

Abstract
Primary sclerosing cholangitis (PSC), first described in the mid-1850’s, is a complex liver that is heterogeneous in its presentation. PSC is characterized by chronic cholestasis, associated with chronic inflammation of the biliary epithelium, resulting in multi-focal bile duct strictures that can affect the entire biliary tree. Chronic inflammation leads to fibrosis involving the hepatic parenchyma and biliary tree, which can lead to cirrhosis and malignancy. The etiology of PSC is not fully understood which in part explains a lack of effective medical therapy for this condition. However, we have begun to better understand the molecular pathogenesis of PSC. The recognition of specific clinical subtypes and their pattern of progression could improve phenotypic and genotypic classification of the disease. We review our current understanding of this enigmatic disorder and discuss important topics for future studies.
Keywords: cholestatic liver disease, cholangiocarcinoma, autoimmune disease, inflammatory bowel disease
Epidemiology and Natural History
Prevalence rates for PSC in North America and Europe range from 6 to 16 cases per 100,000 inhabitants.1–3 Studies of population-based cohorts have estimated incidence of PSC in many North American locations to be approximately 1 per 100,000 persons.3, 4 The incidence of PSC appears similar in North American and Northern European countries.5 However, the incidence and prevalence vary worldwide, with lower estimates reported from Asia and Southern Europe.6, 7 Although PSC may be an uncommon disease, the reported incidence has increased over time.5
The median age of patients diagnosed with PSC is 41 y, and it appears to be more common among men.5 Patients are often diagnosed incidentally and nearly 50% are asymptomatic. Despite being asymptomatic at the time of diagnosis, patients with PSC have shorter average times of survival, compared to matched controls from the general population.8 Five years after diagnosis of asymptomatic PSC, approximately 22% show clinical symptoms, and after 6 y, up to 76% have some evidence of disease progression (biochemical, symptomatic, or radiographic).8, 9 Fatigue is often present at the time of diagnosis. Other presenting signs and symptoms include abdominal pain (37%), jaundice (30%), and fever (17%).9 When patients are symptomatic upon presentation, the median time of survival until death or liver transplantation is 9 y (compared to 12–18 y for all PSC patients, regardless of symptoms).8–10
PSC can present during child-bearing years, yet little is known about PSC progression during pregnancy or its effects on the fetus and mother. In a case series, fertility was not reduced among women with PSC, nor was there a higher proportion of fetal loss or adverse fetal outcomes, compared to the general population. Although no serious maternal outcomes were noted, there was an increase in liver enzymes among 20% of pregnant women with PSC.11
The major risk factor for development of PSC is inflammatory bowel disease (IBD). Sixty to eighty percent of patients with PSC have concurrent IBD (typically ulcerative colitis, UC), and about 4% of patients with UC have coexisting PSC.12, 13 Individuals with PSC are most frequently male, and in rare cases, have a family history of the disease. Smoking has been reported to protect against PSC, even after controlling for underlying UC.5, 14, 15
Several prognostic scores for PSC have been developed. The revised Mayo risk score is based on a combination of patient’s age; levels of bilirubin, aspartate aminotransferase, and albumin; and the presence of variceal bleeding. These factors can predict survival times, without the need for liver biopsy.16 However, the use of prognostic models is not recommended for management of individual patients with PSC, given the significant variations in disease course.10, 12 Complications related to PSC such as intermittent episodes of cholangitis can occur in 10%–15% of patients.17 Portal hypertension and cirrhosis, metabolic bone disease, associated malignancies, and co-existing conditions such as IBD add to the disease burden.
Diagnosis of PSC
Serum Markers
An increased serum level of alkaline phosphatase is the most common biochemical abnormality detected in patients with PSC. In some cases, it is the only biochemical alteration observed, such as in patients with intrahepatic or diffuse involvement with PSC.12 However, the level of alkaline phosphatase can vary throughout the disease course and may be normal.18 Although serum levels of aminotransferase are frequently normal, in some patients they are reduced to 3–4-fold below the upper limit of normal.12 Higher values indicate acute biliary obstruction or even an overlap syndrome with autoimmune hepatitis (AIH). Serum levels of total bilirubin are typically normal, and increased only among patients with significant structuring. Serum levels of albumin, international normalized ratios, and platelet counts are typically normal unless cirrhosis and portal hypertension have developed.
Patients with IBD who have increased liver test values which suggest an underlying cholestatic liver disease should immediately raise the suspicion of concurrent PSC. In rare cases, subjects with IBD undergoing computed tomography or magnetic resonance (MR) enterography have been found to have unsuspected intrahepatic bile duct dilatation, even though they had normal results from biochemical analyses of serum samples for liver function. Subsequent MR cholangiopancreatography (MRCP) identified changes associated with PSC in these subjects.
PSC is associated with a high proportion of non-specific auto-antibodies.19 Unlike primary biliary cirrhosis, there is no diagnostic serologic test that is specifically associated with PSC. Serologic tests might be useful for patients suspected of having PSC and AIH, or immunoglobulin (Ig)G4-associated cholangitis (IAC) in association with autoimmune pancreatitis (AIP).
Imaging
Cholangiography is the best way to identify patients with PSC (Figure 1). The classic features include multi-focal anular structuring within the intrahepatic and/or extrahepatic bile ducts, with alternating normal or slightly dilated segments.20 Typically there is diffuse involvement. However, up to 25% of individuals have only intrahepatic disease.20 Although endoscopic retrograde cholangiopancreatography (ERCP) is used to evaluate symptomatic patients with suspected biliary obstruction or cholangiocarcinoma, MR cholangiopancreatography (MRCP) has largely replaced ERCP as a diagnostic tool, due to improvements in imaging and software-processing technology. Pooled analyses have reported good to excellent diagnostic performance for MRCP, compared to ERCP, for detection of PSC.21 MRCP is noninvasive, avoids radiation, and is more cost effective than ERCP in diagnosis.22, 23 With stronger magnetic fields and availability of 3D image reprocessing, the ability to visualize 3rd- and 4th-order intrahepatic ducts is now possible, which improves the sensitivity of MRCP when no extrahepatic biliary strictures are present. It is reasonable to consider patient referral if quality MRCP images are not available, to exclude or confirm the presence of PSC.
Figure 1.
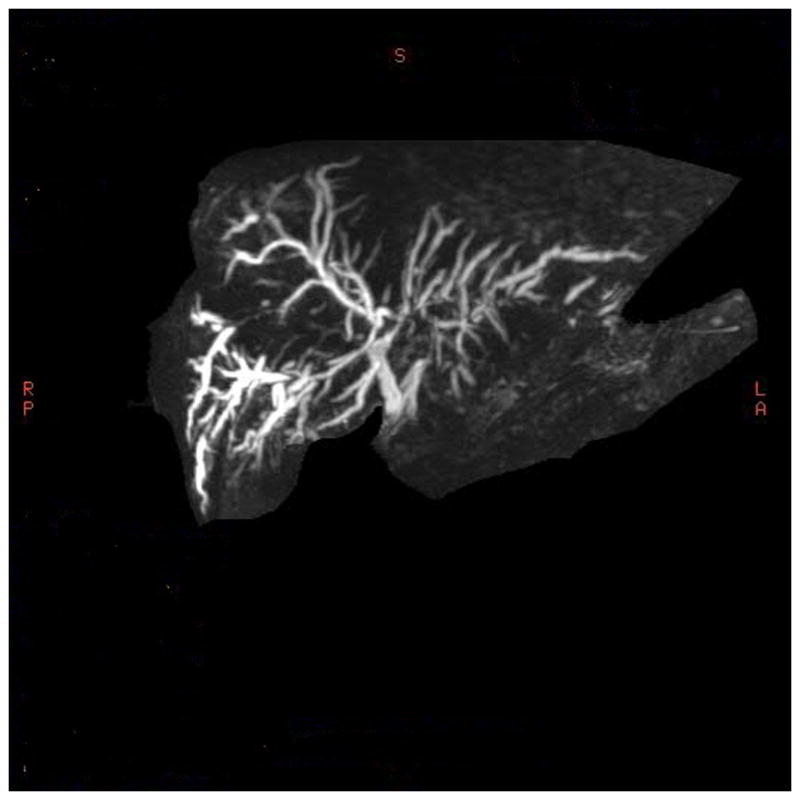
A 3D Post-Processing Image From MR Cholangiography
Diffusely dilated intrahepatic ducts with multifocal narrowing with diffuse ductal wall thickening and enhancement.
Liver Histology
Typically, a liver biopsy is not required to diagnose PSC, unless small-duct PSC is suspected or if there are concerns that a patient also has AIH. Histologic features of PSC are often nonspecific and prone to sampling variations, due to the heterogeneous involvement of the biliary tree.24 Unfortunately, the classic description of concentric ductal fibrosis (“onion skinning”) involving bile ducts within portal tract areas is rarely encountered in clinical practice (Figure 2).25 Use of histologic analysis to determine the stage of liver fibrosis requires a specialized scoring system (Batts-Ludwig).26 However, noninvasive methods to assess fibrosis and cirrhosis, such as elastography imaging, are emerging as useful tools for subjects with PSC.27
Figure 2.
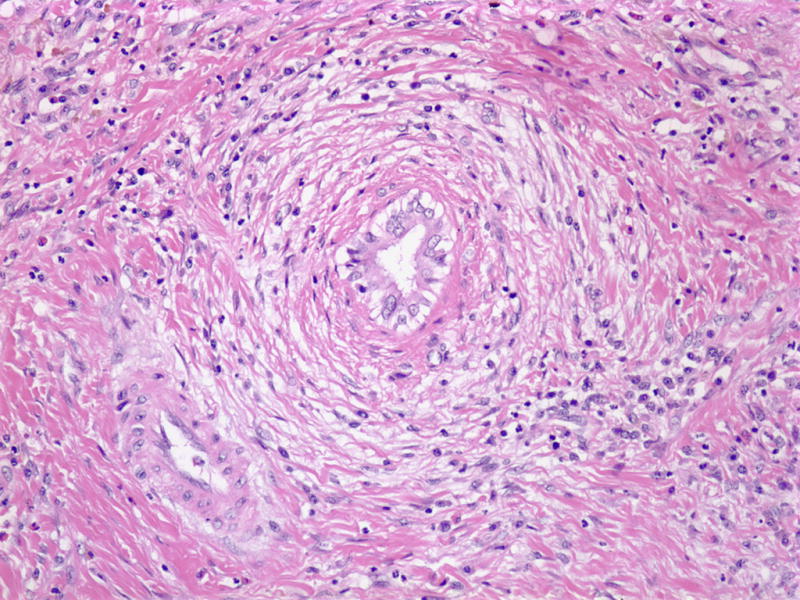
Histologic Features of PSC
Concentric ductal fibrosis.
Distinguishing IAC from PSC
IAC was identified during seminal studies to codify the diagnosis and treatment for AIP. IAC and AIP are each manifestations of a systemic inflammatory condition associated with IgG4 production and deposition within the biliary system and pancreas, respectively28, 29 IAC is often diagnosed based on the HISORt criteria (Table 1). 30 However, other criteria have recently been proposed.31 Although biliary strictures can develop in patients with PSC or IAC, IAC appears to have distinct clinical, biochemical and histologic features from PSC (Table 1). Some individuals believed to have traditional PSC could actually have IAC, so it is recommended that serum levels of IgG4 are measured and cross-sectional imaging is used to evaluate the pancreas.12
Table 1.
Diagnostic Classification of Typical PSC, Small-Duct PSC, AIH-PSC, and IAC
| Cholangiography | Pathology | Diagnosis | Management a | Other Features | |
|---|---|---|---|---|---|
| Typical PSC |
|
|
|
|
|
| Small-Duct PSC |
|
|
|
|
|
| AIH-PSC |
|
|
|
|
|
| IAC |
|
|
|
|
|
Referral for liver transplantation when appropriate is key component of management for all PSC subtypes.
At this time we recommend surveillance for CRN in the setting of IBD, GBN, CCA and HCC (in those with cirrhosis) regardless if they have classic PSC or another PSC subtype.
Abbreviations: PSC (primary sclerosing cholangitis); AIH (autoimmune hepatitis); IAC (IgG4 associated cholangitis); AIP (autoimmune pancreatitis); LPC (lymphoplasmacytic); CRN (colorectal neoplasia); CCA (cholangiocarcinoma); GBN (gallbladder neoplasia); HCC (hepatocellular carcinoma); hpf (high-power field).
