

Abstract
The recent approvals of anticancer therapeutic agents targeting the histone deacetylases and DNA methyltransferases have highlighted the important role that epigenetics plays in human diseases, and suggested that the factors controlling gene expression are novel drug targets. Protein arginine deiminase 4 (PAD4) is one such target because its effects on gene expression parallel those observed for the histone deacetylases. We demonstrated that F- and Cl-amidine, two potent PAD4 inhibitors, display micromolar cytotoxic effects towards several cancerous cell lines (HL-60, MCF7 and HT-29); no effect was observed in noncancerous lines (NIH 3T3 and HL-60 granulocytes). These compounds also induced the differentiation of HL-60 and HT29 cells. Finally, these compounds synergistically potentiated the cell killing effects of doxorubicin. Taken together, these findings suggest PAD4 inhibition as a novel epigenetic approach for the treatment of cancer, and suggest that F- and Cl-amidine are candidate therapeutic agents for this disease.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-010-0480-x) contains supplementary material, which is available to authorized users.
Keywords: Protein arginine deiminase, Haloacetamidine, Inhibition, HL-60, Epigenetics, Citrulline
Introduction
Over the last decade, there has been increased recognition that epigenetics plays an important role in both gene regulation and human disease [1]. Heritable changes in gene expression are thought to arise from at least two phenomena, i.e. histone modifications and DNA methylation. For example, the posttranslational modification of nucleosomal proteins (e.g. histones H3 and H4) contributes to gene regulation via either a direct effect on chromatin structure or through the altered recruitment of additional protein cofactors [2]. These effects ultimately lead to changes in the local chromatin structure surrounding a gene, resulting in increases or decreases in the transcription of that gene [2]. The methylation of CpG islands in DNA is also thought to contribute to gene regulation by altering the binding of transcription factors directly and/or by facilitating the binding of methyl-CpG-binding domain proteins [1]. These latter proteins can then recruit histone-modifying enzymes and chromatin-remodeling enzymes to effect transcriptional repression. In cancer, many gene promoters that control the expression of tumor suppressor proteins are abnormally hypermethylated [1]. This hypermethylation is thought to lead to the recruitment of corepressor complexes that, via the actions of the histone deacetylases (HDACs) and other chromatin-modifying/remodeling enzymes, alter the local structure of chromatin to make it refractory to transcription. Inhibition of these processes has attracted considerable interest, especially with the recent approval of 5-aza-2-deoxycytidine (decitabine) and suberoylanilide hydroxamic acid (SAHA), DNA methyltransferase and HDAC inhibitors, respectively, for the treatment of myelodysplastic syndromes and cutaneous T cell lymphoma [1, 3].
Recently, protein arginine deiminase 4 (PAD4) has emerged as a novel transcriptional corepressor [4–8]. This enzyme is recruited to the promoters of genes regulated by the estrogen receptor, thyroid receptor, and p53, where it catalyzes the posttranslational modification of arginine residues (to form citrulline) in histones H2A, H3, and H4 (Fig. 1). This modification correlates with the decreased expression of a subset of genes regulated by these transcription factors. The fact that PAD4, like the HDACs, is a transcriptional corepressor suggests this enzyme as a target for the development of a novel epigenetic cancer therapy. This argument is further bolstered by the observation that PAD4 is overexpressed in numerous malignant cancers (e.g. breast, colon, bladder, lung and ovarian cancers, metastatic carcinomas, and many others), but not in benign tumors [9, 10].
Fig. 1.

a Reaction catalyzed by PAD enzymes. b Structure of PAD4 inhibitors, F- and Cl-amidine
Under normal circumstances, PAD4 exists as an intracellular protein, but in patients with malignant tumors, PAD4 can be detected in the plasma [8]. Although the mechanism by which PAD4 is released into the extracellular milieu remains unknown, circumstantial evidence of this process does exist. For example, in rheumatoid arthritis, where dysregulated PAD activity is also observed [4, 11], a number of extracellular proteins, including fibrin, are deiminated [12]. With respect to cancer, antithrombin appears to be an extracellular PAD substrate, as evidenced by the fact that serum levels of citrullinated antithrombin are elevated in patients with malignant cancers, coincident with increased expression of serum PAD4 (8). This correlation is noteworthy because, once citrullinated, antithrombin can no longer inhibit thrombin, which would be expected to lead to dysregulated thrombin activity (increased thrombin activity, which is a hallmark of cancer, is thought to contribute to angiogenesis, hyperplasia, and metastasis [13, 14]).
The overexpression of PAD4 within cells may also contribute to tumorigenesis through a variety of mechanisms. For example, higher levels of PAD4 are associated with increased cytokeratin deimination [9]. This finding is significant because citrullinated cytokeratin is resistant to caspase-mediated cleavage, an effect that potentially alters the apoptotic pathway [9]. Additionally, the histone deiminating activity of PAD4 has been shown to downregulate the expression of a number of p53-dependent genes, including p21, PUMA, and GADD45 [5, 6]. It is particularly noteworthy that the identification of a role for PAD4 in the regulation of p53-mediated transcription relied on the use of Cl-amidine (Fig. 1), a highly potent PAD4 inhibitor developed in our laboratory [15]. These studies have also revealed that Cl-amidine induces, in a p53-dependent manner, the expression of the tumor suppressor protein OKL38 (pregnancy induced growth inhibitor) in tumor cell lines (e.g. MCF7 cells) by decreasing PAD4 citrullination of OKL38 promoter-associated nucleosomes [5]. Similar results have been obtained by PAD4 siRNA knock-down [5]. Significantly, Cl-amidine treatment and siRNA knock-down of PAD4 decreased cell viability and induced apoptosis in a subset of cells [5].
