic growth and endocytosis.
Figure 4. Spartin Functions with Eps15 to Control Synaptic Endocytosis.
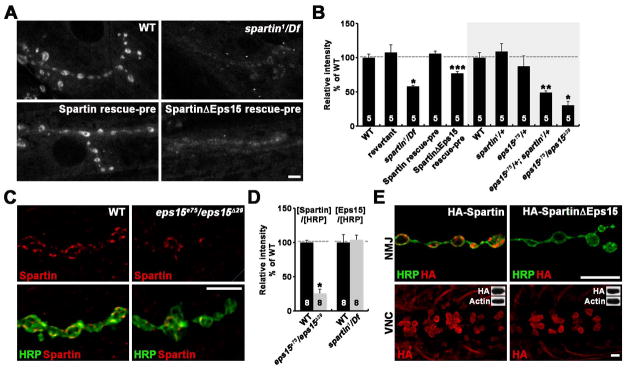
(A and B) Null spartin NMJs show impaired FM1-43FX dye uptake. (A) Representative confocal images of NMJ synapses stimulated with 90 mM K+ in 5 mM Ca2+. Genotypes include wildtype, spartin1/Df, C155-GAL4/+; Df/UAS-HA-spartin,spartin1 (Spartin rescue-pre), and C155-GAL4/+; Df/UAS-HA-spartinΔEps15, spartin1 (SpartinΔEps15 rescue-pre).
(B) Mean intensities of FM1-43FX labeling as percentages of wildtype.
(C–E) Eps15 is required for efficient synaptic localization of Spartin. (C) Single confocal slices of NMJ 6/7 doubly labeled with anti-HRP and anti-Spartin shown for wildtype and eps15 (eps15e75/eps15Δ29) mutants. (D) Quantification of Spartin or Eps15 to HRP level ratios at NMJ 6/7 in genotypes indicated. Values are presented as percentages of wildtype. (E) Single confocal slices of NMJ 6/7 branches and larval ventral nerve cords. Third instar spartin1/Df larvae expressing HA-Spartin or HA-SpartinΔEps15 with C155-GAL4 were double labeled with anti-HA and anti-HRP. Insets show Western blots of the larval CNS. Scale bars: (A and C) 5 μm, (E) 10 μm. *p < 0.001; **p < 0.01; ***p < 0.05.
Drosophila Eps15 plays a critical role in maintaining endocytic proteins at NMJ synapses (Koh et al., 2007). We therefore tested whether Eps15 is also required for synaptic localization of Spartin. We found that Spartin signal intensity normalized to HRP signal intensity was significantly decreased in eps15 mutants compared with controls (p < 0.001) (Figures 4C and 4D), showing that efficient Spartin localization to the NMJ presynaptic terminal depends on Eps15. In the reciprocal experiment, Eps15 localization was not altered in spartin nulls (data not shown). To further substantiate the role of Eps15 in synaptic targeting of Spartin, we compared distribution of HA-Spartin and HA-SpartinΔEps15 in the spartin1/Df background (Figure 4E). Like endogenous Spartin, wildtype HA-Spartin was efficiently targeted to the NMJ synapse. In contrast, HA-SpartinΔEps15 was not enriched at the synapse but was instead detected only in the neuronal soma and within proximal motor axons. Expression levels of both HA fusions were comparable (Figure 4E, insets), suggesting that Eps15 recruits Spartin to the NMJ presynaptic terminal through a Spartin-Eps15 interaction.
Spartin Inhibits BMP Signaling by Endocytic Degradation of BMP Receptor Wit
Recapitulating the spartin null phenotype, elevated BMP signaling by presynaptic co-overexpression of constitutively active Tkv (TkvCA) and wildtype Wit or loss of the inhibitory Smad Dad potently induced synaptic overgrowth with excessive satellite bouton formation (Figures 6B, S3A, and S3B and Table S3) (O’Connor-Giles et al., 2008; Sweeney and Davis, 2002). Postsynaptic Gbb overexpression had the same effect (Figures S3A and S3B), supporting a positive relationship between levels of retrograde BMP signaling and extent of NMJ synaptic growth. We therefore hypothesized that synaptic overgrowth in spartin nulls might be due to defective BMP signaling. To test this hypothesis, we examined genetic interactions between spartin and components of the BMP pathway (Figure 5 and Table S4). We found that removing one copy of the BMP receptor wit, which did not alter NMJ morphology by itself, reversed the synaptic overgrowth phenotype of spartin nulls (Figures 5A and 5B). Moreover, spartin synaptic overgrowth was further suppressed by removing both copies of wit (Figure 5B). To directly test the inhibitory effect of Spartin on BMP signaling, we examined phosphorylated Mad (P-Mad), a well-established readout of BMP signaling (Marques et al., 2002; McCabe et al., 2003). In controls, P-Mad immunoreactivity was detected at NMJ termini and motor neuron nuclei (Figure 5C). The intensity of P-Mad signal at both locations was significantly increased in spartin nulls (p < 0.001) (Figure 5D), showing that Spartin acts as an inhibitor of BMP signaling in motor neurons. These data indicate that synaptic overgrowth in spartin mutants requires BMP signaling and further that Spartin restrains synaptic growth by inhibiting presynaptic BMP signaling.
Figure 6. Spartin and BMP Signaling Regulate dFMRP Expression and Microtubule Stability to Modulate Synaptic Growth.

(A and B) Vinblastine suppresses synaptic overgrowth caused by spartin mutations or elevation of BMP signaling. (A) Confocal images of NMJ 6/7 labeled with anti-HRP shown for spartin1/Df in the absence (−VB) or presence (+VB) of 1 μM vinblastine. (B) Quantification of synaptic structure in the indicated genotypes grown in the absence or presence of vinblastine.
(C and D) Transheterozygous interactions between spartin, dad, and dfmr1. (C) Confocal images of NMJ 6/7 stained with anti-HRP. (D) Quantification of synaptic structure in the indicated genotypes.
(E–G) Spartin and BMP signaling regulates dfmr1 expression in the larval CNS. (E) Quantitation of dfmr1 RNA expression using quantitative real-time PCR (qRT-PCR). Samples were run in triplicate in two independent experiments. rp49 was used as an internal control. (F) Western blot analysis of larval CNS extracts using anti-dFMRP and anti-β-actin. (G) Quantitative analysis by densitometric measurements from 3 independent experiments. β-actin served as an internal control. All comparisons are with wildtype unless indicated (*p < 0.001; **p < 0.01; n.s., not significant). Scale bar, 50 μm.
See also Figure S4 and Tables S5 and S6.
Figure 5. Spartin Restrains Synaptic Growth by Downregulating BMP Signaling.