Based on findings from retrospective studies, it is not clear whether some patients with PSC and increased serum levels of IgG4 actually have IAC or a unique subtype of PSC.32, 33 For example, Bjornsson et al. reviewed data from 285 patients diagnosed with classic PSC; 12% had increased serum levels of IgG4 (>140 mg/dL). However, only 17% of these individuals had pancreatic involvement suggestive of IAC or AIP, and few subjects were assessed by histologic analysis to identify tissue infiltration with IgG4. Mendes et al. found that patients with features of typical PSC and increased serum levels of IgG4 had a shorter time to liver transplantation, compared to patients with normal IgG4 levels, indicating a more aggressive disease course.33
Conversely, 23% of liver explants from patients undergoing liver transplantation for PSC had infiltration with IgG4 positive cells—these patients had a more aggressive disease course and increased risk for recurrent PSC. Interestingly, an estimated 50% of subjects with increased serum levels of IgG4 had normal did not have IgG4 positive tissue.34 Importantly, some patients with PSC and ductal cholangiocarcinoma also have increased levels of total IgG4. Increased production of IgG4 might therefore indicate a more aggressive disease course, rather than serve as marker of patients who should be considered for treatment with corticosteroids.35 Therefore, it is important to carefully exclude the presence of malignancy.30 Future studies should help clarify the prevalence and outcomes of PSC patients with increased levels of IgG4 who do not meet the criteria for IAC.
IBD in Patients with PSC
IBD is a common feature of PSC.36 Patients diagnosed with PSC should undergo colonoscopy and biopsies to determine if they have IBD—even when there are no symptoms.12 A recent study found that the chronologic order of diagnosis has switched in past decades. In a recent cohort (2003–2007), most patients were diagnosed with PSC first; in an earlier cohort (1993–1997), most patients were diagnosed with IBD first.37
Most patients with PSC and IBD have chronic UC (PSC-UC). PSC-UC may have a different phenotype than UC. Several studies have reported that pancolitis is more common in patients with PSC-UC. Although early studies associated with PSC-UC with backwash ileitis or rectal sparing, these findings have not always been reproduced.38–40 Furthermore, subjects with PSC and IBD can have minimal endoscopic activity despite the presence of more active histologic inflammation.41 Those with PSC-UC have an increased cumulative risk for colorectal neoplasia, compared to individuals with UC alone, an increased risk for of pouchitis, and a higher risk of peristomal varices following proctocolectomy with ileostomy.42–44
PSC can be diagnosed following a colectomy just as IBD can develop after liver transplantation.36, 45 Similarly, patients diagnosed with PSC without IBD can still develop this disease and colorectal cancer, at a later time. Thus, a colonoscopy every 5 y following the diagnosis of PSC to exclude or confirm the development of IBD has been suggested.46
Recent observations have suggested an inverse prognostic relationship between PSC and IBD. Progressive PSC requiring a transplant appears to be associated with UC that is more quiescent and less likely to require a colectomy. In contrast, individuals without progressive PSC were found to have an increased need for colectomy, more severe histologic inflammation and increased risk of colorectal neoplasia.47,48 These observations indirectly support a model of PSC and IBD pathogenesis that involves aberrant lymphocyte homing.47
PSC Subtypes
Small-Duct PSC
Among cases with suspected PSC and normal cholangiography, liver biopsy analysis is recommended, to rule out small-duct PSC.12 Nearly 6% of patients with chronic cholestasis, a normal cholangiogram, and IBD have concurrent small-duct PSC.49 Individuals with small-duct PSC have symptoms and laboratory results similar to those of subjects with classic PSC.49 However, patients with small-duct PSC survive longer and have a lower cumulative risk for cholangiocarcinoma than patients with large-duct involvement.50, 51 An estimated 20% of subjects with small-duct PSC develop large-duct PSC (intra or extrahepatic) over a 7–10 y period.50, 51
Overlap Between AIH and PSC
AIH-PSC has been reported to occur in 6% or less of patients with PSC but the true prevalence is unknown.52, 53 Similar to classic PSC, IBD is often associated with AIH-PSC.54, 55 The simplified International Autoimmune Hepatitis Group scoring system can aid in diagnosis of AIH, but it is not recommended for diagnosis of AIH-PSC.56 AIH-PSC should be suspected for patients with abnormal cholangiograms that indicate PSC, biochemical features of AIH (increased antibody titers or levels of transaminases), lymphoplasmacytic portal-based infiltrates, and significant interface hepatitis (based on liver biopsy analysis), or for patients with AIH that becomes refractory to therapy.12, 55 Patients with AIH-PSC could have shorter survival times (or time to liver transplantation), compared to patients with AIH alone or other overlap syndromes such as AIH and Primary Biliary Cirrhosis.52
Pathogenesis
The pathogenic mechanisms of PSC are incompletely understood, but the process is likely multifactorial. A number of hypotheses have emerged; studies of these could clarify pathologic mechanisms and identify potential therapeutic targets. PSC likely occurs in genetically susceptible individuals, perhaps after exposure to environmental triggers. These could initiate a series of events that involve complex interactions between the innate and adaptive immune systems, ultimately leading to lymphocyte migration, cholangiocyte damage, and progressive fibrosis. Several important observations, coupled with the strong association between certain human leukocyte antigen (HLA) haplotypes and frequency of concurrent extrahepatic autoimmune disorders, support the concept that PSC is an immune-mediated phenomenon.57
Genetic Predisposition
Patients who have a first-degree relative with PSC have a 9- to 39-fold increase in risk for the development of the disease. 14 In the past 2 decades, candidate gene studies associated specific HLAs with PSC, including the haplotypes HLA-DRB1*1501-DQB1*0602, HLA-DRB1*1301-DQB1* 0603, and HLA-A1-B8-DRB1*0301-DQB1*0201.58–61
Recent, genome-wide association studies have also reported that the strongest genetic risk to PSC lies within the MHC, where the heritability represented by these loci is twice that of all other risk loci combined. 62 Several other loci have been associated with PSC; these contain genes that regulate immune self-recognition and adaptive immunity. 63 To date, 12 non-MHC risk loci have been associated with PSC. Most of these loci have a stronger association with PSC than IBD, indicating the overlapping yet unique genetic architecture between PSC and IBD.64 However, the picture of genetic predisposition to PSC is not complete; large genetic studies are underway to identify variants associated with PSC and to better determine its association with IBD.
Bacteria, Molecular Patterns and the Innate Immune System
Translocation of microbial flora across an inflamed, permeable gut with subsequent activation of the immune system and inflammation of the biliary tree is a hypothesized mechanism for the development of PSC.65 Small intestinal bacterial overgrowth and introduction of bacterial antigens to the portal circulation cause pericholangitis in animal models.66–68 However, studies in humans have suggested portal venous bacteremia is uncommon in UC.69 Blood samples from patients with PSC have staining patterns for atypical perinuclear anti-neutrophil cytoplasmic antibodies (p-ANCA) that differ from those of patients with other vasculitides.70 Atypical p-ANCA appears to cross react with human β-tubulin isotype 5 and the bacterial protein FtsZ, which is expressed by intestinal flora.71 PSC patients might therefore have an aberrant immune response to intestinal microbes.
Although some antibiotics have been shown to reduce serum levels of alkaline phosphatase and Mayo Risk scores, the long-term effects of antibiotics on PSC progression are not clear.72, 73 Growing interest in the relationship between the human microbiome and chronic disease will undoubtedly lead to studies in patients with PSC.
Normally, biliary epithelial cells are exposed to common intestinal pathogen-associated molecular patterns such as lipopolysaccharide and lipoteichoic acid. However, exposure to lipopolysaccharide may disrupt tight junctions in colonic and biliary epithelial cells, through Toll-like receptor (TLR)4-dependent mechanisms.74, 75 Alteration of such barriers could expose cholangiocytes to a variety of substances, such as bile acids, that could promote injury and inflammation. Disruption of cholangiocyte tight junctions is an important step in the development of PSC in animal models.76, 77 For example, mice with altered cholangiocyte tight junctions leak bile acid into the portal tract. This leads to an inflammatory response that involves CD8+ and CD4+ T cells and upregulation of tumor necrosis factor (TNF)α, transforming growth factor β1, and interleukin (IL)1β. This inflammatory infiltrate causes myofibroblast activation and fibrosis.76
Despite exposure to such common pathogen-associated molecular patterns, the innate immune system of patients without PSC does not appear to be as upregulated by these endotoxins.78, 79 For example, in liver explants from patients with PSC, biliary epithelial cells express higher levels of TLR, nucleotide-binding oligomerization domain, the MyD88/IRAK complex, TNFα, interferon (INF)γ, and IL8 than cells from individuals without PSC. Early-stage PSC samples express lower levels of IL8, TNFα, and TLR than late-stage samples. After repeated exposure to endotoxins, biliary epithelial cells from patients with PSC continued to secrete high levels of IL8, indicating a lack of tolerance to repeated endotoxin exposure. This hyper-responsiveness could be mediated by increases in IFNγ and TNFα, which stimulate TLR4-mediated intake of endotoxin by biliary epithelial cells and ongoing TLR4 signaling in patients with PSC.78 In addition, pathogens could stimulate TLR5 or TLR7 to induce T-helper (Th)17 cells, which produce IL17, in PSC patients.80
These findings indicate that patients with PSC have an increased immune response to intestinal endotoxins, which could contribute to chronic biliary inflammation. The common association between bacteria and dominant biliary strictures could further promote this response in advanced stages.81 Some researchers have proposed that the innate immune system is also involved in the early stages of PSC development. This process could involve activation of macrophages, natural killer cells, and dendritic cells by pathogen-associated molecular patterns via pattern recognition receptors such as TLRs.82
Adhesion Molecules and Lymphocyte Recruitment
The interaction between adhesion molecules and lymphocyte recruitment to the liver is emerging as an important step in the pathogenesis of PSC. Inflammatory mediators appear to upregulate a variety of adhesion molecules during development of PSC, including intercellular adhesion molecules, vascular cell adhesion molecule-1 (VCAM-1), and mucosal addressin cellular adhesion molecule 1 (MAdCAM-1).83 Typically, MAdCAM-1 is expressed in the mucosal vessels of the intestine. However, under conditions of inflammation, it can be expressed by hepatic endothelium.84, 85 Patients with PSC have also been observed to have altered expression of chemokines such as CCL 25, CCL 28, CXCL12, and CXCL 16. Upregulation of CCL 25 and 28 leads to activation of α4β7 integrins, which increases lymphocyte binding to MAdCAM-1. CCL 28 also appears to activate α4β1 integrin and increase its adhesion to VCAM-1, which is primarily expressed in the portal and sinusoidal endothelial cells of the liver. Once lymphocytes have entered the portal tract, CXCL12 and CXCL16 may facilitate lymphocyte binding to the bile duct epithelium.83
The observation that PSC can still develop after colectomy, and IBD can still develop after liver transplantation has led some to suggest that aberrant homing of lymphocytes between the intestine and liver could be involved in the pathogenesis of PSC.86 In this hypothesis, activated intestinal lymphocytes undergo enterohepatic circulation and persist as memory cells that cause hepatic inflammation. Chemokines and adhesion molecules that are shared by the intestine and liver could contribute to binding of immune cells at both sites.86 Vascular adhesion protein (VAP)1 is found in endothelial cells of the liver and in the mucosal vessels. In patients with IBD, the chronic inflammation appears to upregulate VAP-1 expression in intestinal venules.87 Activation of VAP-1 has been shown to increase the expression of MAdCAM-1 in hepatic vessels, which ultimately promotes recruitment of effector lymphocytes to the liver.88 However, upregulation of hepatic MAdCAM-1 has been described in other chronic liver disease, so it might not be a specific feature of PSC.84
Long-lived memory cells that are primed in the intestine and circulate to the liver might lead to PSC after colectomy. Interestingly, it appears that α4β7+CCR9+ CD8+ T cells that infiltrate the liver in PSC appear to be primed by dendritic cells in the intestine, rather than antigen-presenting cells in the liver, through a process that requires retinoic acid.89 These observations support a role for aberrant lymphocyte trafficking in the pathogenesis of PSC. Further studies are needed to clarify these findings and investigate the roles of specific adhesion molecules in the pathogenesis of PSC.
Antibodies and T Cells
A high proportion of patients with PSC have nonspecific autoantibodies19, as well as autoantibodies that bind to biliary epithelial cells.90 These biliary autoantibodies have been shown to bind biliary epithelial cells and activate the innate immune system by inducing expression of transcription factor ERK1/2 and upregulating TLRs, leading to production of inflammatory cytokines.91 The autoantibodies also increase expression of IL6 and adhesion molecules such as CD44, and could thereby promote lymphocyte proliferation, Ig production, and cell adhesion.90, 91
An influx of lymphocytes, particularly T cells, has been observed in the portal tracts of PSC patients.83 In addition to the portal area, the proportion of circulating T cells, such as γδ+ T cells, is increased and also co-express CD45RO and IL2, indicating an activated memory phenotype.82 Regulatory T cells are believed to participate in the pathogenesis of a variety of autoimmune diseases. The observation that patients with PSC-UC have a higher proportion of CD4+CD25+ regulatory T cells than patients with only UC warrants further study.92 Th17 cells have also been implicated in autoimmune diseases.93 Compared to patients with autoimmune hepatitis or fatty liver disease, livers of patients with PSC contain higher number of Th17 cells—possibly a response to microbes.80
Transporter Defects
Multidrug resistance protein (MDR)3 is an ATP-binding cassette transporter that is required for biliary excretion of phosphatidylcholine.94 Defects in MDR3 have been associated with several cholestatic syndromes, in addition to drug-induced cholestasis.95 Mdr2 (the rodent analogue to MDR3) knockout mice have features of sclerosing cholangitis.76 Decreased bile concentrations of phosphatidylcholine might increase the toxicity of other bile acids.76 Patients with cystic fibrosis may develop biliary cholangiographic features analogous to those of patients with PSC.96 Variants and functional mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) have been described in PSC patients.97 However, it is not clear how mutations in MDR3 and CFTR contribute to PSC in the majority of patients.