The observation that Cl-amidine decreases cell viability prompted us to further investigate this phenomenon. We report here that Cl-amidine, and the related PAD4 inhibitor, F-amidine, display low micromolar cytotoxicity towards a number of tumor-derived cell lines; this cytotoxicity, however, was not observed in normal cell lines. Furthermore, both compounds induced the differentiation of HL-60 cells, a leukemic cell line, as demonstrated by decreased expression of myeloperoxidase and increased expression of PAD4, CD38, and p21. We also found that F- and Cl-amidine synergistically potentiate the cell-killing effects of the anticancer drug doxorubicin. The fact that inhibition of the HDACs also leads to cellular differentiation, and potentiates the cytotoxic effects of standard chemotherapeutics, strongly suggests that PAD4 inhibition, like HDAC inhibition [3], represents a novel epigenetic approach to the treatment of cancer.
Materials and methods
Chemicals
F-Amidine and Cl-amidine were synthesized as previously described [15–17]. All-trans retinoic acid (ATRA) and tetradecanoyl phorbol acetate were obtained from Sigma. SAHA was obtained from ChemieTek. Cell culture media (DMEM, RPMI 1640, and fetal bovine serum) were obtained from ThermoScientific. ATRA, and F- and Cl-amidine were dissolved in phosphate buffered saline (PBS).
Cell lines and cell culture
HL-60 human promyelocytic leukemia cells were cultured in RPMI 1640. MCF7, NIH-3T3 and HT-29 cells were cultured in DMEM. In both cases the medium was supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin and the cells were grown in an incubator at 37°C under an atmosphere containing 5% CO2.
Cytotoxicity
Cell viability was determined using the CellTiter 96 nonradioactive cell proliferation assay (Promega). Each condition was evaluated in triplicate. A 100% killing control using 1% Triton was also included. When possible, EC50 values were determined by fitting the dose response data to Eq. 1,
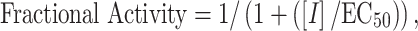 |
1 |
using GraFit (version 5.0.11) [18], where [I] is the concentration of inhibitor (e.g. doxorubicin) and EC50 is the concentration of inhibitor that yields half-maximal cell survival. Detailed methods are described in the Supplementary material.
Western blotting
Membranes were probed with a polyclonal anti-PAD4 antibody (Abcam ab38772), a monoclonal anti-MYO antibody (Abcam, ab45977), a monoclonal anti-p21 antibody (Sigma, p1484), a monoclonal alkaline phosphatase antibody (Abcam, ab54778), or a polyclonal anti-actin antibody (Abcam, ab1801). The methods are described in detail in the Supplementary material.
F- and Cl-amidine decrease in vivo PAD4 activity
MCF7 cells (about 5 × 105) were added to each well of a 12-well plate in phenol red-free DMEM containing 10% charcoal-stripped fetal bovine serum. Cells were incubated in the plate at 37°C in an atmosphere containing 5% CO2. After 48 h the medium was removed and replaced with Locke’s solution (0.15 M NaCl, 5 mM KCl, 5 mM HEPES, 2 mM CaCl2, 0.1% glucose, pH 7.3). F- or Cl-amidine (100 μM) were added to the cells and incubated for 15 min before addition of estrogen (0.1 μM). HL-60 cells (1×106 ml/cell) were treated with ATRA (1 μM final concentration) for 48 h at 37°C, 5% CO2. Cells were split into 12 well plates and treated with 2 mM CaCl2 and either F or Cl-amidine (100 μM). After 15 min at 37°C in an atmosphere containing 5% CO2, the calcium ionophore A23187 (4 μM) was added. In both cases, cells were harvested after 15 min treatment with the stimuli, rinsed with cold PBS and lysed with SDS lysis buffer (2% SDS, 62.5 mM Tris, pH 6.8, 10% glycerol). Proteins were separated by SDS-PAGE and transferred to a nitrocellulose membrane for Western blot analysis. Membranes were blocked with 5% nonfat dried milk in TBST for 1 h at room temperature, and subsequently probed with polyclonal anti-citrulline H3 antibody (Abcam, ab5103) or polyclonal anti-actin (Abcam, ab1801).
Labeling of live cells
HL-60 cells (about 5 × 105) were added to each well of a 12-well plate and treated for 48 h with ATRA (1 μM), Cl-amidine (100 nM), F-amidine (100 nM), or PBS. The cells were harvested by centrifugation and then resuspended in fresh medium (50 μl). Rh-6-(F-araNAD) was then added to a final concentration of 5 μM. After incubation at room temperature for 5 min, the cells were washed with cold medium (1 ml, three times) and then resuspended in cold medium (50 μl). Cells (15 μl) were applied to a microscope slide and confocal images were acquired with a Zeiss LSM 510 META confocal microscope system (we acknowledge the help of Dr. John Fuseler of the Instrumentation Resource Facility at University of South Carolina School of Medicine).
Myeloperoxidase activity
Myeloperoxidase activity was assayed as previously described [19] on crude cell extracts prepared from HL-60 cells treated with 1 μM ATRA, 1 μM Cl-amidine, 1 μM F-amidine, or PBS. The methods are described in detail in the Supplementary material.
Quantitative real-time PCR
Subsequent to treatment with ATRA, F-amidine, Cl-amidine, or PBS, the total RNA was extracted from HL-60 cells using the RNeasy Mini Kit (Qiagen). Equal amounts of RNA were used to prepare first-stand cDNA using the Verso cDNA synthesis kit (Thermo Scientific). The PCR was performed in triplicate using the B-R SYBR green SuperMix (Quanta Biosciences). The amplification program consisted of denaturation at 95°C for 3 min, followed by 40 cycles at 95°C for 15 s and 60°C for 45 s on an iCycler quantitative real-time PCR (RT-PCR) system (BioRad). Gene-specific primers for p21, PAD4, and GAPDH have previously been described [6, 20]. The sequences of these primers are provided in the Supplementary material.
Statistics
Experiments were repeated at least three times and the statistical significance of differences between samples were evaluated using Student’s t test, where p < 0.05 was considered significant.