(A and B) Synaptic overgrowth in spartin nulls depends on BMP signaling. (A) Confocal images of NMJ 6/7 labeled with anti-HRP shown for wildtype, spartin1/Df, and witA12,spartin1/+,Df larvae. (B) Quantification of synaptic structure in indicated genotypes.
(C and D) Spartin is required for P-Mad accumulation in motor neurons. (C) Confocal images of NMJ 6/7 and ventral nerve cord (VNC) labeled for both anti-P-Mad and anti-HRP (NMJ) or anti-Elav (VNC) shown for wildtype and spartin1/Df. (D) Quantification of P-Mad to HRP level ratio (NMJ) and P-Mad to Elav level ratio (VNC).
(E–G) Spartin causes redistribution of Wit from cell surface to lysosome in S2R+ cells. (E) Spartin-dependent internalization of Wit. S2R+ cells transfected with pAc-Myc-wit in the absence (control) or presence of spartin dsRNA treated with cycloheximide for 5 hrs to block new protein synthesis. Surface Myc-Wit protein was labeled with anti-Myc before permeabilization. Total Myc-Wit protein was labeled with anti-Wit after permeabilization.
(F) Quantification of surface-to-total Myc-Wit ratio. Values are presented as percentages of mock-treated controls. (G) S2R+ cells were transfected with pAc-spin-GFP and pAc-Myc-Wit along with or without pAc-HA-spartin were treated with cycloheximide as described in (E). Distribution of Spin-GFP, HA-Spartin, and total Myc-Wit was visualized with anti-GFP, anti-HA, and anti-Wit, respectively. Arrowheads indicate intracellular puncta structures double labeled for Myc-Wit and Spin-GFP.
(H and I) Spartin regulates Wit levels at the NMJ. (H) Confocal images of NMJ 6/7 triple labeled with anti-Wit, anti-HRP, and anti-HA are shown for indicated genotypes. (I) Quantification of the ratio of mean Wit to HRP fluorescence intensities. Values are percentages of the C155-GAL4/+; UAS-wit/+ control. Scale bars: (A) 50 μm, (C) 10 μm, (E, G, and H) 5 μm. *p < 0.001; n.s., not significant.
See also Figure S3 and Tables S3 and S4.
In support of a Spartin role in synaptic endocytosis (Figure 4), spartin interacts with numerous mutations impairing endocytosis (e.g., dap160, endo and synj) during synaptic growth regulation (Figure S3C and Table S3). We therefore hypothesized that Spartin might be required for the endocytic internalization of surface BMP receptors. To test this hypothesis, we transfected a Myc-Wit construct into S2 cells and monitored surface Myc-Wit by immunostaining for the extracellular Myc-tag before cellular permeabilization (Figure 5E). Total Myc-Wit was also monitored by immunostaining for Wit after permeabilization of cells. Robust Myc-Wit expression occurred at the cell surface and in discrete intracellular puncta (Figure 5E). The ratio of surface to intracellular Myc-Wit levels was strongly increased in cells treated with spartin dsRNA compared with mock-treated controls, while Spartin overexpression had the opposite effect (Figure 5F), suggesting that Spartin stimulates the internalization of Wit to limit BMP signaling. To explore this mechanism, we investigated the effect of Spartin overexpression on the subcellular distribution of internalized Wit. In control S2 cells expressing the late endosomal/lysosomal marker Spinster (Spin)-GFP and Myc-Wit, only a small proportion of intracellular Myc-Wit puncta overlapped with Spin-GFP (Figure 5G). However, when HA-Spartin was additionally over-expressed, Myc-Wit puncta strongly overlapped with Spin-GFP, although HA-Spartin did not colocalize with Spin-GFP (Figure 5G). These data suggest that Spartin promotes endosomal trafficking of Wit to lysosomes for degradation.
Finally, we tested whether Spartin regulates Wit in vivo. Available anti-Wit antibodies fail to reproducibly detect any specific signals in wildtype synaptic boutons. To circumvent this limitation, we took advantage of UAS-wit overexpression in neurons using C155-GAL4. In control boutons overexpressing Wit, anti-Wit signals were clearly detected in a punctate pattern (Figure 5H). Levels of synaptic Wit were increased by ~34% in spartin mutants compared with control (Figure 5I). Conversely, co-overexpression of HA-Spartin decreased Wit levels by ~26% (Figure 5I). These data support the conclusion that Spartin acts presynaptically to downregulate Wit.
BMP Signaling and Spartin Regulate dFMRP and Futsch/MAP1B Expression
Disruption of BMP signaling reduces Drosophila Futsch/MAP1B levels and microtubule stability in motor axons and NMJ terminals (Ellis et al., 2010; Wang et al., 2007). Given the role for Spartin in inhibiting BMP signaling, we investigated whether spartin mutants affect Futsch levels to alter microtubule stability. We found that both Futsch levels and stable microtubules were significantly increased in spartin mutants compared with control (Figures S4A–S4D). Moreover, Futsch accumulation in spartin mutants was strongly suppressed by heterozygosity and hemizygosity for wit (Figure S4D), supporting a model in which Spartin inhibits BMP signaling to limit presynaptic Futsch levels and microtubule stablility. Disruption of BMP signaling also reportedly impairs fast axonal transport (Aberle et al., 2002; Ellis et al., 2010; Wang et al., 2007). However, the distribution of mitochondrial Mito-GFP and vesicular Syt in motor axons and NMJ terminals appeared normal in spartin mutants (Figures S4E-S4H), suggesting that axonal transport is not impaired by Spartin loss.
Futsch-dependent microtubule stability regulates NMJ synaptic growth, with hypomorphic futschK68 mutants exhibiting significant undergrowth (Roos et al., 2000). Consistently, loss or reduction of futsch function suppressed the synaptic overgrowth defect in spartin mutants, with transheterozygous interactions between futsch and mutations of BMP pathway components (Figure S4I and Table S5). Taken together, these results suggest that Futsch acts downstream of Spartin and BMP signaling to regulate NMJ growth. To independently assess the involvement of microtubule stability, we next tested effects of the microtubule-severing drug vinblastine (Jordan et al., 1992). Vinblastine fed at low concentration (1 μM) did not affect synaptic structure in wildtype animals but suppressed the NMJ overgrowth phenotype of spartin nulls (Figures 6A and 6B and Table S6). This treatment also restored Futsch and stable microtubules to control levels (Figure S4J). Similarly, synaptic overgrowth caused by presynaptic expression of TkvCA, or loss of Dad, was also significantly rescued by vinblastine treatment (Figure 6B). Collectively, these data are consistent with the model in which Spartin and BMP signaling regulate synaptic growth by modulating presynaptic Futsch levels to control microtubule stability.