Animal Models
A reliable and reproducible single animal model for PSC is needed. A classification scheme for animal models was eloquently described by Pollheimer et al. In these animals, cholangitis is induced by enteric bacteria cell wall components, infectious agents (such as Cryptosporidium parvum), biliary obstruction, chemicals (such as lithocholic acid), knockout of genes such as Mdr2 or Cftr (in mice), or primary biliary and endothelial cell injury.57 Mice injected with death receptor 5 agonists have also provided insight into apoptosis in cholestatic liver disease.98 Given the heterogeneity of PSC and the many factors that contribute to its pathogenesis, a single animal model will be challenging to develop. Consequently, multiple models may be required to examine the various aspects of PSC development.
Pharmacologic Agents
UDCA
All randomized controlled trials of agents designed to prevent PSC progression have produced negative results, despite promising results from open-label precursor studies. The most commonly studied agent is ursodeoxycholic acid (UDCA), which significantly slows progression of other chronic biliary diseases, such as primary biliary cirrhosis (Table 2).99–103 A European study did not demonstrate increased survival times of patients with PSC given 17–23 mg/kg/day UDCA, compared to placebo.102 However, this study was underpowered and concerns were raised about noncompliance among subjects given the test article. Unexpectedly, a North American study was stopped early because 28–30 mg/kg/day UDCA increased the risk of disease progression 2-fold, compared with placebo; study endpoints included cirrhosis, varices, cholangiocarcinoma, liver transplantation, or death. 103 The increase in adverse events appeared to primarily occur in patients with early-stage disease compared to similar patients in the placebo group.104 Furthermore, high-dose UDCA was associated with an increased risk for colorectal neoplasia among patients with UC, providing further evidence for the toxicity of UDCA at this weight-based dosing range.105
Table 2.
Key Randomized Placebo-Controlled Trials of UDCA for PSC
| Year (reference) | Dose UDCA | Follow up | Number of Patients | Outcomes |
|---|---|---|---|---|
| 1997 (99) | 13–15 mg/kg/day | 2.2 years | 105 |
|
| 2001 (100) | 20 mg/kg/day | 2 years | 26 |
|
| 2001 (101) | 25–30 mg/kg/day | 1 year | 30 |
|
| 2005 (102) | 17–23 mg/kg/day | 5 years | 219 |
|
| 2009 (103) | 28–30 mg/kg/day | 6 years | 150 |
|
A meta-analysis of 8 trials determined that UDCA did not slow PSC progression.106 Although there is no clear role for UDCA therapy this time, the safety profile of moderate-dose UDCA (17–23 mg/kg/day) indicates that it could be worth further examination in prospective trials. The American Association for the Study of Liver Diseases (AASLD) recommends against UDCA therapy for PSC, whereas the European Association for the Study of Liver Diseases does not recommend for or against treatment with UDCA, based on the limited data.12, 107
Immunosuppressive Agents
Corticosteroids, etanercept, tacrolimus, cyclosporine, azathioprine, methotrexate, and infliximab have not demonstrated clinical benefits for subjects with PSC.108, 109 Immunosuppressants therefore have no role in the treatment of classic PSC and are not recommended.12
Individuals with evidence for AIH-PSC should be treated with immunosuppressive therapies, as recommended for AIH. Treatment with azathioprine can reduce corticosteroid-related adverse effects. Dosing schedules and duration of therapy are similar to those for AIH.12 The recent interest in budesonide for AIH will likely result in its use for AIH-PSC, yet its efficacy for this condition is unknown.110 There is no clear role for UDCA in the treatment of AIH-PSC.
Endoscopic Therapy
New or worsening symptoms in patients with PSC typically warrant investigation to exclude a dominant extrahepatic biliary stricture. A dominant stricture is defined as a stenosis ≤ 1.5 mm in the common bile duct or ≤1 mm in the hepatic duct.12 When present, it should raise the concern for cholangiocarcinoma. Although the prevalence of dominant strictures in patients with PSC is unknown, it has been approximated at 50%111 Symptomatic dominant strictures are less common, forming in approximately 10%–30% of patients.111, 112 Bacteria are often found in bile from patients with a dominate stricture (more than 40% of cases), in contrast to those without dominant strictures.81
Expansion of dominant strictures by dilatation alone or dilation and stent placement can provide long-term biliary drainage and reduce symptoms.112 However, there are no data from randomized, controlled studies to compare these methods. Importantly, the combination of stenting and dilation was associated with more complications than dilation alone in a large retrospective study.112 Biliary endoprosthetic stent placement should therefore be reserved for cases in which dilatation is unable to maintain lumen patency. 112 The required duration of stenting varies; 6–8 weeks is probably the safest interval to avoid superimposed complications such as cholangitis. Some patients require stenting with regular exchanges for as long as 6–12 months before strictures resolve.
Oral antibiotics for a minimum of 5 days after dilatation and/or stenting can reduce the risk for cholangitis.12 Brush cytology and/or biopsy samples should be obtained to exclude the possibility of cholangiocarcinoma.12 Post-ERCP pancreatitis and cholangitis have been reported in 5%–7% and 1% of patients, respectively.113, 114 Factors that predict post-ERCP complications include the presence of cirrhosis, Crohn’s disease, AIH, level of operator experience, and the performance of a sphincterotomy or biliary dilation.113 Although therapeutic endoscopy can reduce symptoms and prevent and treat cholangitis, its role in modifying disease progression is unclear.
Liver Transplantation
PSC is a leading indication for liver transplantation in some Scandinavian countries, and it is the fifth-most frequent indication for liver transplantation in the United States.115 With 1- and 5-year rates of survival exceeding 90% and 80%, respectively, patients with PSC have among the most successful outcomes following liver transplantation.116 Typically, a Roux-en-Y choledochojejunostomy is the method of choice for biliary reconstruction during liver transplant.117
Indications for transplants from deceased or living donors are similar to those for other chronic liver diseases, including complications of portal hypertension that are refractory to medical therapies. Patients with disease-related complications such as refractory pruritus and recurrent bacterial cholangitis increasingly receive liver transplants from living donors, because of their low priority for livers from deceased donors, based on Model for End-Stage Liver Disease (MELD) scores.
PSC patients are more likely to receive a transplant from a living donor than patients with other chronic liver diseases—this trend has increased since the MELD score came into use.118 However, the practice of referral for liver transplantation from a living donor or granting exception points for a perceived increased risk in mortality secondary to cholangitis was questioned by a study that did not find an increased risk of waitlist removal for death or clinical deterioration secondary to cholangitis.119
Individuals who have undergone liver transplantation for PSC have an increased risk for early (within 30 days) acute cellular rejection, which typically responds to systemic corticosteroids and does not appear to affect graft survival.116 Although chronic ductopenic rejection is becoming less common, it can occur and has deleterious effects on patient and graft survival.116 Other complications include hepatic artery thrombosis, anastomotic biliary stricture, and recurrent disease.
Liver Transplantation for Cholangiocarcinoma
Specific patients with perihilar cholangiocarcinoma, recognized through exceptional criteria established by the United Network of Organ Sharing, can receive a liver from a deceased donor. Previously considered an absolute contra-indication, patients with unresectable early-stage (stage I–II) peri-hilar cholangiocarcinoma that is 3 cm or less might benefit from liver transplantation.12, 120 These individuals receive neoadjuvant therapy with external beam radiation, radiosensitizing chemotherapy, endoscopic brachytherapy, and oral capecitabine before exploratory laparotomy, to verify their candidacy for liver transplantation.120 The 5 y rate of recurrence-free survival was 65% when this method was used at dozen transplant centers—all had similar survival rates.120 More patients with PSC appeared to remain free of recurrence compared to those without PSC, though this was not statistically significant.120
Diagnosis and Management of Recurrent PSC
PSC is estimated to recur in 20%–25% of recipients over a 10 y period after liver transplantation.12 The definition of recurrent PSC requires the exclusion of other confounding events that could cause non-anastomotic biliary strictures, including incompatible blood types, cytomegalovirus infection, and hepatic artery thrombosis.121 Typically, PSC recurs more than 90 days after liver transplantation.122 Interestingly, a large study showed that patients with IBD and an intact colon were at increased risk for PSC recurrence.122
Increasing serum levels of alkaline phosphatase can indicate PSC recurrence; the diagnosis is secured when cholangiography shows features typical of PSC in the original liver.121, 122 Histologic analysis has a secondary role in diagnosis, as it may be performed to exclude rejection. Notably, ductopenia can accompany recurrent PSC in the absence of rejection, making cholangiography necessary.122, 123 No medical therapy has been shown to prevent PSC recurrence or stop disease progression. Hepatic re-transplantation is successful for specific patients with recurrent PSC, but the disease can continue to recur.
Cancer Surveillance in Patients with PSC
Cholangiocarcinoma occurs in 1%–2% of patients annually following a diagnosis of PSC, with a lifetime risk of 5%–10%.124 Cholangiocarcinoma is frequently detected within the first 1–3 years after the initial diagnosis of PSC.125 The presence of advanced fibrosis is not required for cholangiocarcinoma development, unlike hepatocellular carcinoma, which is typically found in conjunction with cirrhosis. Increased levels of bilirubin, duration of IBD, and history of IBD-associated colorectal neoplasia have been proposed as risk factors.124, 126, 127
Cholangiocarcinoma is difficult to diagnose—particularly in trying to distinguish a benign dominant stricture from ductal cholangiocarcinoma. Often the diagnosis requires the use of multiple serum, imaging, and endoscopic tests over time. Furthermore, there are no risk stratification criteria to identify a high-risk group within the PSC population for enhanced surveillance. In the absence of systematic data, a rational approach to cholangiocarcinoma surveillance has been proposed and is currently in use at the Mayo Clinic.128 This approach involves annual MR imaging and MRCP or ultrasound examinations, plus measurement of serum carbohydrate antigen 19-9 (CA19-9). ERCP assessment is reserved for individuals with increasing levels of CA-19-9 and/or imaging evidence for dominant strictures. 128 Although tests for serum levels of CA 19-9 alone are not sensitive or specific enough to facilitate detection of cholangiocarcinoma, values above 130 U/L in the absence of bacterial cholangitis detect cholangiocarcinoma with 79% sensitivity and 98% specificity.129 The value of measuring serum CA 19-9 to detect asymptomatic or early-stage cholangiocarcinoma, however, has never been proven. The advantage of combining serum CA 19-9 measurements with imaging is therefore to improve our ability to detect worrisome extrahepatic strictures that could be cholangiocarcinomas. Alternatively, a normal level of CA 19-9 with evidence of dominant structuring would also raise suspicion for cholangiocarcinoma.
The efficacy of this approach, with several recall mechanisms for enhanced surveillance based on ERCP cytology results, is currently under investigation at the Mayo Clinic. It should be noted that the 2010 AASLD guidelines for PSC do not comment on cholangiocarcinoma screening. However, they do propose diagnostic imaging and assessment of cholangiocarcinoma by measurement of CA19-9 and imaging if a clinical indication develops (deterioration in either clinical status or liver test results).12
Cytology and Fluorescence in situ hybridization (FISH)
Routine brush cytology analysis alone detects cholangiocarcinoma with low levels of sensitivity (40% or less), although the specificity is nearly 100%.12 FISH has been shown to be more sensitive than, and as specific as cytology.130 FISH requires a probe set that is applied to subpopulations of cells with chromosome amplifications, to assess aneusomy. In the presence on aneusomy, the additional copies of chromosomes detected by the FISH probes (typically targeting peri-centromeric regions of chromosomes 3, 7, and 17 and band 9p21) are highlighted; manual counting allows one to describe the extent of aneusomy.131 At the Mayo Clinic, FISH results are assigned to 1 of 3 categories: negative, trisomy (10 or more cells show 3 copies of chromosomes 7 or 3, and 1 or 2 copies of the other probes) or tetrasomy (10 or more cells show 4 copies of all probes), and polysomy (5 or more cells have gained 2 or more chromosomes).131
In a large study of 235 PSC patients, individuals with polysomy (compared to those with trisomy or tetrasomy or negative FISH results) had similar outcomes to those with proven cholangiocarcinoma (Figure 3).131 Among patients with PSC and a dominant stricture, the presence of polysomy identified those with an eventual diagnosis of cholangiocarcinoma with 88% specificity.131 The value of FISH was confirmed by the finding that 75% of individuals with persistent polysomy in more than one test develop cholangiocarcinoma after 3 years.132 Proteomic analyses of urine and bile, along with a variety of serologic markers, are being examined to aid in the diagnosis of cholangiocarcinoma.133–135
Figure 3.