Additive model (O/E ratios)
The effect of the combination of F- and Cl-amidine with doxorubicin on the cytotoxicity of HL-60 cells was assessed using an additive model [21, 22]. The expected value (E) was determined by multiplying together the effect on cell proliferation by each agent alone. The observed value (O) was the actual effect on cell proliferation when the agents were combined.
Results
Effect of F- and Cl-amidine on cell viability
Because we previously demonstrated that Cl-amidine could decrease cell viability at high concentrations (200 μM) [6], we examined the dose dependence of this effect on MCF7 cells, a breast adenocarcinoma cell line, using the MTT assay. Treatment of MCF7 cells with Cl-amidine at doses as low as 500 nM decreased viability by up to 30% after 24 h (Table 1, Fig. 2). Somewhat surprisingly, however, this decrease in cell viability, while dose-dependent, was rather modest at higher concentrations of Cl-amidine: a 200-fold increase in Cl-amidine concentration only yielded an additional 20% decline in cell viability. Similar trends were obtained with F-amidine, a less potent PAD4 inhibitor, but the effect on cell viability was limited to a reduction of about 20%, and remained relatively constant over the entire concentration regime tested (100 nM to 100 μM).
Table 1.
Cytotoxicity of F- and Cl-amidine
| Cell line | Cl-amidine | F-amidine | ||
|---|---|---|---|---|
| Approximate EC50 | Survival (%)a | Approximate EC50 | Survival (%)a | |
| HL-60 | 0.25 | 38 | 0.5 | 79 |
| HL-60 granulocytes | – | 100 | – | 100 |
| MCF7 | 0.05 | 50 | 0.5 | 77 |
| HT-29 | 1 | 59 | 1 | 91 |
| NIH 3T3 | – | 100 | – | 100 |
aBased on cell viability at the maximum agent concentration tested
Fig. 2.

Cytotoxic effects of F- and Cl-amidine. a MCF7 cells were incubated with various concentrations of F- and Cl-amidine over the course of 24 h, and cell viability was determined by a standard MTT assay. Each data point represents the average of three trials (p < 0.05). Data were normalized to controls treated with PBS only. b Cytotoxic effects of Cl-amidine and ATRA on human HL-60 and HL-60 granulocytes. Cell viability was measured, using a standard MTT assay, after a 24-h incubation with the inhibitors. Each data point represents the average of three trials (p < 0.05). Data were normalized to controls treated with PBS only. c Levels of citrullinated histone H3 in MCF7 cells after treatment with F- and Cl-amidine. See “Materials and methods” section for experimental details. d Levels of citrullinated histone H3 in HL-60 granulocytes after treatment with F- and Cl-amidine. See “Materials and methods” section for experimental details
To ascertain whether or not this result was a general phenomenon, the effects of both F- and Cl-amidine on the viability of a number of cancerous (i.e. human promyelocytic leukemia HL-60 and colon adenocarcinoma HT-29 cells) and noncancerous cell lines (i.e. mouse fibroblast NIH-3T3 and HL-60 granulocytes) were examined (Table 1). With respect to the cancerous cell lines, the results of each study mimicked those obtained with the MCF7 cells. For example, nanomolar concentrations of Cl-amidine decreased the viability of HL-60 cells by up to 40% (Table 1, Fig. 2b). As with the MCF7 cells, higher concentrations of Cl-amidine (up to 100 μM) did not further decrease cell viability to zero. Near identical results were obtained with HT-29 cells (Table 1, Fig. S1). Similar trends were observed with F-amidine; however, the effect on cell viability was less profound, and typically limited to a 10% reduction in cell viability (Table 1). Interestingly, F- and Cl-amidine had little to no effect on the viability of HL-60 granulocytes, a differentiated form of HL-60 cells (Table 1, Fig. 2), suggesting that these compounds are not cytotoxic to noncancerous cell lines. Bolstering this possibility is the fact that F- and Cl-amidine do not affect the viability of NIH-3T3 fibroblasts.
To confirm that the effects of F- and Cl-amidine correlate with decreased PAD activity, we examined whether these agents could decrease histone H3 citrullination in both HL-60 and MCF7 cells in response to the calcium ionophore A23187 and estrogen, respectively. Both of these compounds decreased the levels of citrullinated histone H3 (Fig. 2c, d). These results are consistent with previous reports that the F- and Cl-amidine decrease histone citrullination [5, 6, 15, 17, 23], and indicate that the effects on cell viability are due to their ability to inhibit PAD activity.
F- and Cl-amidine mediated differentiation of HL-60 cells
To explain the observation that F- and Cl-amidine did not decrease cell viability completely, we considered the possibility that these two compounds were differentiating the cancerous cell lines into noncancerous ones. At least partially consistent with such a model is the fact that treatment of HL-60 cells with ATRA, which is known to differentiate HL-60 cells into HL-60 granulocytes [24], yielded a similar dose-dependent decrease in cell viability (Fig. 2b). To further examine this phenomenon, we set out to determine if F- and Cl-amidine do indeed trigger the differentiation of HL-60 cells. We chose this cell line for our studies because it is a well-established system with multiple known markers of differentiation. These markers include decreased myeloperoxidase expression and increased expression of CD38, a cell surface glycoprotein that catalyzes the synthesis of cyclic ADP ribose, and p21. In addition to these differentiation markers, we also examined the expression levels of PAD4 because this protein was originally identified as a protein whose expression is elevated upon the differentiation of HL-60 cells [24].