How do Spartin and BMP signaling regulate presynaptic Futsch? dFMRP has been shown to restrain synaptic growth by repressing the expression of Futsch (Coffee et al., 2010; Zhang et al., 2001). We confirmed that dfmr1 null mutants display an increase in overall and satellite bouton number (Figure 6D), recapitulating defects caused by loss of Spartin and elevated BMP signaling. To test the possible link, we assayed transheterozygous interactions between dfmr1 and spartin or dad. In heterozygous spartin1/+, dadJ1e4/+, or dfmr1Δ50M/+ animals, overall and satellite bouton number were at wildtype levels (Figure 6D), however both parameters of synaptic growth were significantly increased in transheterozygous spartin1/+; dfmr1Δ50M/+ and dfmr1Δ50M/+; dadJ1e4/+ (Figures 6C and 6D and Table S6). We next assayed whether Spartin and BMP signaling regulate dfmr1 expression. In the larval CNS (brain and ventral nerve cord), dfmr1 levels were significantly decreased by loss of Spartin or Dad (Figure 6E). Moreover, postsynaptic overexpression of Gbb similarly decreased dfmr1 expression, whereas loss of Wit had the opposite effect of elevating dfmr1 expression (Figure 6E). Consistently, dFMRP protein levels were likewise affected by all genetic manipulations (Figures 6F and 6G). Together, these results strongly argue that both Spartin and BMP signaling modulate synaptic growth primarily through regulation of dfmr1 expression.
Adult spartin Mutants Display Reduced Locomotor Activity and Neurodegeneration
The HSP clinical hallmark is progressive lower limb spasticity and weakness severely affecting movement. We therefore tested whether Drosophila spartin mutants similarly exhibit movement dysfunction. Climbing assays were done on adult flies (20 days) by measuring vertical distance climbed in a graduated cylinder within 30 seconds. Compared with revertant controls, spartin nulls displayed significantly reduced climbing ability (control: 18.4 ± 0.4 cm; spartin1/Df: 10.6 ± 1.1 cm, p < 0.001) (Figures 7A and 7B). This movement defect was rescued by either neuronal expression of Spartin or by feeding adult animals with 1 μM vinblastine (Figure 7B), linking the movement phenotype to microtubule stability in neurons.
Figure 7. Loss of spartin Function in Neurons Causes Movement Defects and Progressive Neurodegeneration.

(A and B) Loss of neural Spartin severely impairs adult locomotor activity. (A) Distribution of the distance climbed by 20-day-old revertant, spartin1/Df, and C155-GAL4/+; Df/UAS-HA- spartin,spartin1 (Spartin rescue) measured over 30 sec in a vertical graduated cylinder after the mechanical shock of 10-sec vortexing. (B) Average distance climbed by 20-day-old revertant, spartin1/Df, and C155-GAL4/+; Df/UAS-HA-spartin,spartin1 (Spartin rescue) grown in the absence (−VB) or presence (+VB) of 1 μM vinblastine.
(C–G) Loss of neuronal Spartin causes age-dependent neurodegeneration. (C) H&E-stained frontal sections (5 μm thick) of brains from 20-day-old wildtype and spartin1/Df animals. Vacuoles are marked by white arrows. (D) Quantification of vacuoles with diameter greater than 5 μm in wildtype and spartin1/Df brains at different ages. (E) Quantification of brain vacuoles in 20-day-old wildtype, revertant, spartin1/Df, C155-GAL4/+; Df/UAS-HAspartin, spartin1 (Spartin rescue), and C155-GAL4/+; Df/UAS-Myc-spartin-human,spartin1 (human Spartin rescue). (F) Confocal slices of 20-day-old spartin1/Df brains double labeled with TUNEL or anti-Caspase-3 and anti-Elav or anti-Repo. (G) Quantification of TUNEL-positive cells in 3 consecutive, middle frontal sections (10 μm thick) of 20-day-old brains from the indicated genotypes. The sample size in (D), (E), and (G) was n = 10 animals for each genotype. All comparisons are with wildtype unless indicated (*p < 0.001; ***p < 0.05). Scale bars: (C) 50 μm, (F) 10 μm.
We next tested whether Spartin loss is associated with neurodegeneration, which is manifested by progressive vacuolization in the adult brain (Muqit and Feany, 2002). At eclosion (< 2 days), spartin nulls were not significantly different from wildtype or revertant controls with respect to anatomical and histological organization of the brain (data not shown). However, progressively aged spartin mutants exhibited numerous brain neuropil vacuoles (Figure 7C). At 10 and 20 days of age, the average number of vacuoles in spartin nulls was significantly increased by 3.6-fold and 5.1-fold, respectively, compared with controls (Figure 7D). The brains of 30- and 40-day-old spartin flies exhibited even more vacuolization, while age-matched controls were well preserved. To characterize neurodegeneration, apoptotic cell death was assessed in 20-day old brains. While no TUNEL- or caspase-3-positive cells were present in control brains, spartin nulls strongly labeled for both TUNEL and caspase-3 colocalized with neuronal marker anti-Elav but not with glial marker anti-Repo (Figures 7F). Neuronal expression of Drosophila or human Spartin in spartin nulls prevented brain vacuolization and apoptosis at 20 days of age (Figures 7E and 7G).
Spartin/BMP Control of Microtubule Stability Critical for Neuron Viability
To explore the Spartin mechanism preventing neurodegeneration, we assayed phenotypes of spartin mutants in BMP signaling and microtubule stability. We found that the levels of P-Mad, Futsch and acetylated α-tubulin were elevated ~54–137% in spartin brains compared to wildtype (Figures S5A-S5C), suggesting Spartin acts as an inhibitor of BMP signaling. Importantly, most TUNEL- or caspase-3-positive cells in spartin mutant brains were strongly labeled for P-Mad (Figure S5A). To test the hypothesis that abnormal elevation of BMP signaling is responsible for spartin-induced neurodegeneration, we first investigated whether genetic upregulation of BMP signaling itself enhances neurodegeneration. We overexpressed UAS-tkvCA or UAS-gbb only in adult neurons using elav-GeneSwitch-GAL4 (Gatto and Broadie, 2009; Osterwalder et al., 2001), and observed strongly induced brain vacuolization at 20 day of age (Figure 8A). Double staining of UAS-tkvCA or -gbb brains with TUNEL and anti-Elav revealed that elevated BMP signaling colocalized with neuronal cell death (Figures 8B and 8C). Indeed, cell death occurred only in neurons with elevated levels of P-Mad (Figure 8D), supporting a cell-autonomous role for elevated BMP signaling in neurodegeneration. We then examined whether genetic manipulations to downregulate BMP signaling could rescue spartin-induced neurodegeneration. Removing one copy of wit, which had no effect on brain vacuolization in a wildtype background, strongly suppressed brain vacuolization in the spartin null (Figure 8E). Moreover, reduction of BMP signaling by neuronal overexpression of Dad also caused complete rescue (Figure 8E). These data suggest that elevated BMP signaling is responsible for spartin-induced neurodegeneration.