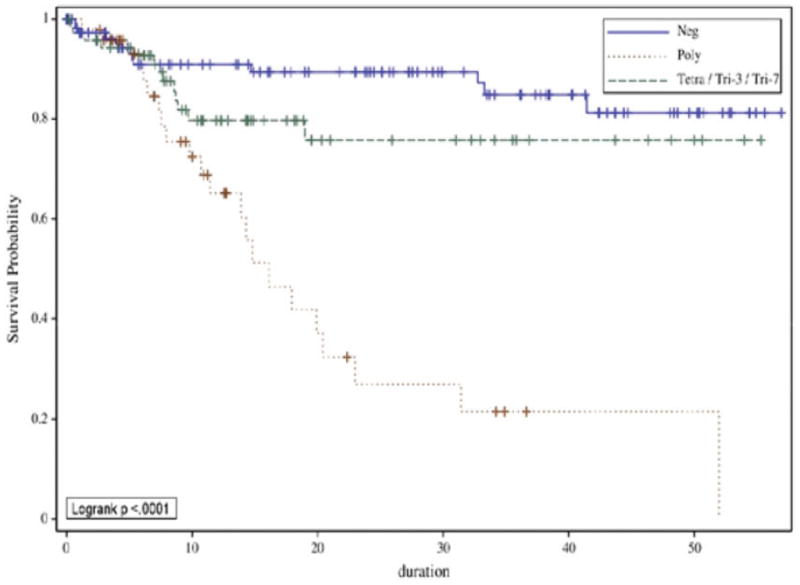
Survival of Patients with PSC with Polysomy, Trisomy 3/7 or Tetrasomy, or no Chromosome Amplifications, Based on FISH Analysis.
Reused with permission from Lindor and colleagues.131
Gallbladder Neoplasia
Gallbladder disease such as cholelithiasis is common among patients with PSC. 136 In addition, PSC patients have an increased risk of gallbladder neoplasia. Although the exact prevalence of neoplasia is not known, one series described among PSC patients that underwent a cholecystectomy, 14% had a gallbladder mass which was malignant in 57% of cases.137 Current AASLD guidelines recommend an annual ultrasound to detect lesions in the gallbladder, and cholecystectomy if lesions are detected, regardless of size.12 Despite this recommendation, the likelihood of malignancy in small gallbladder polyps is unclear. One study did not find neoplasia in polyps less than 0.8 cm but reported 40% morbidity associated with cholecystectomy in PSC patients—particularly among those with advanced liver disease.138 Although there have been rare reports of small polyps harboring malignancies, physicians should weigh the risks and benefits of a cholecystectomy (vs monitoring) for patients with small polyps and advanced liver disease from PSC, without other high-risk features.15
Colorectal Neoplasia
The combination of PSC and UC is associated with a 4-fold increase in risk of colorectal neoplasia (CRN), compared to UC alone.42 PSC patients with Crohn’s disease also have an increased risk of CRN.139 It is common to detect CRN shortly after the 2 conditions are discovered.140
Colonoscopic Surveillance
With the initial diagnosis of PSC in subjects with IBD, immediate and annual surveillance colonoscopy and biopsy analysis, throughout the colon, is recommended.12, 140 The risk is not reduced by liver transplantation, so annual surveillance should continue following this procedure.12 Surveillance biopsies provide limited information, because only a small surface area of the entire colon is examined. Researchers are therefore investigating the ability of other modalities, such as chromoendoscopy, to increase rates of neoplasia detection in this population. In clinical practice, we increase the frequency of surveillance and use chromoendoscopy for patients with indefinite or low-grade dysplasia (LGD) who elect not to undergo colectomy. Raised areas that are suspected to be neoplastic should be removed (if possible), and surrounding biopsies should be collected to evaluate for colitis and endoscopically flat neoplasia.
Following ileal–anal pouch anastomosis in patients with PSC-IBD, the pouch is still at risk for cancer. 141 Some clinicians have proposed annual surveillance of the pouch for patients with PSC, although there is little evidence to support this strategy.142
Indications for Colectomy
Indications for colectomy in PSC-IBD patients include cancer, high-grade dysplasia, and unresectable LGD.128, 143 Although few data are available from patients with PSC-IBD about sporadic adenomas (identified by endoscopic examination, raised lesions outside regions of colitis, with normal surrounding tissue based on biopsy analysis), it may be reasonable to follow these individuals with endoscopic examinations—the typical practice for patients with only UC, particularly if only a small, single sporadic adenoma is detected.144 Notably, these lesions are rarely encountered, due to the high prevalence of pancolitis.38 The fate of unifocal or multi-focal LGDs (flat or raised) in regions of colitis are not well defined in PSC-IBD patients. However, the 1 y cumulative incidence of high-grade dysplasia or cancer in patients with PSC-UC and a diagnosis of flat or adenoma-like dysplasia is 25%, whereas multifocal LGD was associated with neoplasia recurrence or progression.145 These findings support the recommendation of colectomy for patients with multi-focal LGD that is flat and found in areas of colitis.
Evidence for UDCA as Chemoprevention Agent
UDCA has been proposed to decrease the risk of CRN in patients with PSC and UC,146, 147 based on results from retrospective studies. However, these studies had inconsistent results, so UDCA has not been recommended a chemopreventative agent for patients with PSC-IBD.12, 105, 148–150
Future Directions
Novel Pharmacological Therapies
Thus far, antibodies against TNF have been ineffective in patients with PSC.108, 151 The role of biologic agents such as ustekinumab and vedolizumab in the treatment of IBD is being investigated, and their role in treatment of PSC-IBD remains to be seen. 152, 153 Given the possible role of lymphocyte trafficking in the pathogenesis of PSC, monoclonal antibodies that alter this process could have a potential therapeutic benefit.
Janus kinase (JAK) is a tyrosine kinase component of signaling pathways for many cytokines.154 Tofacitinib is a JAK inhibitor that decreases signaling by inflammatory cytokines, T-cell differentiation, and lipopolysaccharide-induced innate immune responses.155–157 JAK kinase signaling is therefore a therapeutic target for many autoimmune and inflammatory disorders.154 Recently, tofacitinib was examined for the treatment of UC.155
Inhibiting progressive fibrosis is an attractive target, and tyrosine kinase receptors appear to have important roles in fibrosis.158 Imatinib is a tyrosine kinase inhibitor that has anti-fibrotic properties in livers of animal models.159 In the clinic, it is used to treat gastrointestinal stromal tumors and chronic myelogenous leukemia. Imatinib has been reported to have hepatotoxic effects, and the safety of this agent in PSC patients is unknown.160 Other agents with potential anti-fibrotic properties include angiotensin receptor blockers, colchicine, and pentoxifylline.161, 162 However colchicine and pentoxifylline have not shown benefit in patients with PSC.163, 164 The combination of all-trans retinoic acid and UDCA has been shown to reduce bile duct proliferation and fibrosis in animal models.165 The efficacy of this combination is under investigation.
Farnesoid X receptor (FXR) is a nuclear receptor that regulates bile acid homeostasis.166, 167 When activated, FXR limits hepatic bile acid accumulation and upregulates bilirubin and phospholipid export pumps. FXR activation is also involved in anti-bacterial defense and protects against inflammation.167 Obeticholic acid is an FXR agonist under investigation for primary biliary cirrhosis. FXR agonists have potent choleretic activity, so in patients with downstream obstructions (biliary strictures), they might worsen liver disease by increasing biliary pressure.166 Studies of FXR agonists in patients with PSC should therefore be performed with great care.
24-norUrsodeoxycholic acid is a derivative of UDCA.168 It has been shown to improve liver test results and histologic features, compared to UDCA, in animal models of PSC.169 Phase 2 clinical trials are underway to determine the efficacy of this agent in PSC patients.
The therapeutic potential of the above agents is largely speculative. Preclinical and small pilot studies are needed to investigate their mechanisms of action, safety, and potential efficacy.
Validation of Common and Novel Clinical Trial Endpoints
PSC is a rare, slowly progressive disease, so it is a challenge to design clinical trials powered to capture differences in relevant clinical endpoints. Validated markers of disease progression are important. A retrospective study associated normalization of alkaline phosphatase level with an improved prognosis.18 Another retrospective study associated reduction of alkaline phosphatase levels to below 1.5-fold the upper limit of normal with better outcomes including a decrease in the development of cholangiocarcinoma.170 Additional biomarkers would improve the sensitivity and specificity of algorithms that predict disease progression and outcome of these subjects. Other approaches to monitor disease progression and response to therapy include MR and CT cholangiography and measurements of liver stiffness with elastography.
Conclusions
PSC is a rare but important cholestatic liver disease that reduces patients survival and quality of life. Management of patients involves early recognition of the disorder, implementation of routine screening protocols to identify complications (Figure 4), and treating comorbid conditions. In the absence of effective medical therapy for the disease itself, treatment centers on endoscopic management and referral for liver transplantation, when necessary.
Figure 4.
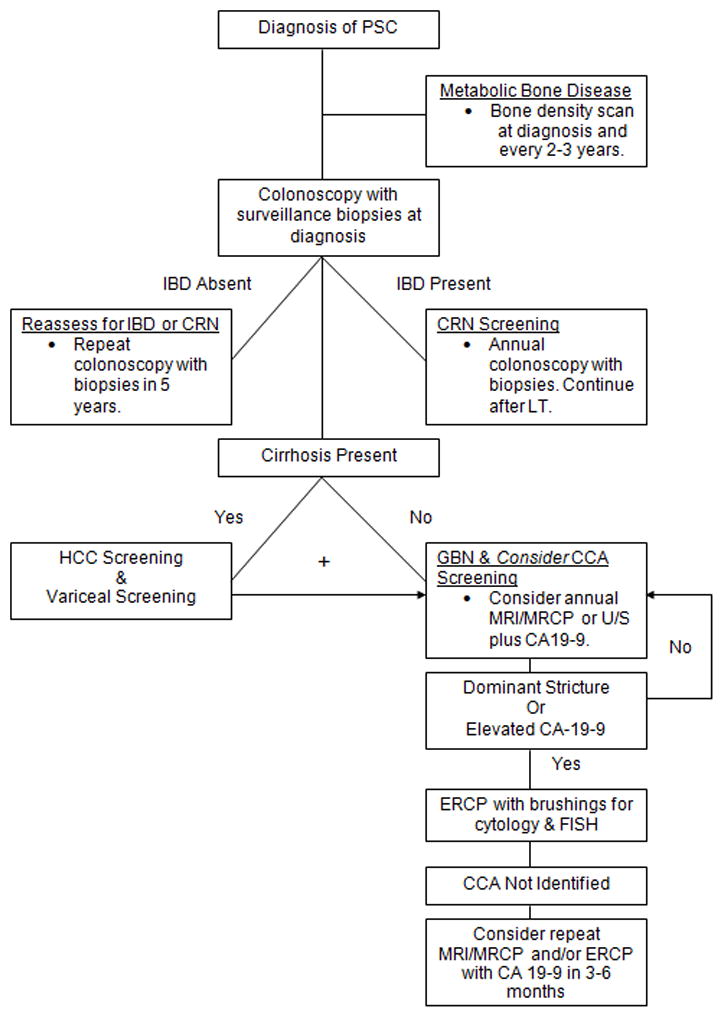
Surveillance of Co-Morbid Conditions in Patients with PSC.
Abbreviations: PSC (primary sclerosing cholangitis); IBD (inflammatory bowel disease); CRN (colorectal neoplasia); HCC (hepatocellular carcinoma); GBN (gallbladder neoplasia); CCA (cholangiocarcinoma); ERCP (endoscopic retrograde cholangiopancreatography); MRCP (magnetic resonance cholangiopancreatography); FISH (fluorescence in situ hybridization).
Although our understanding of PSC and its comorbid conditions has improved, much is left to learn about its pathogenesis. Animal models and genomic-based approaches will increase our understanding of the pathophysiology, and hopefully lead to new therapeutic strategies.
Acknowledgments
Grant Support: Supported by NIH grants DK 56924 (JAT), DK 84960 (KNL), DK 63947 (GJG).