Myeloperoxidase expression and activity
The effects of F- and Cl-amidine on myeloperoxidase expression levels were first examined as a function of time (0–48 h) and dose (100 nM, 1 μM, or 100 μM F- or Cl-amidine). ATRA, a known inducer of differentiation along the granulocytic lineage, served as a positive control. As expected, ATRA treatment resulted in decreased myeloperoxidase expression after 12–48 h (Fig. 3a). Similar results were obtained with both F- and Cl-amidine (Fig. 3a). In addition to changes in expression, changes in myeloperoxidase activity are also associated with the differentiation of HL-60 cells into HL-60 granulocytes; therefore we also examined the effects of F- and Cl-amidine on the activity of this enzyme. ATRA again served as the positive control. Myeloperoxidase activity was monitored over the same 48-h time period using concentrations of F- and Cl-amidine that yielded near maximal effects on both cell viability and the reduction in myeloperoxidase expression (i.e. 1 μM). Treatment of cells with either F- or Cl-amidine led to a significant (p < 0.001, as compared to PBS vehicle controls) reduction in myeloperoxidase activity after 48 h that was comparable to that obtained with an equivalent concentration of ATRA (Fig. 3b). This result is consistent with our hypothesis that these compounds trigger the differentiation of HL-60 cells into HL-60 granulocytes.
Fig. 3.

F- and Cl-amidine induce the differentiation of HL-60 cells. a HL-60 cells were exposed to ATRA, or F-, or Cl-amidine over the course of 48 h with samples taken after 6, 12, 24, and 48 h. ATRA served as the positive control. PBS was used as the vehicle control. Myeloperoxidase and PAD4 protein levels were determined by Western blotting. b Myeloperoxidase activity in HL-60 cells in response to incubation with ATRA, and F- and Cl-amidine after 24 and 48 h. Specific activity is defined as the rate of formation of 1 mol of tetraguaiacol from 1 mol guaiacol and 4 mol of H2O2. Significant decreases in myeloperoxidase activity, as compared to the PBS vehicle control, were observed after 48 h of incubation (*p < 0.05). c Relative PAD4 mRNA levels, as determined by quantitative RT-PCR, in response to treatment with ATRA, and F-, and Cl-amidine for 48 h
PAD4 expression
To further validate these findings, the effects of F- and Cl-amidine on PAD4 expression levels were measured. As shown in Fig. 3a, the F- and Cl-amidine treatments caused a dose- and time-dependent increase in PAD4 expression. For example, at the highest doses of F- and Cl-amidine (e.g. 100 μM), PAD4 expression was apparent after only 6 h of treatment; with lower doses (e.g. 100 nM) increased levels of this enzyme were observed only after 24 or 48 h. Parallel results were obtained with 1 μM ATRA. These results clearly indicate that, like ATRA, the treatment of HL-60 cells with F- and Cl-amidine, leads to increased PAD4 expression. Potential reasons for the increased expression of PAD4 in response to PAD4 inhibition are described below in the Discussion.
Because ATRA has previously been shown to increase PAD4 mRNA levels [24], the effects of ATRA, and F-, and Cl-amidine on the levels of this transcript were determined by quantitative RT-PCR. Consistent with the Western blot data, higher quantities of PAD4 mRNA were observed in cells treated with ATRA, or F-, or Cl-amidine (Fig. 3c). Although it is unclear whether the increased amounts of PAD4 mRNA are due to transcriptional or posttranscriptional effects, the fact that PAD4 is known to act as a transcriptional corepressor [5–8] may suggest that this enzyme has the capacity to inhibit its own expression. Follow-up studies to distinguish between these two possibilities will undoubtedly provide new insights into the regulation of this physiologically important enzyme.
CD38 expression
To further examine the possibility that F- and Cl-amidine induce the differentiation of HL-60 cells, the cell surface levels of CD38 were measured using rhodamine-tagged arabinosyl 2′-fluoro-2′-deoxy NAD (Rh-6-F-araNAD, Fig. 4a), a recently described CD38-specific probe that covalently modifies this enzyme at Glu226 [25] (Fig. 4b). For these experiments, live HL-60 cells were treated with the vehicle control (i.e. PBS), ATRA, or F-, or Cl-amidine for 48 h. The cells were subsequently treated with Rh-6-F-araNAD and imaged by confocal fluorescence microscopy. As expected, the PBS-treated controls were not labeled by Rh-6-F-araNAD, as HL-60 cells do not express CD38 (Fig. 4c). In contrast, cells treated with ATRA, our positive control, were labeled by this compound, indicating that these cells were successfully differentiated. Similar effects were noted for cells treated with F- and Cl-amidine (Fig. 4c). These results are consistent with the observed changes in the expression of myeloperoxidase and PAD4, and are consistent with our hypothesis that F- and Cl-amidine can trigger the differentiation of HL-60 cells.
Fig. 4.

a Structure of Rh-6(F-araNAD). b Mechanism-based labeling of CD38 with Rh-6(F-araNAD). c Labeling of CD38 in live HL-60 cells after treatment with ATRA, or F-, or Cl-amidine for 48 h. PBS was used as the vehicle control. Cells were treated with 5 μM Rh-6(F-araNAD) for 5 min and visualized under a confocal microscope. The top images show the bright-field view and the bottom images are the corresponding captures of the rhodamine fluorescence
F- and Cl-amidine induce p21 expression
To begin to investigate the molecular basis for the differentiating effects of F- and Cl-amidine, the levels of p21 mRNA and protein were examined in HL-60 cells by RT-PCR. These experiments were performed for several reasons: (1) p21 is a well-characterized cyclin-dependent kinase inhibitor that is known to play an important role in the growth arrest and terminal differentiation of HL-60 cells [26–29]; (2) Cl-amidine has previously been shown to induce p21 expression in U2OS cells and p53+/+ HCT116 cells [6]; and (3) HDAC inhibition upregulates p21 expression [30], suggesting that F- and Cl-amidine might also increase the expression of this gene in HL-60 cells. For these experiments, the effects of F- and Cl-amidine (1 μM) on p21 mRNA levels were determined by quantitative RT-PCR. Both of these agents led to increased expression of p21 mRNA (Fig. 5a). To verify that the increased mRNA levels correlated with increased p21 protein, the effects of F- and Cl-amidine (1 μM) on p21 protein levels were determined as a function of time. Both compounds clearly induced p21 expression within 6 h of their addition (Fig. 5b), suggesting that PAD4 inhibition leads to increased p21 expression, which subsequently triggers growth arrest and differentiation.