Figure 8. Spartin Regulates Neuronal Survival by Modulating BMP Signaling and Microtubule Stability.

(A–D) Genetic upregulation of BMP signaling or Futsch causes neuronal cell death in the adult brain. (A) Quantification of vacuoles in 20-day-old brains of the indicated genotypes. (B) Confocal slices of 20-day-old brains stained with TUNEL and anti-Elav are shown for elav-GS-GAL4/UAS-tkvCA (Tkv OE), elav-GS-GAL4/UAS-gbb (Gbb OE), and C155-GAL4/futschEP1419 (Futsch OE). Scale bar, 10 μm. (C) Quantification of TUNEL-positive cells observed in 3 consecutive, middle frontal sections (10 μm thick) of 20-day-old brains from the indicated genotypes. (D) Confocal slices of 20-day-old brains labeled with TUNEL and anti-P-Mad. Scale bar, 10 μm.
(E–G) Quantification of vacuoles in 20-day-old brains of the indicated genotypes. (E and F) spartin genetically interacts with BMP pathway components (E) or futsch (F). (G) Administration of vinblastine (1 μM) ameliorates brain vacuolization caused by spartin mutations, genetic elevation of BMP signaling, or Futsch neuronal overexpression.
(H) Model for Spartin/BMP signaling-dependent regulation of synaptic growth and neuronal cell survival.
In (A), (C), and (G), flies carrying both elav-GS-GAL4 and either UAS-tkvCA or UAS-gbb were fed with the progesterone analog RU486 starting right after eclosion. The sample size was n = 10 animals for each genotype. All comparisons are with wildtype or elav-GS-GAL4 control unless indicated (*p < 0.001; n.s., not significant).
See also Figure S5.
We also investigated whether the observed upregulation of Futsch levels is responsible for the neurodegeneration in the spartin mutants. To test this hypothesis, we examined genetic interactions between spartin and futsch in the adult brain. We observed strong neurodegeneration caused by overexpressing Futsch only in neurons (Figures 8A-8C). Notably, animals heterozygous for hypomorphic futschK68 also show neurodegeneration (Figure 8F), suggesting that a precise level of Futsch activity is required for neuronal maintenance. We found heterozygosity for futschK68 rescued spartin neurodegeneration, while neuronal overexpression of Futsch in spartin caused a strongly enhanced phenotype (Figure 8F). Furthermore, the Futsch overexpression phenotype was slightly, but significantly, suppressed by Spartin overexpression (Figure 8F). These genetic data are consistent with the model in which Spartin acts to maintain neuronal cell survival by regulating Futsch levels.
Finally, we tested whether increased microtubule stability is responsible for the neurodegeneration in spartin mutants or caused by elevated BMP signaling and Futsch levels. We found that feeding adult flies with 1 μM vinblastine, which had no effect on brain anatomy, significantly ameliorated neurodegeneration caused by spartin mutation or neuronal overexpression of Gbb and TkvCA (Figure 8G). Vinblastine treatment also partially, but significantly, suppressed brain vacuolization in animals overexpressing Futsch, while it caused a significantly enhanced phenotype in animals heterozygous for futschK68 (Figure 8G). Taken together, these data indicate that Spartin/BMP signaling-dependent regulation of Futsch and microtubule stability is critical for maintaining neuronal cell survival in the aging brain.
DISCUSSION
Troyer syndrome is caused by loss-of-function spartin mutations, but the mechanistic bases of the progressive ataxia and neurodegeneration are largely unknown. Here, we generate a Drosophila disease model to investigate in vivo Spartin functions and uncover the pathogenic mechanisms of Troyer syndrome. We provide evidence that Spartin inhibits neuronal BMP signaling by regulating the endocytic internalization and subsequent endosomal trafficking of the type II BMP receptor Wit. BMP signaling in turn regulates the dFMRP translational regulator controlling Futsch expression to modulate neuronal microtubule stability, which controls both synaptogenesis and neuronal survival. This study is the first demonstration of Spartin function in the nervous system, and suggests a mechanistic basis linking Fragile X and Troyer syndromes.
Our studies provide compelling evidence that Spartin acts with Eps15 to regulate synaptic vesicle endocytosis and structural growth. First, spartin null NMJs exhibit overgrowth with excessive satellite bouton formation and impairment of FM1-43FX dye uptake, similar to defects in eps15 mutants (Koh et al., 2007), suggesting a functional relationship between Spartin and Eps15. Second, Spartin interacts physically with Eps15. Third, Spartin and Eps15 strongly colocalize in the presynaptic terminal. Fourth, spartin and eps15 show transheterozygous interactions during synaptic growth and FM1-43FX uptake, indicating function in the same pathway. Finally, the Eps15-binding domain in Spartin is also essential for its synaptic localization and function. Thus, Spartin is involved in both synaptic growth and vesicle endocytosis regulation through an essential interaction with Eps15.
Is there a relationship between Spartin roles in synaptic growth and endocytosis? Previous Drosophila NMJ studies have shown that abnormal elevation of BMP signaling, via mutations impairing BMP receptor internalization, causes synaptic overgrowth with supernumerary satellite bouton formation (O’Connor-Giles et al., 2008; Wang et al., 2007). Since satellite bouton formation is similarly excessive in spartin mutants, it was attractive to hypothesize that Spartin restrains synaptic growth by promoting endocytic downregulation of surface BMP receptors. Consistently, presynaptic P-Mad levels are elevated in spartin mutants, and genetic interaction experiments show that synaptic overgrowth in spartin mutants is dependent on BMP signaling. Most directly, we demonstrate that Spartin promotes internalization of type II BMP receptor Wit, and subsequent sorting to lysosomes. Similarly, mammalian Spartin regulates internalization and degradative endosomal sorting of EGFR (Bakowska et al., 2007). Our findings thus reveal that Spartin function in receptor-mediated endocytosis is conserved between Drosophila and mammals.