Abbreviations
- PSC
primary sclerosing cholangitis
- IBD
inflammatory bowel disease
- UC
ulcerative colitis
- AIH
autoimmune hepatitis
- ERCP
endoscopic retrograde cholangiopancreatography
- MRCP
magnetic resonance cholangiopancreatography
- IAC
IgG4-associated sclerosing cholangitis
- AIP
autoimmune pancreatitis
- VCAM-1
vascular cell adhesion molecule-1
- M MAdCAM-1
mucosal addressin cellular adhesion molecule 1
- MDR3
multidrug resistance protein 3
- CFTR
Cystic fibrosis transmembrane conductance regulator
- CA 19-9
carbohydrate antigen 19-9
- HLA
human leukocyte antigen
- MELD
Model for End-Stage Liver Disease
- FISH
fluorescence in situ hybridization
- CRN
colorectal neoplasia
- LGD
low-grade dysplasia
- IL
interleukin
- JAK
janus kinase
- FXR
farnesoid X receptor
Footnotes
Disclosures: The authors have nothing to disclose
Transcript Profile: None.
Writing Assistance: Not utilized.
Author Contributions: All authors were involved in the drafting and critical revision of the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Berdal JE, Ebbesen J, Rydning A. Incidence and prevalence of autoimmune liver diseases. Tidsskrift for den Norske laegeforening: tidsskrift for praktisk medicin, ny raekke. 1998;118:4517–9. [PubMed] [Google Scholar]
- 2.Lindkvist B, Benito de Valle M, Gullberg B, Bjornsson E. Incidence and prevalence of primary sclerosing cholangitis in a defined adult population in Sweden. Hepatology. 2010;52:571–7. doi: 10.1002/hep.23678. [DOI] [PubMed] [Google Scholar]
- 3.Bambha K, Kim WR, Talwalkar J, Torgerson H, Benson JT, Therneau TM, Loftus EV, Jr, Yawn BP, Dickson ER, Melton LJ., 3rd Incidence, clinical spectrum, and outcomes of primary sclerosing cholangitis in a United States community. Gastroenterology. 2003;125:1364–9. doi: 10.1016/j.gastro.2003.07.011. [DOI] [PubMed] [Google Scholar]
- 4.Kaplan GG, Laupland KB, Butzner D, Urbanski SJ, Lee SS. The burden of large and small duct primary sclerosing cholangitis in adults and children: a population-based analysis. The American journal of gastroenterology. 2007;102:1042–9. doi: 10.1111/j.1572-0241.2007.01103.x. [DOI] [PubMed] [Google Scholar]
- 5.Molodecky NA, Kareemi H, Parab R, Barkema HW, Quan H, Myers RP, Kaplan GG. Incidence of primary sclerosing cholangitis: a systematic review and meta-analysis. Hepatology. 2011;53:1590–9. doi: 10.1002/hep.24247. [DOI] [PubMed] [Google Scholar]
- 6.Escorsell A, Pares A, Rodes J, Solis-Herruzo JA, Miras M, de la Morena E. Epidemiology of primary sclerosing cholangitis in Spain. Spanish Association for the Study of the Liver. Journal of hepatology. 1994;21:787–91. doi: 10.1016/s0168-8278(94)80240-8. [DOI] [PubMed] [Google Scholar]
- 7.Ang TL, Fock KM, Ng TM, Teo EK, Chua TS, Tan JY. Clinical profile of primary sclerosing cholangitis in Singapore. Journal of gastroenterology and hepatology. 2002;17:908–13. doi: 10.1046/j.1440-1746.2002.02835.x. [DOI] [PubMed] [Google Scholar]
- 8.Porayko MK, Wiesner RH, LaRusso NF, Ludwig J, MacCarty RL, Steiner BL, Twomey CK, Zinsmeister AR. Patients with asymptomatic primary sclerosing cholangitis frequently have progressive disease. Gastroenterology. 1990;98:1594–602. doi: 10.1016/0016-5085(90)91096-o. [DOI] [PubMed] [Google Scholar]
- 9.Broome U, Olsson R, Loof L, Bodemar G, Hultcrantz R, Danielsson A, Prytz H, Sandberg-Gertzen H, Wallerstedt S, Lindberg G. Natural history and prognostic factors in 305 Swedish patients with primary sclerosing cholangitis. Gut. 1996;38:610–5. doi: 10.1136/gut.38.4.610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ponsioen CY, Vrouenraets SM, Prawirodirdjo W, Rajaram R, Rauws EA, Mulder CJ, Reitsma JB, Heisterkamp SH, Tytgat GN. Natural history of primary sclerosing cholangitis and prognostic value of cholangiography in a Dutch population. Gut. 2002;51:562–6. doi: 10.1136/gut.51.4.562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wellge BE, Sterneck M, Teufel A, Rust C, Franke A, Schreiber S, Berg T, Gunther R, Kreisel W, Zu Eulenburg C, Braun F, Beuers U, Galle PR, Lohse AW, Schramm C. Pregnancy in primary sclerosing cholangitis. Gut. 2011;60:1117–21. doi: 10.1136/gut.2010.228924. [DOI] [PubMed] [Google Scholar]
- 12.Chapman R, Fevery J, Kalloo A, Nagorney DM, Boberg KM, Shneider B, Gores GJ. Diagnosis and management of primary sclerosing cholangitis. Hepatology. 2010;51:660–78. doi: 10.1002/hep.23294. [DOI] [PubMed] [Google Scholar]
- 13.Olsson R, Danielsson A, Jarnerot G, Lindstrom E, Loof L, Rolny P, Ryden BO, Tysk C, Wallerstedt S. Prevalence of primary sclerosing cholangitis in patients with ulcerative colitis. Gastroenterology. 1991;100:1319–23. [PubMed] [Google Scholar]
- 14.Bergquist A, Lindberg G, Saarinen S, Broome U. Increased prevalence of primary sclerosing cholangitis among first-degree relatives. Journal of hepatology. 2005;42:252–6. doi: 10.1016/j.jhep.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 15.Mitchell SA, Thyssen M, Orchard TR, Jewell DP, Fleming KA, Chapman RW. Cigarette smoking, appendectomy, and tonsillectomy as risk factors for the development of primary sclerosing cholangitis: a case control study. Gut. 2002;51:567–73. doi: 10.1136/gut.51.4.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim WR, Therneau TM, Wiesner RH, Poterucha JJ, Benson JT, Malinchoc M, LaRusso NF, Lindor KD, Dickson ER. A revised natural history model for primary sclerosing cholangitis. Mayo Clinic proceedings Mayo Clinic. 2000;75:688–94. doi: 10.4065/75.7.688. [DOI] [PubMed] [Google Scholar]
- 17.Lee YM, Kaplan MM. Management of primary sclerosing cholangitis. The American journal of gastroenterology. 2002;97:528–34. doi: 10.1111/j.1572-0241.2002.05585.x. [DOI] [PubMed] [Google Scholar]
- 18.Stanich PP, Bjornsson E, Gossard AA, Enders F, Jorgensen R, Lindor KD. Alkaline phosphatase normalization is associated with better prognosis in primary sclerosing cholangitis. Digestive and liver disease: official journal of the Italian Society of Gastroenterology and the Italian Association for the Study of the Liver. 2011;43:309–13. doi: 10.1016/j.dld.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Angulo P, Peter JB, Gershwin ME, DeSotel CK, Shoenfeld Y, Ahmed AE, Lindor KD. Serum autoantibodies in patients with primary sclerosing cholangitis. Journal of hepatology. 2000;32:182–7. doi: 10.1016/s0168-8278(00)80061-6. [DOI] [PubMed] [Google Scholar]
- 20.MacCarty RL, LaRusso NF, Wiesner RH, Ludwig J. Primary sclerosing cholangitis: findings on cholangiography and pancreatography. Radiology. 1983;149:39–44. doi: 10.1148/radiology.149.1.6412283. [DOI] [PubMed] [Google Scholar]
- 21.Dave M, Elmunzer BJ, Dwamena BA, Higgins PD. Primary sclerosing cholangitis: meta-analysis of diagnostic performance of MR cholangiopancreatography. Radiology. 2010;256:387–96. doi: 10.1148/radiol.10091953. [DOI] [PubMed] [Google Scholar]
- 22.Kaltenthaler E, Vergel YB, Chilcott J, Thomas S, Blakeborough T, Walters SJ, Bouchier H. A systematic review and economic evaluation of magnetic resonance cholangiopancreatography compared with diagnostic endoscopic retrograde cholangiopancreatography. Health technology assessment. 2004;8:iii, 1–89. doi: 10.3310/hta8100. [DOI] [PubMed] [Google Scholar]
- 23.Talwalkar JA, Angulo P, Johnson CD, Petersen BT, Lindor KD. Cost-minimization analysis of MRC versus ERCP for the diagnosis of primary sclerosing cholangitis. Hepatology. 2004;40:39–45. doi: 10.1002/hep.20287. [DOI] [PubMed] [Google Scholar]
- 24.Scheuer PJ. Ludwig Symposium on biliary disorders--part II. Pathologic features and evolution of primary biliary cirrhosis and primary sclerosing cholangitis. Mayo Clinic proceedings Mayo Clinic. 1998;73:179–83. doi: 10.4065/73.2.179. [DOI] [PubMed] [Google Scholar]
- 25.Burak KW, Angulo P, Lindor KD. Is there a role for liver biopsy in primary sclerosing cholangitis? The American journal of gastroenterology. 2003;98:1155–8. doi: 10.1111/j.1572-0241.2003.07401.x. [DOI] [PubMed] [Google Scholar]
- 26.Ludwig JLN, Wiesner RH. Primary sclerosing cholangitis. Contemporary issues in surgical pathology: liver pathology. New York: Churchill Livingstone; 1986. pp. 193–213. [Google Scholar]
- 27.Corpechot C, El Naggar A, Poujol-Robert A, Ziol M, Wendum D, Chazouilleres O, de Ledinghen V, Dhumeaux D, Marcellin P, Beaugrand M, Poupon R. Assessment of biliary fibrosis by transient elastography in patients with PBC and PSC. Hepatology. 2006;43:1118–24. doi: 10.1002/hep.21151. [DOI] [PubMed] [Google Scholar]
- 28.Kojima E, Kimura K, Noda Y, Kobayashi G, Itoh K, Fujita N. Autoimmune pancreatitis and multiple bile duct strictures treated effectively with steroid. Journal of gastroenterology. 2003;38:603–7. [PubMed] [Google Scholar]
- 29.Bjornsson E, Chari ST, Smyrk TC, Lindor K. Immunoglobulin G4 associated cholangitis: description of an emerging clinical entity based on review of the literature. Hepatology. 2007;45:1547–54. doi: 10.1002/hep.21685. [DOI] [PubMed] [Google Scholar]
- 30.Ghazale A, Chari ST, Zhang L, Smyrk TC, Takahashi N, Levy MJ, Topazian MD, Clain JE, Pearson RK, Petersen BT, Vege SS, Lindor K, Farnell MB. Immunoglobulin G4-associated cholangitis: clinical profile and response to therapy. Gastroenterology. 2008;134:706–15. doi: 10.1053/j.gastro.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 31.Ohara H, Okazaki K, Tsubouchi H, Inui K, Kawa S, Kamisawa T, Tazuma S, Uchida K, Hirano K, Yoshida H, Nishino T, Ko SB, Mizuno N, Hamano H, Kanno A, Notohara K, Hasebe O, Nakazawa T, Nakanuma Y, Takikawa H. Clinical diagnostic criteria of IgG4-related sclerosing cholangitis 2012. Journal of hepato-biliary-pancreatic sciences. 2012;19:536–42. doi: 10.1007/s00534-012-0521-y. [DOI] [PubMed] [Google Scholar]
- 32.Bjornsson E, Chari S, Silveira M, Gossard A, Takahashi N, Smyrk T, Lindor K. Primary sclerosing cholangitis associated with elevated immunoglobulin G4: clinical characteristics and response to therapy. American journal of therapeutics. 2011;18:198–205. doi: 10.1097/MJT.0b013e3181c9dac6. [DOI] [PubMed] [Google Scholar]
- 33.Mendes FD, Jorgensen R, Keach J, Katzmann JA, Smyrk T, Donlinger J, Chari S, Lindor KD. Elevated serum IgG4 concentration in patients with primary sclerosing cholangitis. The American journal of gastroenterology. 2006;101:2070–5. doi: 10.1111/j.1572-0241.2006.00772.x. [DOI] [PubMed] [Google Scholar]
- 34.Zhang L, Lewis JT, Abraham SC, Smyrk TC, Leung S, Chari ST, Poterucha JJ, Rosen CB, Lohse CM, Katzmann JA, Wu TT. IgG4+ plasma cell infiltrates in liver explants with primary sclerosing cholangitis. The American journal of surgical pathology. 2010;34:88–94. doi: 10.1097/PAS.0b013e3181c6c09a. [DOI] [PubMed] [Google Scholar]