Fig. 5.

p21 protein and mRNA levels. HL-60 cells were treated with F- and Cl-amidine (1 μM final concentration) and p21 protein and mRNA expression levels were monitored. a Relative expression levels of p21 mRNA, as determined by RT-PCR, in response to treatment with F- or Cl-amidine for 48 h. b p21 protein levels in response to F- and Cl-amidine were monitored over a 48-h time period
Differentiation of HT-29 cells
To explore the generality of this phenomenon, we examined whether F- and Cl-amidine could also trigger the differentiation of HT-29 cells, a colon adenocarcinoma-derived cell line. For these experiments, HT-29 cells were treated with either F- or Cl-amidine (1 μM final concentration), and after 48 h the levels of alkaline phosphatase, a known marker of HT-29 differentiation [31], were measured. SAHA, a class I/II HDAC inhibitor, served as the positive control as this compound is known to trigger the differentiation of several cancerous cell lines, including HT-29 cells [32]. As depicted in Fig. S2, SAHA, and F- and Cl-amidine all induced the expression of alkaline phosphatase, consistent with the differentiation of this cell line, thus suggesting that PAD inhibition can lead to the differentiation of multiple cancerous cell lines.
F- and Cl-amidine potentiate the cytotoxicity of doxorubicin
Given that PAD4 inhibition parallels the effects of HDAC inhibition, i.e. both trigger cellular differentiation and induce p21 expression, we were curious to see whether these similarities extended further. Therefore, we examined whether F- and Cl-amidine could potentiate the effects of doxorubicin, an antineoplastic agent that is known to potentiate the effects of HDAC inhibitors such as SAHA [33, 34]. For these experiments, the effect of doxorubicin on HL-60 cell survival was determined in the absence and presence of increasing concentrations of F- and Cl-amidine. As is apparent in Fig. 6, increasing concentrations of both compounds caused the dose response curves to shift to the left, resulting in a dose-dependent decrease in the EC50 for doxorubicin (Table 2). Consistent with the fact that Cl-amidine is the more potent inhibitor, this compound decreased the EC50 to a greater extent (18-fold at the highest dose tested, i.e. 100 μM Cl-amidine) than F-amidine (5-fold). This level of enhancement is gratifyingly consistent with the relative potencies of the two compounds (i.e. k inact/K I = 13,000 vs. 3,000 M−1min−1 or about 4.3-fold [15, 17]). In addition to decreasing the EC50 of doxorubicin, both compounds also decreased cell survival to background levels, i.e. viable cells were undetectable (Fig. 6). As with the EC50 values, Cl-amidine had a greater effect on cell viability than F-amidine. For example, with as little as 1 μM of Cl-amidine, viable cells were undetectable at higher levels of doxorubicin; 10 μM of F-amidine was required to achieve a similar level of cell killing.
Fig. 6.

Effects of F-amidine (a) and Cl-amidine (b) in combination with doxorubicin on cell viability of HL-60 cells. Cell viability was measured using a standard MTT assay, after a 24 h incubation with the inhibitors. Each data point represents the average of three trials (p < 0.05). The dashed curve for 100 μM Cl-amidine represents an estimated nonlinear curve. The combination of F-amidine (c) and Cl-amidine (d) with doxorubicin resulted in synergistic killing of HL-60 cells. Synergistic, additive, and subadditive effects of the combination therapy were determined by a comparison of the O/E ratios, where an O/E ratio < 0.8 is considered synergistic, an O/E ratio in the range 0.8–1.2 is additive, and an O/E ratio >1.2 is subadditive
Table 2.
Cytotoxicity of F- and Cl-amidine in combination with doxorubicin in HL-60 cells
| Treatment | ECa50 | Survival (%)b |
|---|---|---|
| Doxorubicin | 2.50 ± 1.16 | 12.0 ± 4.4 |
| Doxorubicin + 1 μM Cl-amidine | 1.42 ± 0.02 | ND |
| Doxorubicin + 10 μM Cl-amidine | 1.29 ± 0.04 | ND |
| Doxorubicin + 100 μM Cl-amidine | 0.14 ± 0.09 | ND |
| Doxorubicin + 1 μM F-amidine | 2.33 ± 1.74 | 8.14 ± 5.10 |
| Doxorubicin + 10 μM F-amidine | 1.30 ± 0.66 | ND |
| Doxorubicin + 100 μM F-amidine | 0.49 ± 0.25 | ND |
ND no viable cells were detected as defined by lack of absorbance above the 100% killing control.
aValues determined by fitting the dose response data to Eq. 1
bBased on cell viability at the maximum agent concentration tested
An additive model [21, 22] was used to determine if the combined effects of F- or Cl-amidine and doxorubicin were subadditive, additive, or synergistic. This model predicts that two drugs are subadditive when the O/E ratio is 1.2, additive when the O/E ratio is between 0.8 and 1.2, and synergistic when the O/E ratio is < 0.8. The results of these analyses (Fig. 6) indicate that as the concentration of doxorubicin is increased, F- and Cl-amidine potentiate cell killing in a synergistic and dose-dependent fashion.
Discussion
Given the mechanistic similarities between PAD4 and the HDACs (both act as transcriptional corepressors and downregulate the expression of a subset of overlapping genes, e.g. p21), we were intrigued to find that Cl-amidine, a PAD4 inhibitor, and PAD4 siRNA knockdown, decreased cell viability and induced apoptosis in a subset of cells [5, 6]. These results suggest that PAD4 inhibitors, like HDAC inhibitors, could represent a novel epigenetic cancer therapy. Further bolstering this possibility is the recent demonstration that PAD4 interacts and cooperates with HDAC1 to downregulate the expression of estrogen receptor-regulated genes [35].