Null spartin mutants exhibit reduced neurotransmitter release. This finding was surprising, given that disruption of BMP signaling also impairs neurotransmitter release (Aberle et al., 2002; Marques et al., 2002; McCabe et al., 2004; McCabe et al., 2003; Rawson et al., 2003). One possible explanation is that chronic impairment in synaptic vesicle endocytosis and/or disruption of proteins required for vesicle release may impair neurotransmitter release, overwhelming the effect of elevated BMP signaling. In support of this idea, neurotransmitter release is also impaired by loss of Dap160 (Marie et al., 2004), another endocytic molecule that functions with Eps15 at the NMJ (Koh et al., 2007). Another possibility is that Spartin-mediated regulation of BMP signaling plays a prime role in regulating synaptic growth, but not synaptic function. This idea may be supported by our observation that neuronal overexpression of Spartin decreases synaptic structural growth, but has no effect on neurotransmitter release. A similarly separable role in synaptic growth versus function has been described for the Nemo (Nmo) kinase, another negative regulator of BMP signaling (Merino et al., 2009).
Drosophila NMJ studies have revealed a direct link between microtubule stability and synaptic growth. Loss of microtubule-severing Spastin, whose human ortholog is mutated in ~40% of autosomal dominant HSP patients, causes accumulation of stable microtubules and increase in overall and satellite bouton numbers (Orso et al., 2005; Sherwood et al., 2004; Trotta et al., 2004). In contrast, loss of Futsch/MAP1B disrupts synaptic microtubule stability and reduces bouton number (Roos et al., 2000). Since synaptic levels of Futsch are regulated by BMP signaling (Ellis et al., 2010), an interesting question was whether Futsch and the microtubule cytoskeleton are major targets for BMP signaling in regulating synaptic growth. Here, we show that synaptic growth defects caused by elevated BMP signaling are completely suppressed by futsch mutations, or treatment with the microtubule-destabilizing agent vinblastine. Based on these results, we propose that Futsch-dependent regulation of microtubule stability is a major target for BMP signaling in the regulation of synaptic growth (Figure 8H).
BMP signaling regulation of MAPs has also been suggested in mammalian systems. BMP2, BMP6 and BMP7 promote dendrite formation by increasing microtubule-associated protein-2 (MAP2) expression in cultured mammalian neurons (Guo et al., 1998; Withers et al., 2000). Moreover, BMP7 induces microtubule stabilization by activating Jun N-terminal kinase (JNK) (Podkowa et al., 2010), which in turn regulates microtubule stability through MAP2 phosphorylation (Bjorkblom et al., 2005). Here, we show that Drosophila FMRP, a known translational repressor of futsch mRNA (Zhang et al., 2001), acts downstream of Spartin and BMP signaling to modulate Futsch/MAP1B levels. Null dfmr1 with spartin (or dad) show transheterozygous interactions at the NMJ, showing that they act in the same pathway. Furthermore, loss of Spartin or genetic elevation of BMP signaling reduces the levels of dfmr1 transcript, whereas disruption of BMP signaling by wit mutations has the opposite effect of elevating dfmr1 expression. Taken together, these data strongly indicate that dFMRP regulates Futsch levels downstream of Spartin function and BMP signaling. Nevertheless, it remains to be determined whether BMP signaling reduces dfmr1 expression by directly repressing the dfmr1 promotor, or exerts its effect by activating transcription of other target genes, which negatively control stability and/or processing of the dfmr1 transcript (Figure 8H).
Increasing evidence suggests a connection between elevated BMP signaling and the HSP disease state. Spichthyin (Spict), Drosophila ortholog of HSP protein NIPA1, inhibits signaling by regulating BMP receptor trafficking (Wang et al., 2007). Subsequent studies in mammals confirm that NIPA1 inhibits BMP signaling by promoting endocytosis and lysosomal degradation of BMP receptors (Tsang et al., 2009). Other HSP proteins, including Atlastin, Spastin and Spartin, have similarly act as BMP inhibitors in mammalian and zebrafish systems (Fassier et al., 2010; Tsang et al., 2009), suggesting that elevated BMP signaling is a common pathological defect in at least several HSPs. Importantly, our findings support a causative role for elevated BMP signaling in spartin-induced neurodegeneration. First, we found progressive, age-dependent neurodegeneration in spartin null adult brains. Second, elevation of BMP signaling achieved by expressing either Gbb or constitutively active Tkv, similarly induces neuronal cell death. Third, neurodegeneration in spartin mutants is ameliorated by overexpressing the BMP inhibitor Dad specifically in neurons. These data suggest that neurodegeneration in spartin mutants is largely due to elevated BMP signaling.
A final point of interest is the role of dysregulated microtubule stability in the neurodegeneration caused by Spartin loss and elevated BMP signaling. We show that neurodegeneration in spartin mutants is strongly suppressed by heterozygosity of futsch, which can itself induce neurodegeneration. In addition, feeding adults with the microtubule-destabilizing drug vinblastine rescues neurodegeneration caused by spartin mutations and elevated BMP signaling. These results indicate that Spartin/BMP-mediated regulation of microtubule stability via Futsch is critical for the maintenance of the nervous system. Defects in microtubule stability and other microtubule-dependent processes (e.g., axonal transport) have been proposed as pathogenic mechanisms of many neurodegenerative diseases, including HSPs (Falnikar and Baas, 2009). However, despite elevated microtubule stability in spartin mutants, axonal transport appears normal. Similarly, axonal transport is not impaired by loss of Spict, which also negatively regulates BMP-dependent microtubule stability (Wang et al., 2007). Thus, Spartin/BMP-mediated regulation of neuronal microtubule stability, not related to transport per se, appears to be the determinant of neuronal viability. It will be interesting in the future to investigate how this microtubule dysregulation induces progressive neurodegeneration in the adult CNS.
EXPERIMENTAL PROCEDURES
Fly stocks
The w1118 strain was used as the wildtype control. An EP insertion in the spartin locus (G8635) was obtained from GenExel (Republic of Korea) and imprecisely excised to produce spartin1. For description of other strains and culture conditions, see Supplemental Experimental Procedures.
Molecular Biology
For detection of surface and internalized Wit receptor in S2R+ cells, the Myc epitope was introduced into full-length wit cDNA via PCR-based mutagenesis. The tag was inserted in-frame, immediately downstream of the putative signal peptide sequence of Wit, and a Myc-wit fragment was subcloned into the pAc5.1 (Invitrogen) vector to generate pAc-Myc-wit. To determine the subcellular distribution of internalized Wit receptor, full-length spinster (spin) cDNA generated from Drosophila mRNA by RT-PCR was subcloned into the pAc-EGFP vector to produce pAc-spin-EGFP.
Details on generation of other plasmids as well as analysis of spartin and dfmr1 transcripts by reverse-transcription PCR and quantitative real-time PCR appear in Supplemental Experimental Procedures.
Cell Transfection, Double-Stranded RNA Interference, and Quantification of Surface Wit Receptor
Transient transfection of plasmid DNA, double-stranded RNA interference, and quantification of surface Wit receptor in S2R+ cells were performed as detailed in Supplemental Experimental Procedures.