- 35.Oseini AM, Chaiteerakij R, Shire AM, Ghazale A, Kaiya J, Moser CD, Aderca I, Mettler TA, Therneau TM, Zhang L, Takahashi N, Chari ST, Roberts LR. Utility of serum immunoglobulin G4 in distinguishing immunoglobulin G4-associated cholangitis from cholangiocarcinoma. Hepatology. 2011;54:940–8. doi: 10.1002/hep.24487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Verdonk RC, Dijkstra G, Haagsma EB, Shostrom VK, Van den Berg AP, Kleibeuker JH, Langnas AN, Sudan DL. Inflammatory bowel disease after liver transplantation: risk factors for recurrence and de novo disease. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2006;6:1422–9. doi: 10.1111/j.1600-6143.2006.01333.x. [DOI] [PubMed] [Google Scholar]
- 37.Sinakos E, Samuel S, Enders F, Loftus EV, Jr, Sandborn WJ, Lindor KD. Inflammatory Bowel Disease in Primary Sclerosing Cholangitis: A Robust yet Changing Relationship. Inflammatory bowel diseases. 2013;19:1004–1009. doi: 10.1097/MIB.0b013e3182802893. [DOI] [PubMed] [Google Scholar]
- 38.Loftus EV, Jr, Harewood GC, Loftus CG, Tremaine WJ, Harmsen WS, Zinsmeister AR, Jewell DA, Sandborn WJ. PSC-IBD: a unique form of inflammatory bowel disease associated with primary sclerosing cholangitis. Gut. 2005;54:91–6. doi: 10.1136/gut.2004.046615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boonstra K, van Erpecum KJ, van Nieuwkerk KM, Drenth JP, Poen AC, Witteman BJ, Tuynman HA, Beuers U, Ponsioen CY. Primary sclerosing cholangitis is associated with a distinct phenotype of inflammatory bowel disease. Inflammatory bowel diseases. 2012;18:2270–6. doi: 10.1002/ibd.22938. [DOI] [PubMed] [Google Scholar]
- 40.Joo M, Abreu-e-Lima P, Farraye F, Smith T, Swaroop P, Gardner L, Lauwers GY, Odze RD. Pathologic features of ulcerative colitis in patients with primary sclerosing cholangitis: a case-control study. The American journal of surgical pathology. 2009;33:854–62. doi: 10.1097/PAS.0b013e318196d018. [DOI] [PubMed] [Google Scholar]
- 41.Jorgensen KK, Grzyb K, Lundin KE, Clausen OP, Aamodt G, Schrumpf E, Vatn MH, Boberg KM. Inflammatory bowel disease in patients with primary sclerosing cholangitis: clinical characterization in liver transplanted and nontransplanted patients. Inflammatory bowel diseases. 2012;18:536–45. doi: 10.1002/ibd.21699. [DOI] [PubMed] [Google Scholar]
- 42.Soetikno RM, Lin OS, Heidenreich PA, Young HS, Blackstone MO. Increased risk of colorectal neoplasia in patients with primary sclerosing cholangitis and ulcerative colitis: a meta-analysis. Gastrointestinal endoscopy. 2002;56:48–54. doi: 10.1067/mge.2002.125367. [DOI] [PubMed] [Google Scholar]
- 43.Penna C, Dozois R, Tremaine W, Sandborn W, LaRusso N, Schleck C, Ilstrup D. Pouchitis after ileal pouch-anal anastomosis for ulcerative colitis occurs with increased frequency in patients with associated primary sclerosing cholangitis. Gut. 1996;38:234–9. doi: 10.1136/gut.38.2.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wiesner RH, LaRusso NF, Dozois RR, Beaver SJ. Peristomal varices after proctocolectomy in patients with primary sclerosing cholangitis. Gastroenterology. 1986;90:316–22. doi: 10.1016/0016-5085(86)90926-1. [DOI] [PubMed] [Google Scholar]
- 45.Chapman RW, Arborgh BA, Rhodes JM, Summerfield JA, Dick R, Scheuer PJ, Sherlock S. Primary sclerosing cholangitis: a review of its clinical features, cholangiography, and hepatic histology. Gut. 1980;21:870–7. doi: 10.1136/gut.21.10.870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fevery J, Henckaerts L, Van Oirbeek R, Vermeire S, Rutgeerts P, Nevens F, Van Steenbergen W. Malignancies and mortality in 200 patients with primary sclerosering cholangitis: a long-term single-centre study. Liver international: official journal of the International Association for the Study of the Liver. 2012;32:214–22. doi: 10.1111/j.1478-3231.2011.02575.x. [DOI] [PubMed] [Google Scholar]
- 47.Marelli L, Xirouchakis E, Kalambokis G, Cholongitas E, Hamilton MI, Burroughs AK. Does the severity of primary sclerosing cholangitis influence the clinical course of associated ulcerative colitis? Gut. 2011;60:1224–8. doi: 10.1136/gut.2010.235408. [DOI] [PubMed] [Google Scholar]
- 48.Navaneethan U, Venkatesh PG, Mukewar S, Lashner BA, Remzi FH, McCullough AJ, Kiran RP, Shen B, Fung JJ. Progressive primary sclerosing cholangitis requiring liver transplantation is associated with reduced need for colectomy in patients with ulcerative colitis. Clinical gastroenterology and hepatology: the official clinical practice journal of the American Gastroenterological Association. 2012;10:540–6. doi: 10.1016/j.cgh.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 49.Angulo P, Maor-Kendler Y, Lindor KD. Small-duct primary sclerosing cholangitis: a long-term follow-up study. Hepatology. 2002;35:1494–500. doi: 10.1053/jhep.2002.33202. [DOI] [PubMed] [Google Scholar]
- 50.Bjornsson E, Olsson R, Bergquist A, Lindgren S, Braden B, Chapman RW, Boberg KM, Angulo P. The natural history of small-duct primary sclerosing cholangitis. Gastroenterology. 2008;134:975–80. doi: 10.1053/j.gastro.2008.01.042. [DOI] [PubMed] [Google Scholar]
- 51.Bjornsson E, Boberg KM, Cullen S, Fleming K, Clausen OP, Fausa O, Schrumpf E, Chapman RW. Patients with small duct primary sclerosing cholangitis have a favourable long term prognosis. Gut. 2002;51:731–5. doi: 10.1136/gut.51.5.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Al-Chalabi T, Portmann BC, Bernal W, McFarlane IG, Heneghan MA. Autoimmune hepatitis overlap syndromes: an evaluation of treatment response, long-term outcome and survival. Alimentary pharmacology & therapeutics. 2008;28:209–20. doi: 10.1111/j.1365-2036.2008.03722.x. [DOI] [PubMed] [Google Scholar]
- 53.Kaya M, Angulo P, Lindor KD. Overlap of autoimmune hepatitis and primary sclerosing cholangitis: an evaluation of a modified scoring system. Journal of hepatology. 2000;33:537–42. doi: 10.1034/j.1600-0641.2000.033004537.x. [DOI] [PubMed] [Google Scholar]
- 54.Abdalian R, Dhar P, Jhaveri K, Haider M, Guindi M, Heathcote EJ. Prevalence of sclerosing cholangitis in adults with autoimmune hepatitis: evaluating the role of routine magnetic resonance imaging. Hepatology. 2008;47:949–57. doi: 10.1002/hep.22073. [DOI] [PubMed] [Google Scholar]
- 55.Trivedi PJ, Hirschfield GM. Review article: overlap syndromes and autoimmune liver disease. Alimentary pharmacology & therapeutics. 2012;36:517–33. doi: 10.1111/j.1365-2036.2012.05223.x. [DOI] [PubMed] [Google Scholar]
- 56.Boberg KM, Chapman RW, Hirschfield GM, Lohse AW, Manns MP, Schrumpf E. Overlap syndromes: the International Autoimmune Hepatitis Group (IAIHG) position statement on a controversial issue. Journal of hepatology. 2011;54:374–85. doi: 10.1016/j.jhep.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 57.Pollheimer MJ, Halilbasic E, Fickert P, Trauner M. Pathogenesis of primary sclerosing cholangitis. Best practice & research Clinical gastroenterology. 2011;25:727–39. doi: 10.1016/j.bpg.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Spurkland A, Saarinen S, Boberg KM, Mitchell S, Broome U, Caballeria L, Ciusani E, Chapman R, Ercilla G, Fausa O, Knutsen I, Pares A, Rosina F, Olerup O, Thorsby E, Schrumpf E. HLA class II haplotypes in primary sclerosing cholangitis patients from five European populations. Tissue antigens. 1999;53:459–69. doi: 10.1034/j.1399-0039.1999.530502.x. [DOI] [PubMed] [Google Scholar]
- 59.Farrant JM, Doherty DG, Donaldson PT, Vaughan RW, Hayllar KM, Welsh KI, Eddleston AL, Williams R. Amino acid substitutions at position 38 of the DR beta polypeptide confer susceptibility to and protection from primary sclerosing cholangitis. Hepatology. 1992;16:390–5. doi: 10.1002/hep.1840160217. [DOI] [PubMed] [Google Scholar]
- 60.Mehal WZ, Lo YM, Wordsworth BP, Neuberger JM, Hubscher SC, Fleming KA, Chapman RW. HLA DR4 is a marker for rapid disease progression in primary sclerosing cholangitis. Gastroenterology. 1994;106:160–7. doi: 10.1016/s0016-5085(94)95085-7. [DOI] [PubMed] [Google Scholar]
- 61.Olerup O, Olsson R, Hultcrantz R, Broome U. HLA-DR and HLA-DQ are not markers for rapid disease progression in primary sclerosing cholangitis. Gastroenterology. 1995;108:870–8. doi: 10.1016/0016-5085(95)90463-8. [DOI] [PubMed] [Google Scholar]
- 62.Karlsen TH, Franke A, Melum E, Kaser A, Hov JR, Balschun T, Lie BA, Bergquist A, Schramm C, Weismuller TJ, Gotthardt D, Rust C, Philipp EE, Fritz T, Henckaerts L, Weersma RK, Stokkers P, Ponsioen CY, Wijmenga C, Sterneck M, Nothnagel M, Hampe J, Teufel A, Runz H, Rosenstiel P, Stiehl A, Vermeire S, Beuers U, Manns MP, Schrumpf E, Boberg KM, Schreiber S. Genome-wide association analysis in primary sclerosing cholangitis. Gastroenterology. 2010;138:1102–11. doi: 10.1053/j.gastro.2009.11.046. [DOI] [PubMed] [Google Scholar]
- 63.Melum E, Franke A, Schramm C, Weismuller TJ, Gotthardt DN, Offner FA, Juran BD, Laerdahl JK, Labi V, Bjornsson E, Weersma RK, Henckaerts L, Teufel A, Rust C, Ellinghaus E, Balschun T, Boberg KM, Ellinghaus D, Bergquist A, Sauer P, Ryu E, Hov JR, Wedemeyer J, Lindkvist B, Wittig M, Porte RJ, Holm K, Gieger C, Wichmann HE, Stokkers P, Ponsioen CY, Runz H, Stiehl A, Wijmenga C, Sterneck M, Vermeire S, Beuers U, Villunger A, Schrumpf E, Lazaridis KN, Manns MP, Schreiber S, Karlsen TH. Genome-wide association analysis in primary sclerosing cholangitis identifies two non-HLA susceptibility loci. Nature genetics. 2011;43:17–9. doi: 10.1038/ng.728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu JZ, Hov JR, Folseraas T, Ellinghaus E, Rushbrook SM, Doncheva NT, Andreassen OA, Weersma RK, Weismuller TJ, Eksteen B, Invernizzi P, Hirschfield GM, Gotthardt DN, Pares A, Ellinghaus D, Shah T, Juran BD, Milkiewicz P, Rust C, Schramm C, Muller T, Srivastava B, Dalekos G, Nothen MM, Herms S, Winkelmann J, Mitrovic M, Braun F, Ponsioen CY, Croucher PJ, Sterneck M, Teufel A, Mason AL, Saarela J, Leppa V, Dorfman R, Alvaro D, Floreani A, Onengut-Gumuscu S, Rich SS, Thompson WK, Schork AJ, Naess S, Thomsen I, Mayr G, Konig IR, Hveem K, Cleynen I, Gutierrez-Achury J, Ricano-Ponce I, van Heel D, Bjornsson E, Sandford RN, Durie PR, Melum E, Vatn MH, Silverberg MS, Duerr RH, Padyukov L, Brand S, Sans M, Annese V, Achkar JP, Boberg KM, Marschall HU, Chazouilleres O, Bowlus CL, Wijmenga C, Schrumpf E, Vermeire S, Albrecht M, Rioux JD, Alexander G, Bergquist A, Cho J, Schreiber S, Manns MP, Farkkila M, Dale AM, Chapman RW, Lazaridis KN, Franke A, Anderson CA, Karlsen TH. Dense genotyping of immune-related disease regions identifies nine new risk loci for primary sclerosing cholangitis. Nature genetics. 2013 doi: 10.1038/ng.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.O’Mahony CA, Vierling JM. Etiopathogenesis of primary sclerosing cholangitis. Seminars in liver disease. 2006;26:3–21. doi: 10.1055/s-2006-933559. [DOI] [PubMed] [Google Scholar]
- 66.Lichtman SN, Sartor RB. Hepatobiliary injury associated with experimental small-bowel bacterial overgrowth in rats. Immunologic research. 1991;10:528–31. doi: 10.1007/BF02919752. [DOI] [PubMed] [Google Scholar]