Given these parallels, and given the fact that PAD4 is overexpressed in numerous malignant tumors [9, 10], we set out to examine the cytotoxic effects of Cl-amidine, and the related compound F-amidine, in greater detail. As described above, both F- and Cl-amidine decreased the viability of a number of tumor-derived cell lines (i.e. HL-60, HT-29, and MCF7 cells) in a dose-dependent manner. In contrast, no effect was observed in noncancerous cell lines (i.e. HL-60 granulocytes and NIH3T3 cells). Interestingly, while the EC50 values were in the micromolar to submicromolar range, we were unable to achieve complete cell killing. Because SAHA, a general inhibitor of HDAC activity, also triggers the differentiation of a subset of cancer cell lines [3], we considered the possibility that the lack of complete cell killing could be explained if F- and Cl-amidine are, like SAHA, differentiating agents.
To investigate this possibility, the ability of F- and Cl-amidine to differentiate the leukemic HL-60 cell line into HL-60 granulocytes was examined. This system was chosen because it is well characterized and because there are several known differentiation markers, including: increased expression of both PAD4 and CD38, as well as decreased expression of myeloperoxidase. As described above, F- and Cl-amidine do indeed trigger HL-60 differentiation. The differentiation of colon cancer HT-29 cell line was also demonstrated, suggesting that their ability to differentiate tumor-derived cell lines is a general feature of these compounds.
Although the underlying mechanism by which F- and Cl-amidine induce cellular differentiation and decrease cell viability is unknown, it is likely that their ability to inhibit histone deimination [6] plays a role. This is the case because PAD4 acts as a transcriptional corepressor and its inhibition would be expected to alter gene expression, potentially leading to the increased expression of genes that are important for the differentiation of HL-60 cells into HL-60 granulocytes. One such gene is p21. We hypothesized that this might be the case because increased p21 expression is important for the terminal differentiation of HL-60 cells [26–29], and Cl-amidine has previously been shown to induce the expression of this gene in U2OS and HCT116 cells [6]. As described above, both PAD4 inhibitors did indeed increase the expression of p21, at both the mRNA and protein levels, suggesting that the effects of F- and Cl-amidine are mediated, at least in part, by the increased expression of this cell cycle inhibitor. Although not investigated in this study, PAD4 inhibition, and the consequent increase in p21 expression, likely leads to G1/G2 arrest as shRNA knockdown of PAD4 increases the G1 population of p53+/+ HCT116 cells [6]. In support of this notion, HL60 cells treated with either F- or Cl-amidine show decreased expression of H3 phospho S10, a mitotic marker, indicating that the population of cells in mitosis are decreasing, consistent with a G1/G2 arrest (Fig. S3).
In addition to increased expression of p21, PAD4 inhibition may also positively influence gene transcription mediated by retinoic acid receptor (RAR). Such an effect is likely, given that RAR is the molecular target of ATRA and is a member of the nuclear receptor family of transcriptional activators, and PAD4 acts as a transcriptional corepressor for several other members of this family, including the estrogen receptor and thyroid receptor [7, 8]. Although speculative, if PAD4 does indeed act as a transcriptional corepressor for RAR, then inhibition of PAD4 would be expected to mimic the effects of ATRA, thereby activating the expression of additional genes that are involved in initiating the differentiation program.
As described above, the treatment of HL-60 cells with F- or Cl-amidine increased the expression of PAD4. Given that PAD4 was originally described as an enzyme whose expression is induced during the differentiation of HL-60 cells into HL-60 granulocytes and monocytes [24], it is somewhat paradoxical that inhibitors of PAD4 actually enhance its own expression. Nevertheless, we have routinely found that PAD4 is expressed at low, but nonetheless significant, amounts in undifferentiated HL-60 cells. Although it is a formal possibility that the differentiating effects of F- and Cl-amidine are due to the inhibition of an unknown process, we note that these compounds, especially F-amidine, selectively modify PAD4 [36], and a number of studies have validated their use as PAD4 inhibitors in cellulo [5, 6, 15, 17, 23]. Thus, at low levels it appears that this enzyme prevents the normal differentiation of this cell line, presumably via its transcriptional corepressor activity. Although the higher levels of PAD4 in differentiated cells may reflect a compensatory effect to PAD4 inhibition, we note again that PAD4 expression is increased with other differentiating agents, e.g. ATRA and vitamin D [24]. Thus, increased PAD4 expression may reflect a specialized role for the enzyme in granulocytes that is distinct from its role as a transcriptional corepressor. This seems likely given the fact that PAD4 is localized to cytoplasmic granules in these leukocytes [37]. Also consistent with this notion is the fact that high doses of Cl-amidine do not induce PAD4 expression in several different cell lines, including U2OS and HCT116 cells [6].
The fact that PAD4 inhibition parallels the effects of HDAC inhibition, i.e. both trigger cellular differentiation and induce p21 expression, prompted us to examine whether F- and Cl-amidine could, like SAHA [33, 34], potentiate the cytotoxicity of doxorubicin. As described above, increased amounts of both F- and Cl-amidine led to a dose-dependent decrease in the EC50 for doxorubicin. At the highest doses tested (i.e. 100 μM), F- and Cl-amidine decreased the EC50 value by 5- and 18-fold, respectively, values that are consistent with the relative in vitro potencies of the two compounds. In addition to an effect on the EC50, both compounds also synergistically activated the cell killing effects of doxorubicin. As with the EC50 values, Cl-amidine had the greater effect. Importantly, this effect was dependent on the dose of F- or Cl-amidine, i.e. higher doses of these compounds led to greater synergistic killing. In total, these results indicate that F- and Cl-amidine synergistically potentiate the cytotoxic effects of doxorubicin and suggest that these compounds may be useful adjuvant therapies for the treatment of cancer.