Generation of Anti-Spartin Antibody and Binding Experiments
A rat polyclonal antibody against Spartin was generated against a purified GST fusion containing amino acids 102–292 of Spartin. Antisera were affinity purified using the same recombinant protein cross-linked CNBr-activated Sepharose 4 Fast Flow beads (GE Healthcare).
GST pull-down assays and coimmunoprecipitation experiments were performed as described in Supplemental Experimental Procedures.
Immunohistochemistry and Imaging of Larval NMJs
Wandering third-instar larvae were dissected in Ca2+-free HL3 saline and fixed in PBS containing 4% formaldehyde for 30 min as previously described (Nahm et al., 2010a). For additional details on immunostaining and imaging of NMJ synapses, see Supplemental Experimental Procedures.
FM1-43FX Uptake Assays and Electrophysiology
FM1-43FX dye uptake experiments were carried out as described previously (Verstreken et al., 2008). Briefly, wandering third instar larvae were dissected in Ca2+-free HL3 saline. The preparation was washed with fresh HL3 saline and then incubated in HL3 saline containing 90 mM KCl, 5 mM CaCl2, and 4 μM FM1-43FX (Molecular Probes). After dye loading, the samples were washed for 10 min in Ca2+-free HL3 saline and then fixed in 4% formaldehyde in PBS for 30 min, followed by washing with PBS three times. Images were acquired using an FV300 laser-scanning confocal microscope (Olympus) using a Plan Apo 60x 1.4 NA Oil objective. The fluorescence intensity was measured using the FLOUVIEW software (Olympus).
Electrophysiological recordings were carried out as described previously (Rohrbough et al., 1999). See Supplemental Experimental Procedures for details.
Histology, Immunostaining, and TUNEL Staining of Adult Brains
Paraffin sectioning and H&E staining of adult brains were performed following standard protocols. Briefly, adult heads were removed and fixed overnight in 4% formaldehyde in PBS. Paraffin-embedded brains were subjected to serial 5-μm sectioning on a RM2255 microtome (Leica) in a frontal orientation and stained with H&E. To quantify brain vacuolization, the number of vacuoles with diameters > 5 μm was counted for all sections spanning the entire brain.
Apoptotic cell death was assessed by TUNEL staining on cryosections of adult heads. See Supplemental Experimental Procedures for details.
Adult Behavioral Analysis
Adult flies aged for 20 days were tested for locomotor ability in an adult climbing assay (Ellis et al., 2010). For each genotype, 100 individual flies were collected and placed into an empty glass cylinder with a line drawn 2 cm from the bottom. After acclimate to its environment for 5 min, flies were gently vortexed. The number of flies that climbed above the 2-cm mark after 30 sec was counted.
Statistical Analysis
Data are presented as mean ± SEM. The numbers of samples analyzed are indicated inside the bars or in the figure legends. To determine statistical significance, we applied one-way ANOVA followed by post hoc pair-wise comparisons of means using Tukey-Kramer test.
Supplementary Material
Table S1. Quantification of NMJ parameters for experiments in Figure 3B.
Table S2. Quantification of NMJ parameters for experiments in Figure S2D.
Table S3. Quantification of NMJ parameters for experiments in Figure S3.
Table S4. Quantification of NMJ parameters for experiments in Figure 5B.
Table S5. Quantification of NMJ parameters for experiments in Figure S4I.
Table S6. Quantification of NMJ parameters for experiments in Figure 6.
Research Highlights.
HSP gene spartin mutations cause synapse overgrowth and brain neurodegeneration
Spartin inhibits BMP signaling by endocytic degradation of the BMP receptor Wit
FMRP and Futsch/MAP1B are downstream effectors of Spartin and BMP signaling
Restoration of microtubule stability rescues spartin-induced neuronal phenotypes
Acknowledgments
We thank T. Adachi-Yamada, H. Bellen, M. Gonzalez-Gaitan, T. Tabata, K. Wharton, and the Bloomington Stock Center for reagents and fly stocks; S. Choi and F. Yu for helpful discussion. This work was supported by the National Research Foundation of Korea (Nos. 2011-0019228, 2010-0029398, and 2009-0071481) and the Brain Research Center of the 21st Century Frontier Research Program (No. 2011K000281) grants funded by the Ministry of Education, Science and Technology, the Republic of Korea, and NIH R01 grant MH096832 to K.B.
Footnotes
Supplemental Information includes five figures, six tables, and Supplemental Experimental Procedures.
References
- Aberle H, Haghighi AP, Fetter RD, McCabe BD, Magalhaes TR, Goodman CS. wishful thinking encodes a BMP type II receptor that regulates synaptic growth in Drosophila. Neuron. 2002;33:545–558. doi: 10.1016/s0896-6273(02)00589-5. [DOI] [PubMed] [Google Scholar]
- Bakowska JC, Jenkins R, Pendleton J, Blackstone C. The Troyer syndrome (SPG20) protein spartin interacts with Eps15. Biochem Biophys Res Commun. 2005;334:1042–1048. doi: 10.1016/j.bbrc.2005.06.201. [DOI] [PubMed] [Google Scholar]
- Bakowska JC, Jupille H, Fatheddin P, Puertollano R, Blackstone C. Troyer syndrome protein spartin is mono-ubiquitinated and functions in EGF receptor trafficking. Mol Biol Cell. 2007;18:1683–1692. doi: 10.1091/mbc.E06-09-0833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayat V, Jaiswal M, Bellen HJ. The BMP signaling pathway at the Drosophila neuromuscular junction and its links to neurodegenerative diseases. Curr Opin Neurobiol. 2011;21:182–188. doi: 10.1016/j.conb.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettencourt da Cruz A, Schwarzel M, Schulze S, Niyyati M, Heisenberg M, Kretzschmar D. Disruption of the MAP1B-related protein FUTSCH leads to changes in the neuronal cytoskeleton, axonal transport defects, and progressive neurodegeneration in Drosophila. Mol Biol Cell. 2005;16:2433–2442. doi: 10.1091/mbc.E04-11-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkblom B, Ostman N, Hongisto V, Komarovski V, Filen JJ, Nyman TA, Kallunki T, Courtney MJ, Coffey ET. Constitutively active cytoplasmic c-Jun N-terminal kinase 1 is a dominant regulator of dendritic architecture: role of microtubule-associated protein 2 as an effector. J Neurosci. 2005;25:6350–6361. doi: 10.1523/JNEUROSCI.1517-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackstone C, O’Kane CJ, Reid E. Hereditary spastic paraplegias: membrane traffic and the motor pathway. Nat Rev Neurosci. 2011;12:31–42. doi: 10.1038/nrn2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccarelli FD, Proukakis C, Patel H, Cross H, Azam S, Patton MA, Bork P, Crosby AH. The identification of a conserved domain in both spartin and spastin, mutated in hereditary spastic paraplegia. Genomics. 2003;81:437–441. doi: 10.1016/s0888-7543(03)00011-9. [DOI] [PubMed] [Google Scholar]