- 67.Yamada S, Ishii M, Liang LS, Yamamoto T, Toyota T. Small duct cholangitis induced by N-formyl L-methionine L-leucine L-tyrosine in rats. Journal of gastroenterology. 1994;29:631–6. doi: 10.1007/BF02365447. [DOI] [PubMed] [Google Scholar]
- 68.Pollheimer MJ, Trauner M, Fickert P. Will we ever model PSC? – “it’s hard to be a PSC model!”. Clinics and research in hepatology and gastroenterology. 2011;35:792–804. doi: 10.1016/j.clinre.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 69.Palmer KR, Duerden BI, Holdsworth CD. Bacteriological and endotoxin studies in cases of ulcerative colitis submitted to surgery. Gut. 1980;21:851–4. doi: 10.1136/gut.21.10.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Terjung B, Spengler U. Atypical p-ANCA in PSC and AIH: a hint toward a “leaky gut”? Clinical reviews in allergy & immunology. 2009;36:40–51. doi: 10.1007/s12016-008-8088-8. [DOI] [PubMed] [Google Scholar]
- 71.Terjung B, Sohne J, Lechtenberg B, Gottwein J, Muennich M, Herzog V, Mahler M, Sauerbruch T, Spengler U. p-ANCAs in autoimmune liver disorders recognise human beta-tubulin isotype 5 and cross-react with microbial protein FtsZ. Gut. 2010;59:808–16. doi: 10.1136/gut.2008.157818. [DOI] [PubMed] [Google Scholar]
- 72.Farkkila M, Karvonen AL, Nurmi H, Nuutinen H, Taavitsainen M, Pikkarainen P, Karkkainen P. Metronidazole and ursodeoxycholic acid for primary sclerosing cholangitis: a randomized placebo-controlled trial. Hepatology. 2004;40:1379–86. doi: 10.1002/hep.20457. [DOI] [PubMed] [Google Scholar]
- 73.Silveira MG, Torok NJ, Gossard AA, Keach JC, Jorgensen RA, Petz JL, Lindor KD. Minocycline in the treatment of patients with primary sclerosing cholangitis: results of a pilot study. The American journal of gastroenterology. 2009;104:83–8. doi: 10.1038/ajg.2008.14. [DOI] [PubMed] [Google Scholar]
- 74.Sheth P, Delos Santos N, Seth A, LaRusso NF, Rao RK. Lipopolysaccharide disrupts tight junctions in cholangiocyte monolayers by a c-Src-, TLR4-, and LBP-dependent mechanism. American journal of physiology Gastrointestinal and liver physiology. 2007;293:G308–18. doi: 10.1152/ajpgi.00582.2006. [DOI] [PubMed] [Google Scholar]
- 75.Guo S, Al-Sadi R, Said HM, Ma TY. Lipopolysaccharide causes an increase in intestinal tight junction permeability in vitro and in vivo by inducing enterocyte membrane expression and localization of TLR-4 and CD14. The American journal of pathology. 2013;182:375–87. doi: 10.1016/j.ajpath.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fickert P, Fuchsbichler A, Wagner M, Zollner G, Kaser A, Tilg H, Krause R, Lammert F, Langner C, Zatloukal K, Marschall HU, Denk H, Trauner M. Regurgitation of bile acids from leaky bile ducts causes sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology. 2004;127:261–74. doi: 10.1053/j.gastro.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 77.Fickert P, Fuchsbichler A, Marschall HU, Wagner M, Zollner G, Krause R, Zatloukal K, Jaeschke H, Denk H, Trauner M. Lithocholic acid feeding induces segmental bile duct obstruction and destructive cholangitis in mice. The American journal of pathology. 2006;168:410–22. doi: 10.2353/ajpath.2006.050404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mueller T, Beutler C, Pico AH, Shibolet O, Pratt DS, Pascher A, Neuhaus P, Wiedenmann B, Berg T, Podolsky DK. Enhanced innate immune responsiveness and intolerance to intestinal endotoxins in human biliary epithelial cells contributes to chronic cholangitis. Liver international: official journal of the International Association for the Study of the Liver. 2011;31:1574–88. doi: 10.1111/j.1478-3231.2011.02635.x. [DOI] [PubMed] [Google Scholar]
- 79.Medvedev AE, Sabroe I, Hasday JD, Vogel SN. Tolerance to microbial TLR ligands: molecular mechanisms and relevance to disease. Journal of endotoxin research. 2006;12:133–50. doi: 10.1179/096805106X102255. [DOI] [PubMed] [Google Scholar]
- 80.Katt J, Schwinge D, Schoknecht T, Quaas A, Sobottka I, Burandt E, Becker C, Neurath MF, Lohse AW, Herkel J, Schramm C. increased Th17 response to pathogen stimulation in patients with primary sclerosing cholangitis. Hepatology. 2013 doi: 10.1002/hep.26447. [DOI] [PubMed] [Google Scholar]
- 81.Pohl J, Ring A, Stremmel W, Stiehl A. The role of dominant stenoses in bacterial infections of bile ducts in primary sclerosing cholangitis. European journal of gastroenterology & hepatology. 2006;18:69–74. doi: 10.1097/00042737-200601000-00012. [DOI] [PubMed] [Google Scholar]
- 82.Aron JH, Bowlus CL. The immunobiology of primary sclerosing cholangitis. Seminars in immunopathology. 2009;31:383–97. doi: 10.1007/s00281-009-0154-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Borchers AT, Shimoda S, Bowlus C, Keen CL, Gershwin ME. Lymphocyte recruitment and homing to the liver in primary biliary cirrhosis and primary sclerosing cholangitis. Seminars in immunopathology. 2009;31:309–22. doi: 10.1007/s00281-009-0167-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hillan KJ, Hagler KE, MacSween RN, Ryan AM, Renz ME, Chiu HH, Ferrier RK, Bird GL, Dhillon AP, Ferrell LD, Fong S. Expression of the mucosal vascular addressin, MAdCAM-1, in inflammatory liver disease. Liver. 1999;19:509–18. doi: 10.1111/j.1478-3231.1999.tb00084.x. [DOI] [PubMed] [Google Scholar]
- 85.Grant AJ, Lalor PF, Hubscher SG, Briskin M, Adams DH. MAdCAM-1 expressed in chronic inflammatory liver disease supports mucosal lymphocyte adhesion to hepatic endothelium (MAdCAM-1 in chronic inflammatory liver disease) Hepatology. 2001;33:1065–72. doi: 10.1053/jhep.2001.24231. [DOI] [PubMed] [Google Scholar]
- 86.Grant AJ, Lalor PF, Salmi M, Jalkanen S, Adams DH. Homing of mucosal lymphocytes to the liver in the pathogenesis of hepatic complications of inflammatory bowel disease. Lancet. 2002;359:150–7. doi: 10.1016/S0140-6736(02)07374-9. [DOI] [PubMed] [Google Scholar]
- 87.Salmi M, Kalimo K, Jalkanen S. Induction and function of vascular adhesion protein-1 at sites of inflammation. The Journal of experimental medicine. 1993;178:2255–60. doi: 10.1084/jem.178.6.2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liaskou E, Karikoski M, Reynolds GM, Lalor PF, Weston CJ, Pullen N, Salmi M, Jalkanen S, Adams DH. Regulation of mucosal addressin cell adhesion molecule 1 expression in human and mice by vascular adhesion protein 1 amine oxidase activity. Hepatology. 2011;53:661–72. doi: 10.1002/hep.24085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Eksteen B, Mora JR, Haughton EL, Henderson NC, Lee-Turner L, Villablanca EJ, Curbishley SM, Aspinall AI, von Andrian UH, Adams DH. Gut homing receptors on CD8 T cells are retinoic acid dependent and not maintained by liver dendritic or stellate cells. Gastroenterology. 2009;137:320–9. doi: 10.1053/j.gastro.2009.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xu B, Broome U, Ericzon BG, Sumitran-Holgersson S. High frequency of autoantibodies in patients with primary sclerosing cholangitis that bind biliary epithelial cells and induce expression of CD44 and production of interleukin 6. Gut. 2002;51:120–7. doi: 10.1136/gut.51.1.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Karrar A, Broome U, Sodergren T, Jaksch M, Bergquist A, Bjornstedt M, Sumitran-Holgersson S. Biliary epithelial cell antibodies link adaptive and innate immune responses in primary sclerosing cholangitis. Gastroenterology. 2007;132:1504–14. doi: 10.1053/j.gastro.2007.01.039. [DOI] [PubMed] [Google Scholar]
- 92.Kekilli M, Tunc B, Beyazit Y, Kurt M, Onal IK, Ulker A, Haznedaroglu IC. Circulating CD4+CD25+ Regulatory T Cells in the Pathobiology of Ulcerative Colitis and Concurrent Primary Sclerosing Cholangitis. Digestive diseases and sciences. 2013 doi: 10.1007/s10620-012-2511-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annual review of immunology. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 94.Oude Elferink RP, Paulusma CC. Function and pathophysiological importance of ABCB4 (MDR3 P-glycoprotein) Pflugers Archiv: European journal of physiology. 2007;453:601–10. doi: 10.1007/s00424-006-0062-9. [DOI] [PubMed] [Google Scholar]
- 95.Trauner M, Fickert P, Wagner M. MDR3 (ABCB4) defects: a paradigm for the genetics of adult cholestatic syndromes. Seminars in liver disease. 2007;27:77–98. doi: 10.1055/s-2006-960172. [DOI] [PubMed] [Google Scholar]
- 96.Durieu I, Pellet O, Simonot L, Durupt S, Bellon G, Durand DV, Minh VA. Sclerosing cholangitis in adults with cystic fibrosis: a magnetic resonance cholangiographic prospective study. Journal of hepatology. 1999;30:1052–6. doi: 10.1016/s0168-8278(99)80259-1. [DOI] [PubMed] [Google Scholar]
- 97.Sheth S, Shea JC, Bishop MD, Chopra S, Regan MM, Malmberg E, Walker C, Ricci R, Tsui LC, Durie PR, Zielenski J, Freedman SD. Increased prevalence of CFTR mutations and variants and decreased chloride secretion in primary sclerosing cholangitis. Human genetics. 2003;113:286–92. doi: 10.1007/s00439-003-0963-z. [DOI] [PubMed] [Google Scholar]
- 98.Takeda K, Kojima Y, Ikejima K, Harada K, Yamashina S, Okumura K, Aoyama T, Frese S, Ikeda H, Haynes NM, Cretney E, Yagita H, Sueyoshi N, Sato N, Nakanuma Y, Smyth MJ. Death receptor 5 mediated-apoptosis contributes to cholestatic liver disease. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:10895–900. doi: 10.1073/pnas.0802702105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lindor KD. Ursodiol for primary sclerosing cholangitis. Mayo Primary Sclerosing Cholangitis-Ursodeoxycholic Acid Study Group. The New England journal of medicine. 1997;336:691–5. doi: 10.1056/NEJM199703063361003. [DOI] [PubMed] [Google Scholar]
- 100.Mitchell SA, Bansi DS, Hunt N, Von Bergmann K, Fleming KA, Chapman RW. A preliminary trial of high-dose ursodeoxycholic acid in primary sclerosing cholangitis. Gastroenterology. 2001;121:900–7. doi: 10.1053/gast.2001.27965. [DOI] [PubMed] [Google Scholar]
- 101.Harnois DM, Angulo P, Jorgensen RA, Larusso NF, Lindor KD. High-dose ursodeoxycholic acid as a therapy for patients with primary sclerosing cholangitis. The American journal of gastroenterology. 2001;96:1558–62. doi: 10.1111/j.1572-0241.2001.03777.x. [DOI] [PubMed] [Google Scholar]