In summary, the highly potent PAD4 inhibitors, F- and Cl-amidine, exhibit cytotoxic effects in a number of cancer-derived cell lines, but not in noncancerous ones, and trigger the terminal differentiation of both HL-60 and HT-29 cells. Additionally, these compounds synergistically potentiate the cytotoxicity of doxorubicin in a dose-dependent manner. Given the parallels between PAD4 and both the HDACs and the DNA methyltransferases, our results suggest PAD4 as a target for the development of a novel epigenetic cancer therapy, and F- and Cl-amidine as candidate therapies for this disease.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We thank Hening Lin and Hong Jiang for the generous gift of Rh-6(F-araNAD), and Michael Wyatt, Franklin Berger and Lee Ferguson, respectively, for the generous gifts of the HT-29, NIH3T3, and MCF7 cells. We also thank Franklin Berger for providing critical comments on the manuscript. This work was supported by NIH grant GM079357 to PRT.
Conflicts of interest
The authors declare a conflict of interest. The University of South Carolina and P.R.T have a financial interest in F-amidine and Cl-amidine.
Abbreviations
- PAD
Protein Arginine Deiminase
- HDAC
Histone deacetylase
- RA
Rheumatoid arthritis
- ER
Estrogen receptor
- TR
Thyroid receptor
- ATRA
All trans retinoic acid
- RAR
Retinoic acid receptor
References
- 1.Lyko F, Brown R. DNA methyltransferase inhibitors and the development of epigenetic cancer therapies. J Natl Cancer Inst. 2005;97:1498–1506. doi: 10.1093/jnci/dji311. [DOI] [PubMed] [Google Scholar]
- 2.Fischle W, Wang Y, Allis CD. Binary switches and modification cassettes in histone biology and beyond. Nature. 2003;425:475–479. doi: 10.1038/nature02017. [DOI] [PubMed] [Google Scholar]
- 3.Marks PA, Breslow R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotechnol. 2007;25:84–90. doi: 10.1038/nbt1272. [DOI] [PubMed] [Google Scholar]
- 4.Jones JE, Causey CP, Knuckley B, Slack-Noyes JL, Thompson PR. Protein arginine deiminase 4 (PAD4): Current understanding and future therapeutic potential. Curr Opin Drug Discov Devel. 2009;12:616–627. [PMC free article] [PubMed] [Google Scholar]
- 5.Yao H, Li P, Venters BJ, Zheng S, Thompson PR, Pugh BF, Wang Y. Histone Arg modifications and p53 regulate the expression of OKL38, a mediator of apoptosis. J Biol Chem. 2008;283:20060–20068. doi: 10.1074/jbc.M802940200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li P, Yao H, Zhang Z, Li M, Luo Y, Thompson PR, Gilmour DS, Wang Y. Regulation of p53 target gene expression by peptidylarginine deiminase 4. Mol Cell Biol. 2008;28:4745–4758. doi: 10.1128/MCB.01747-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Y, Wysocka J, Sayegh J, Lee YH, Perlin JR, Leonelli L, Sonbuchner LS, McDonald CH, Cook RG, Dou Y, Roeder RG, Clarke S, Stallcup MR, Allis CD, Coonrod SA. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science. 2004;306:279–283. doi: 10.1126/science.1101400. [DOI] [PubMed] [Google Scholar]
- 8.Cuthbert GL, Daujat S, Snowden AW, Erdjument-Bromage H, Hagiwara T, Yamada M, Schneider R, Gregory PD, Tempst P, Bannister AJ, Kouzarides T. Histone deimination antagonizes arginine methylation. Cell. 2004;118:545–553. doi: 10.1016/j.cell.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 9.Chang X, Han J. Expression of peptidylarginine deiminase type 4 (PAD4) in various tumors. Mol Carcinog. 2006;45:183–196. doi: 10.1002/mc.20169. [DOI] [PubMed] [Google Scholar]
- 10.Chang X, Han J, Pang L, Zhao Y, Yang Y, Shen Z. Increased PADI4 expression in blood and tissues of patients with malignant tumors. BMC Cancer. 2009;9:40. doi: 10.1186/1471-2407-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vossenaar ER, Zendman AJ, van Venrooij WJ, Pruijn GJ. PAD, a growing family of citrullinating enzymes: genes, features and involvement in disease. Bioessays. 2003;25:1106–1118. doi: 10.1002/bies.10357. [DOI] [PubMed] [Google Scholar]
- 12.Masson-Bessiere C, Sebbag M, Girbal-Neuhauser E, Nogueira L, Vincent C, Senshu T, Serre G. The major synovial targets of the rheumatoid arthritis-specific antifilaggrin autoantibodies are deiminated forms of the alpha- and beta-chains of fibrin. J Immunol. 2001;166:4177–4184. doi: 10.4049/jimmunol.166.6.4177. [DOI] [PubMed] [Google Scholar]
- 13.Maragoudakis ME, Tsopanoglou NE, Andriopoulou P. Mechanism of thrombin-induced angiogenesis. Biochem Soc Trans. 2002;30:173–177. doi: 10.1042/BST0300173. [DOI] [PubMed] [Google Scholar]
- 14.Wojtukiewicz MZ, Tang DG, Ciarelli JJ, Nelson KK, Walz DA, Diglio CA, Mammen EF, Honn KV. Thrombin increases the metastatic potential of tumor cells. Int J Cancer. 1993;54:793–806. doi: 10.1002/ijc.2910540514. [DOI] [PubMed] [Google Scholar]
- 15.Luo Y, Arita K, Bhatia M, Knuckley B, Lee YH, Stallcup MR, Thompson PR. Inhibitors and inactivators of protein arginine deiminase 4: functional and structural characterization. Biochemistry. 2006;45:11727–11736. doi: 10.1021/bi061180d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Causey CP, Thompson PR. An improved synthesis of haloaceteamidine-based inactivators of protein arginine deiminase 4 (PAD4) Tetrahedron Lett. 2008;49:4383–4385. doi: 10.1016/j.tetlet.2008.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luo Y, Knuckley B, Lee YH, Stallcup MR, Thompson PR. A fluoro-acetamidine based inactivator of protein arginine deiminase 4 (PAD4): design, synthesis, and in vitro and in vivo evaluation. J Am Chem Soc. 2006;128:1092–1093. doi: 10.1021/ja0576233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leatherbarrow RJ. Grafit Ver 5.0. Staines: Erathicus Software; 2004. [Google Scholar]