- Coffee RL, Jr, Tessier CR, Woodruff EA, 3rd, Broadie K. Fragile X mental retardation protein has a unique, evolutionarily conserved neuronal function not shared with FXR1P or FXR2P. Dis Model Mech. 2010;3:471–485. doi: 10.1242/dmm.004598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross HE, McKusick VA. The Troyer syndrome. A recessive form of spastic paraplegia with distal muscle wasting. Arch Neurol. 1967;16:473–485. doi: 10.1001/archneur.1967.00470230025003. [DOI] [PubMed] [Google Scholar]
- Eastman SW, Yassaee M, Bieniasz PD. A role for ubiquitin ligases and Spartin/SPG20 in lipid droplet turnover. J Cell Biol. 2009;184:881–894. doi: 10.1083/jcb.200808041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards TL, Clowes VE, Tsang HT, Connell JW, Sanderson CM, Luzio JP, Reid E. Endogenous spartin (SPG20) is recruited to endosomes and lipid droplets and interacts with the ubiquitin E3 ligases AIP4 and AIP5. Biochem J. 2009;423:31– 39. doi: 10.1042/BJ20082398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis JE, Parker L, Cho J, Arora K. Activin signaling functions upstream of Gbb to regulate synaptic growth at the Drosophila neuromuscular junction. Dev Biol. 2010;342:121–133. doi: 10.1016/j.ydbio.2010.03.012. [DOI] [PubMed] [Google Scholar]
- Falnikar A, Baas PW. Critical roles for microtubules in axonal development and disease. Results Probl Cell Differ. 2009;48:47–64. doi: 10.1007/400_2009_2. [DOI] [PubMed] [Google Scholar]
- Fassier C, Hutt JA, Scholpp S, Lumsden A, Giros B, Nothias F, Schneider-Maunoury S, Houart C, Hazan J. Zebrafish atlastin controls motility and spinal motor axon architecture via inhibition of the BMP pathway. Nat Neurosci. 2010;13:1380–1387. doi: 10.1038/nn.2662. [DOI] [PubMed] [Google Scholar]
- Gatto CL, Broadie K. Temporal requirements of the fragile x mental retardation protein in modulating circadian clock circuit synaptic architecture. Front Neural Circuits. 2009;3:8. doi: 10.3389/neuro.04.008.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Rueger D, Higgins D. Osteogenic protein-1 and related bone morphogenetic proteins regulate dendritic growth and the expression of microtubule-associated protein-2 in rat sympathetic neurons. Neurosci Lett. 1998;245:131–134. doi: 10.1016/s0304-3940(98)00192-x. [DOI] [PubMed] [Google Scholar]
- Hooper C, Puttamadappa SS, Loring Z, Shekhtman A, Bakowska JC. Spartin activates atrophin-1-interacting protein 4 (AIP4) E3 ubiquitin ligase and promotes ubiquitination of adipophilin on lipid droplets. BMC Biol. 2010;8:72. doi: 10.1186/1741-7007-8-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan MA, Thrower D, Wilson L. Effects of vinblastine, podophyllotoxin and nocodazole on mitotic spindles. Implications for the role of microtubule dynamics in mitosis. J Cell Sci. 1992;102(Pt 3):401–416. doi: 10.1242/jcs.102.3.401. [DOI] [PubMed] [Google Scholar]
- Joshi DC, Bakowska JC. SPG20 protein spartin associates with cardiolipin via its plant-related senescence domain and regulates mitochondrial Ca2+ homeostasis. PLoS One. 2011;6:e19290. doi: 10.1371/journal.pone.0019290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Wairkar YP, Daniels RW, DiAntonio A. The novel endosomal membrane protein Ema interacts with the class C Vps-HOPS complex to promote endosomal maturation. J Cell Biol. 2010;188:717–734. doi: 10.1083/jcb.200911126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh TW, Korolchuk VI, Wairkar YP, Jiao W, Evergren E, Pan H, Zhou Y, Venken KJ, Shupliakov O, Robinson IM, et al. Eps15 and Dap160 control synaptic vesicle membrane retrieval and synapse development. J Cell Biol. 2007;178:309–322. doi: 10.1083/jcb.200701030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzini MC, Rajab A, Maynard TM, Mochida GH, Tan WH, Nasir R, Hill RS, Gleason D, Al Saffar M, Partlow JN, et al. Developmental and degenerative features in a complicated spastic paraplegia. Ann Neurol. 2010;67:516–525. doi: 10.1002/ana.21923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie B, Sweeney ST, Poskanzer KE, Roos J, Kelly RB, Davis GW. Dap160/intersectin scaffolds the periactive zone to achieve high-fidelity endocytosis and normal synaptic growth. Neuron. 2004;43:207–219. doi: 10.1016/j.neuron.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Marques G, Bao H, Haerry TE, Shimell MJ, Duchek P, Zhang B, O’Connor MB. The Drosophila BMP type II receptor Wishful Thinking regulates neuromuscular synapse morphology and function. Neuron. 2002;33:529–543. doi: 10.1016/s0896-6273(02)00595-0. [DOI] [PubMed] [Google Scholar]
- McCabe BD, Hom S, Aberle H, Fetter RD, Marques G, Haerry TE, Wan H, O’Connor MB, Goodman CS, Haghighi AP. Highwire regulates presynaptic BMP signaling essential for synaptic growth. Neuron. 2004;41:891–905. doi: 10.1016/s0896-6273(04)00073-x. [DOI] [PubMed] [Google Scholar]
- McCabe BD, Marques G, Haghighi AP, Fetter RD, Crotty ML, Haerry TE, Goodman CS, O’Connor MB. The BMP homolog Gbb provides a retrograde signal that regulates synaptic growth at the Drosophila neuromuscular junction. Neuron. 2003;39:241–254. doi: 10.1016/s0896-6273(03)00426-4. [DOI] [PubMed] [Google Scholar]
- Merino C, Penney J, Gonzalez M, Tsurudome K, Moujahidine M, O’Connor MB, Verheyen EM, Haghighi P. Nemo kinase interacts with Mad to coordinate synaptic growth at the Drosophila neuromuscular junction. J Cell Biol. 2009;185:713–725. doi: 10.1083/jcb.200809127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muqit MM, Feany MB. Modelling neurodegenerative diseases in Drosophila: a fruitful approach? Nat Rev Neurosci. 2002;3:237–243. doi: 10.1038/nrn751. [DOI] [PubMed] [Google Scholar]