- 19.Yamada M, Mori M, Sugimura T. Purification and characterization of small molecular weight myeloperoxidase from human promyelocytic leukemia HL-60 cells. Biochemistry. 1981;20:766–771. doi: 10.1021/bi00507a018. [DOI] [PubMed] [Google Scholar]
- 20.Dong S, Zhang Z, Takahara H. Estrogen-enhanced peptidylarginine deiminase type IV gene (PADI4) expression in MCF-7 cells is mediated by estrogen receptor-alpha-promoted transfactors activator protein-1, nuclear factor-Y, and Sp1. Mol Endocrinol. 2007;21:1617–1629. doi: 10.1210/me.2006-0550. [DOI] [PubMed] [Google Scholar]
- 21.Valeriote F, Lin H. Synergistic interaction of anticancer agents: a cellular perspective. Cancer Chemother Rep. 1975;59:895–900. [PubMed] [Google Scholar]
- 22.Jonsson E, Fridborg H, Nygren P, Larsson R. Synergistic interactions of combinations of topotecan with standard drugs in primary cultures of human tumor cells from patients. Eur J Clin Pharmacol. 1998;54:509–514. doi: 10.1007/s002280050505. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, Hayama R, Leonelli L, Han H, Grigoryev SA, Allis CD, Coonrod SA. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol. 2009;184:205–213. doi: 10.1083/jcb.200806072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakashima K, Hagiwara T, Ishigami A, Nagata S, Asaga H, Kuramoto M, Senshu T, Yamada M. Molecular characterization of peptidylarginine deiminase in HL-60 cells induced by retinoic acid and 1alpha,25-dihydroxyvitamin D(3) J Biol Chem. 1999;274:27786–27792. doi: 10.1074/jbc.274.39.27786. [DOI] [PubMed] [Google Scholar]
- 25.Jiang H, Congleton J, Liu Q, Merchant P, Malavasi F, Lee HC, Hao Q, Yen A, Lin H. Mechanism-based small molecule probes for labeling CD38 on live cells. J Am Chem Soc. 2009;131:1658–1659. doi: 10.1021/ja808387g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang J, Zhao Y, Kauss MA, Spindel S, Lian H. Akt regulates vitamin D3-induced leukemia cell functional differentiation via Raf/MEK/ERK MAPK signaling. Eur J Cell Biol. 2009;88:103–115. doi: 10.1016/j.ejcb.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 27.Zeng YX, el-Deiry WS. Regulation of p21WAF1/CIP1 expression by p53-independent pathways. Oncogene. 1996;12:1557–1564. [PubMed] [Google Scholar]
- 28.Savoysky E, Yoshida K, Ohtomo T, Yamaguchi Y, Akamatsu K, Yamazaki T, Yoshida S, Tsuchiya M. Down-regulation of telomerase activity is an early event in the differentiation of HL60 cells. Biochem Biophys Res Commun. 1996;226:329–334. doi: 10.1006/bbrc.1996.1356. [DOI] [PubMed] [Google Scholar]
- 29.Shankar S, Singh TR, Fandy TE, Luetrakul T, Ross DD, Srivastava RK. Interactive effects of histone deacetylase inhibitors and TRAIL on apoptosis in human leukemia cells: involvement of both death receptor and mitochondrial pathways. Int J Mol Med. 2005;16:1125–1138. [PubMed] [Google Scholar]
- 30.Marks PA. The mechanism of the anti-tumor activity of the histone deacetylase inhibitor, suberoylanilide hydroxamic acid (SAHA) Cell Cycle. 2004;3:534–535. doi: 10.4161/cc.3.5.824. [DOI] [PubMed] [Google Scholar]
- 31.Barnard JA, Warwick G. Butyrate rapidly induces growth inhibition and differentiation in HT-29 cells. Cell Growth Differ. 1993;4:495–501. [PubMed] [Google Scholar]
- 32.Wilson AJ, Byun DS, Popova N, Murray LB, L’Italien K, Sowa Y, Arango D, Velcich A, Augenlicht LH, Mariadason JM. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J Biol Chem. 2006;281:13548–13558. doi: 10.1074/jbc.M510023200. [DOI] [PubMed] [Google Scholar]
- 33.Luong QT, O’Kelly J, Braunstein GD, Hershman JM, Koeffler HP. Antitumor activity of suberoylanilide hydroxamic acid against thyroid cancer cell lines in vitro and in vivo. Clin Cancer Res. 2006;12:5570–5577. doi: 10.1158/1078-0432.CCR-06-0367. [DOI] [PubMed] [Google Scholar]
- 34.Kim MS, Blake M, Baek JH, Kohlhagen G, Pommier Y, Carrier F. Inhibition of histone deacetylase increases cytotoxicity to anticancer drugs targeting DNA. Cancer Res. 2003;63:7291–7300. [PubMed] [Google Scholar]
- 35.Denis H, Deplus R, Putmans P, Yamada M, Metivier R, Fuks F. Functional connection between deimination and deacetylation of histones. Mol Cell Biol. 2009;29:4982–4993. doi: 10.1128/MCB.00285-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luo Y, Knuckley B, Bhatia M, Thompson PR. Activity based protein profiling reagents for protein arginine deiminase 4 (PAD4): synthesis and in vitro evaluation of a fluorescently-labeled probe. J Am Chem Soc. 2006;128:14468–14469. doi: 10.1021/ja0656907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Asaga H, Nakashima K, Senshu T, Ishigami A, Yamada M. Immunocytochemical localization of peptidylarginine deiminase in human eosinophils and neutrophils. J Leukoc Biol. 2001;70:46–51. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.