- Nahm M, Kim S, Paik SK, Lee M, Lee S, Lee ZH, Kim J, Lee D, Bae YC, Lee S. dCIP4 (Drosophila Cdc42-interacting protein 4) restrains synaptic growth by inhibiting the secretion of the retrograde Glass bottom boat signal. J Neurosci. 2010a;30:8138–8150. doi: 10.1523/JNEUROSCI.0256-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahm M, Long AA, Paik SK, Kim S, Bae YC, Broadie K, Lee S. The Cdc42-selective GAP rich regulates postsynaptic development and retrograde BMP transsynaptic signaling. J Cell Biol. 2010b;191:661–675. doi: 10.1083/jcb.201007086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor-Giles KM, Ho LL, Ganetzky B. Nervous wreck interacts with thickveins and the endocytic machinery to attenuate retrograde BMP signaling during synaptic growth. Neuron. 2008;58:507–518. doi: 10.1016/j.neuron.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orso G, Martinuzzi A, Rossetto MG, Sartori E, Feany M, Daga A. Disease-related phenotypes in a Drosophila model of hereditary spastic paraplegia are ameliorated by treatment with vinblastine. J Clin Invest. 2005;115:3026–3034. doi: 10.1172/JCI24694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterwalder T, Yoon KS, White BH, Keshishian H. A conditional tissue-specific transgene expression system using inducible GAL4. Proc Natl Acad Sci USA. 2001;98:12596–12601. doi: 10.1073/pnas.221303298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel H, Cross H, Proukakis C, Hershberger R, Bork P, Ciccarelli FD, Patton MA, McKusick VA, Crosby AH. SPG20 is mutated in Troyer syndrome, an hereditary spastic paraplegia. Nat Genet. 2002;31:347–348. doi: 10.1038/ng937. [DOI] [PubMed] [Google Scholar]
- Podkowa M, Zhao X, Chow CW, Coffey ET, Davis RJ, Attisano L. Microtubule stabilization by bone morphogenetic protein receptor-mediated scaffolding of c-Jun N-terminal kinase promotes dendrite formation. Mol Cell Biol. 2010;30:2241–2250. doi: 10.1128/MCB.01166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawson JM, Lee M, Kennedy EL, Selleck SB. Drosophila neuromuscular synapse assembly and function require the TGF-beta type I receptor saxophone and the transcription factor Mad. J Neurobiol. 2003;55:134–150. doi: 10.1002/neu.10189. [DOI] [PubMed] [Google Scholar]
- Regehr WG, Carey MR, Best AR. Activity-dependent regulation of synapses by retrograde messengers. Neuron. 2009;63:154–170. doi: 10.1016/j.neuron.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renvoise B, Parker RL, Yang D, Bakowska JC, Hurley JH, Blackstone C. SPG20 protein spartin is recruited to midbodies by ESCRT-III protein Ist1 and participates in cytokinesis. Mol Biol Cell. 2010;21:3293–3303. doi: 10.1091/mbc.E09-10-0879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrbough J, Pinto S, Mihalek RM, Tully T, Broadie K. latheo, a Drosophila gene involved in learning, regulates functional synaptic plasticity. Neuron. 1999;23:55–70. doi: 10.1016/s0896-6273(00)80753-9. [DOI] [PubMed] [Google Scholar]
- Roos J, Hummel T, Ng N, Klambt C, Davis GW. Drosophila Futsch regulates synaptic microtubule organization and is necessary for synaptic growth. Neuron. 2000;26:371–382. doi: 10.1016/s0896-6273(00)81170-8. [DOI] [PubMed] [Google Scholar]
- Sherwood NT, Sun Q, Xue M, Zhang B, Zinn K. Drosophila spastin regulates synaptic microtubule networks and is required for normal motor function. PLoS Biol. 2004;2:e429. doi: 10.1371/journal.pbio.0020429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney ST, Davis GW. Unrestricted synaptic growth in spinster-a late endosomal protein implicated in TGF-beta-mediated synaptic growth regulation. Neuron. 2002;36:403–416. doi: 10.1016/s0896-6273(02)01014-0. [DOI] [PubMed] [Google Scholar]
- Trotta N, Orso G, Rossetto MG, Daga A, Broadie K. The hereditary spastic paraplegia gene, spastin, regulates microtubule stability to modulate synaptic structure and function. Curr Biol. 2004;14:1135–1147. doi: 10.1016/j.cub.2004.06.058. [DOI] [PubMed] [Google Scholar]
- Tsang HT, Edwards TL, Wang X, Connell JW, Davies RJ, Durrington HJ, O’Kane CJ, Luzio JP, Reid E. The hereditary spastic paraplegia proteins NIPA1, spastin and spartin are inhibitors of mammalian BMP signalling. Hum Mol Genet. 2009;18:3805–3821. doi: 10.1093/hmg/ddp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstreken P, Ohyama T, Bellen HJ. FM 1-43 labeling of synaptic vesicle pools at the Drosophila neuromuscular junction. Methods Mol Biol. 2008;440:349–369. doi: 10.1007/978-1-59745-178-9_26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Shaw WR, Tsang HT, Reid E, O’Kane CJ. Drosophila spichthyin inhibits BMP signaling and regulates synaptic growth and axonal microtubules. Nat Neurosci. 2007;10:177–185. doi: 10.1038/nn1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Withers GS, Higgins D, Charette M, Banker G. Bone morphogenetic protein-7 enhances dendritic growth and receptivity to innervation in cultured hippocampal neurons. Eur J Neurosci. 2000;12:106–116. doi: 10.1046/j.1460-9568.2000.00889.x. [DOI] [PubMed] [Google Scholar]
- Zhang YQ, Bailey AM, Matthies HJ, Renden RB, Smith MA, Speese SD, Rubin GM, Broadie K. Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell. 2001;107:591–603. doi: 10.1016/s0092-8674(01)00589-x. [DOI] [PubMed] [Google Scholar]
- Zweifel LS, Kuruvilla R, Ginty DD. Functions and mechanisms of retrograde neurotrophin signalling. Nat Rev Neurosci. 2005;6:615–625. doi: 10.1038/nrn1727. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Quantification of NMJ parameters for experiments in Figure 3B.
Table S2. Quantification of NMJ parameters for experiments in Figure S2D.
Table S3. Quantification of NMJ parameters for experiments in Figure S3.
Table S4. Quantification of NMJ parameters for experiments in Figure 5B.
Table S5. Quantification of NMJ parameters for experiments in Figure S4I.
Table S6. Quantification of NMJ parameters for experiments in Figure 6.