- 102.Olsson R, Boberg KM, de Muckadell OS, Lindgren S, Hultcrantz R, Folvik G, Bell H, Gangsoy-Kristiansen M, Matre J, Rydning A, Wikman O, Danielsson A, Sandberg-Gertzen H, Ung KA, Eriksson A, Loof L, Prytz H, Marschall HU, Broome U. High-dose ursodeoxycholic acid in primary sclerosing cholangitis: a 5-year multicenter, randomized, controlled study. Gastroenterology. 2005;129:1464–72. doi: 10.1053/j.gastro.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 103.Lindor KD, Kowdley KV, Luketic VA, Harrison ME, McCashland T, Befeler AS, Harnois D, Jorgensen R, Petz J, Keach J, Mooney J, Sargeant C, Braaten J, Bernard T, King D, Miceli E, Schmoll J, Hoskin T, Thapa P, Enders F. High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology. 2009;50:808–14. doi: 10.1002/hep.23082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Imam MH, Sinakos E, Gossard AA, Kowdley KV, Luketic VA, Edwyn Harrison M, McCashland T, Befeler AS, Harnois D, Jorgensen R, Petz J, Keach J, DeCook AC, Enders F, Lindor KD. High-dose ursodeoxycholic acid increases risk of adverse outcomes in patients with early stage primary sclerosing cholangitis. Alimentary pharmacology & therapeutics. 2011;34:1185–92. doi: 10.1111/j.1365-2036.2011.04863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Eaton JE, Silveira MG, Pardi DS, Sinakos E, Kowdley KV, Luketic VA, Harrison ME, McCashland T, Befeler AS, Harnois D, Jorgensen R, Petz J, Lindor KD. High-dose ursodeoxycholic acid is associated with the development of colorectal neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. The American journal of gastroenterology. 2011;106:1638–45. doi: 10.1038/ajg.2011.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Triantos CK, Koukias NM, Nikolopoulou VN, Burroughs AK. Meta-analysis: ursodeoxycholic acid for primary sclerosing cholangitis. Alimentary pharmacology & therapeutics. 2011;34:901–10. doi: 10.1111/j.1365-2036.2011.04822.x. [DOI] [PubMed] [Google Scholar]
- 107.EASL Clinical Practice Guidelines: management of cholestatic liver diseases. Journal of hepatology. 2009;51:237–67. doi: 10.1016/j.jhep.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 108.Hommes DW, Erkelens W, Ponsioen C, Stokkers P, Rauws E, van der Spek M, ten Kate F, van Deventer SJ. A double-blind, placebo-controlled, randomized study of infliximab in primary sclerosing cholangitis. Journal of clinical gastroenterology. 2008;42:522–6. doi: 10.1097/MCG.0b013e3181662426. [DOI] [PubMed] [Google Scholar]
- 109.Cullen SN, Chapman RW. Review article: current management of primary sclerosing cholangitis. Alimentary pharmacology & therapeutics. 2005;21:933–48. doi: 10.1111/j.1365-2036.2005.02407.x. [DOI] [PubMed] [Google Scholar]
- 110.Manns MP, Woynarowski M, Kreisel W, Lurie Y, Rust C, Zuckerman E, Bahr MJ, Gunther R, Hultcrantz RW, Spengler U, Lohse AW, Szalay F, Farkkila M, Prols M, Strassburg CP. Budesonide induces remission more effectively than prednisone in a controlled trial of patients with autoimmune hepatitis. Gastroenterology. 2010;139:1198–206. doi: 10.1053/j.gastro.2010.06.046. [DOI] [PubMed] [Google Scholar]
- 111.Stiehl A, Rudolph G, Kloters-Plachky P, Sauer P, Walker S. Development of dominant bile duct stenoses in patients with primary sclerosing cholangitis treated with ursodeoxycholic acid: outcome after endoscopic treatment. Journal of hepatology. 2002;36:151–6. doi: 10.1016/s0168-8278(01)00251-3. [DOI] [PubMed] [Google Scholar]
- 112.Kaya M, Petersen BT, Angulo P, Baron TH, Andrews JC, Gostout CJ, Lindor KD. Balloon dilation compared to stenting of dominant strictures in primary sclerosing cholangitis. The American journal of gastroenterology. 2001;96:1059–66. doi: 10.1111/j.1572-0241.2001.03690.x. [DOI] [PubMed] [Google Scholar]
- 113.Alkhatib AA, Hilden K, Adler DG. Comorbidities, sphincterotomy, and balloon dilation predict post-ERCP adverse events in PSC patients: operator experience is protective. Digestive diseases and sciences. 2011;56:3685–8. doi: 10.1007/s10620-011-1830-8. [DOI] [PubMed] [Google Scholar]
- 114.Ismail S, Kylanpaa L, Mustonen H, Halttunen J, Lindstrom O, Jokelainen K, Udd M, Farkkila M. Risk factors for complications of ERCP in primary sclerosing cholangitis. Endoscopy. 2012;44:1133–8. doi: 10.1055/s-0032-1325677. [DOI] [PubMed] [Google Scholar]
- 115.Bjoro K, Brandsaeter B, Foss A, Schrumpf E. Liver transplantation in primary sclerosing cholangitis. Seminars in liver disease. 2006;26:69–79. doi: 10.1055/s-2006-933565. [DOI] [PubMed] [Google Scholar]
- 116.Graziadei IW, Wiesner RH, Marotta PJ, Porayko MK, Hay JE, Charlton MR, Poterucha JJ, Rosen CB, Gores GJ, LaRusso NF, Krom RA. Long-term results of patients undergoing liver transplantation for primary sclerosing cholangitis. Hepatology. 1999;30:1121–7. doi: 10.1002/hep.510300501. [DOI] [PubMed] [Google Scholar]
- 117.Welsh FK, Wigmore SJ. Roux-en-Y Choledochojejunostomy is the method of choice for biliary reconstruction in liver transplantation for primary sclerosing cholangitis. Transplantation. 2004;77:602–4. doi: 10.1097/01.tp.0000113807.74942.d2. [DOI] [PubMed] [Google Scholar]
- 118.Goldberg DS, French B, Thomasson A, Reddy KR, Halpern SD. Current trends in living donor liver transplantation for primary sclerosing cholangitis. Transplantation. 2011;91:1148–52. doi: 10.1097/TP.0b013e31821694b3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Goldberg DS, Camp A, Martinez-Camacho A, Forman L, Fortune B, Reddy KR. Risk of waitlist mortality in patients with primary sclerosing cholangitis and bacterial cholangitis. Liver transplantation: official publication of the American Association for the Study of Liver Diseases and the International Liver Transplantation Society. 2013;19:250–8. doi: 10.1002/lt.23587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Darwish Murad S, Kim WR, Harnois DM, Douglas DD, Burton J, Kulik LM, Botha JF, Mezrich JD, Chapman WC, Schwartz JJ, Hong JC, Emond JC, Jeon H, Rosen CB, Gores GJ, Heimbach JK. Efficacy of neoadjuvant chemoradiation, followed by liver transplantation, for perihilar cholangiocarcinoma at 12 US centers. Gastroenterology. 2012;143:88–98. e3. doi: 10.1053/j.gastro.2012.04.008. quiz e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Graziadei IW, Wiesner RH, Batts KP, Marotta PJ, LaRusso NF, Porayko MK, Hay JE, Gores GJ, Charlton MR, Ludwig J, Poterucha JJ, Steers JL, Krom RA. Recurrence of primary sclerosing cholangitis following liver transplantation. Hepatology. 1999;29:1050–6. doi: 10.1002/hep.510290427. [DOI] [PubMed] [Google Scholar]
- 122.Fosby B, Karlsen TH, Melum E. Recurrence and rejection in liver transplantation for primary sclerosing cholangitis. World journal of gastroenterology: WJG. 2012;18:1–15. doi: 10.3748/wjg.v18.i1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Adeyi O, Fischer SE, Guindi M. Liver allograft pathology: approach to interpretation of needle biopsies with clinicopathological correlation. Journal of clinical pathology. 2010;63:47–74. doi: 10.1136/jcp.2009.068254. [DOI] [PubMed] [Google Scholar]
- 124.Bergquist A, Ekbom A, Olsson R, Kornfeldt D, Loof L, Danielsson A, Hultcrantz R, Lindgren S, Prytz H, Sandberg-Gertzen H, Almer S, Granath F, Broome U. Hepatic and extrahepatic malignancies in primary sclerosing cholangitis. Journal of hepatology. 2002;36:321–7. doi: 10.1016/s0168-8278(01)00288-4. [DOI] [PubMed] [Google Scholar]
- 125.Fevery J, Verslype C, Lai G, Aerts R, Van Steenbergen W. Incidence, diagnosis, and therapy of cholangiocarcinoma in patients with primary sclerosing cholangitis. Digestive diseases and sciences. 2007;52:3123–35. doi: 10.1007/s10620-006-9681-4. [DOI] [PubMed] [Google Scholar]
- 126.Broome U, Lofberg R, Veress B, Eriksson LS. Primary sclerosing cholangitis and ulcerative colitis: evidence for increased neoplastic potential. Hepatology. 1995;22:1404–8. doi: 10.1002/hep.1840220511. [DOI] [PubMed] [Google Scholar]
- 127.Boberg KM, Bergquist A, Mitchell S, Pares A, Rosina F, Broome U, Chapman R, Fausa O, Egeland T, Rocca G, Schrumpf E. Cholangiocarcinoma in primary sclerosing cholangitis: risk factors and clinical presentation. Scandinavian journal of gastroenterology. 2002;37:1205–11. doi: 10.1080/003655202760373434. [DOI] [PubMed] [Google Scholar]
- 128.Razumilava N, Gores GJ, Lindor KD. Cancer surveillance in patients with primary sclerosing cholangitis. Hepatology. 2011;54:1842–52. doi: 10.1002/hep.24570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Levy C, Lymp J, Angulo P, Gores GJ, Larusso N, Lindor KD. The value of serum CA 19-9 in predicting cholangiocarcinomas in patients with primary sclerosing cholangitis. Digestive diseases and sciences. 2005;50:1734–40. doi: 10.1007/s10620-005-2927-8. [DOI] [PubMed] [Google Scholar]
- 130.Kipp BR, Stadheim LM, Halling SA, Pochron NL, Harmsen S, Nagorney DM, Sebo TJ, Therneau TM, Gores GJ, de Groen PC, Baron TH, Levy MJ, Halling KC, Roberts LR. A comparison of routine cytology and fluorescence in situ hybridization for the detection of malignant bile duct strictures. The American journal of gastroenterology. 2004;99:1675–81. doi: 10.1111/j.1572-0241.2004.30281.x. [DOI] [PubMed] [Google Scholar]
- 131.Bangarulingam SY, Bjornsson E, Enders F, Barr Fritcher EG, Gores G, Halling KC, Lindor KD. Long-term outcomes of positive fluorescence in situ hybridization tests in primary sclerosing cholangitis. Hepatology. 2010;51:174–80. doi: 10.1002/hep.23277. [DOI] [PubMed] [Google Scholar]
- 132.Barr Fritcher EG, Kipp BR, Voss JS, Clayton AC, Lindor KD, Halling KC, Gores GJ. Primary sclerosing cholangitis patients with serial polysomy fluorescence in situ hybridization results are at increased risk of cholangiocarcinoma. The American journal of gastroenterology. 2011;106:2023–8. doi: 10.1038/ajg.2011.272. [DOI] [PubMed] [Google Scholar]
- 133.Sandanayake NS, Sinclair J, Andreola F, Chapman MH, Xue A, Webster GJ, Clarkson A, Gill A, Norton ID, Smith RC, Timms JF, Pereira SP. A combination of serum leucine-rich alpha-2-glycoprotein 1, CA19-9 and interleukin-6 differentiate biliary tract cancer from benign biliary strictures. British journal of cancer. 2011;105:1370–8. doi: 10.1038/bjc.2011.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lankisch TO, Metzger J, Negm AA, Vosskuhl K, Schiffer E, Siwy J, Weismuller TJ, Schneider AS, Thedieck K, Baumeister R, Zurbig P, Weissinger EM, Manns MP, Mischak H, Wedemeyer J. Bile proteomic profiles differentiate cholangiocarcinoma from primary sclerosing cholangitis and choledocholithiasis. Hepatology. 2011;53:875–84. doi: 10.1002/hep.24103. [DOI] [PubMed] [Google Scholar]
- 135.Metzger J, Negm AA, Plentz RR, Weismuller TJ, Wedemeyer J, Karlsen TH, Dakna M, Mullen W, Mischak H, Manns MP, Lankisch TO. Urine proteomic analysis differentiates cholangiocarcinoma from primary sclerosing cholangitis and other benign biliary disorders. Gut. 2013;62:122–30. doi: 10.1136/gutjnl-2012-302047. [DOI] [PubMed] [Google Scholar]
- 136.Brandt DJ, MacCarty RL, Charboneau JW, LaRusso NF, Wiesner RH, Ludwig J. Gallbladder disease in patients with primary sclerosing cholangitis. AJR American journal of roentgenology. 1988;150:571–4. doi: 10.2214/ajr.150.3.571. [DOI] [PubMed] [Google Scholar]
- 137.Buckles DC, Lindor KD, Larusso NF, Petrovic LM, Gores GJ. In primary sclerosing cholangitis, gallbladder polyps are frequently malignant. The American journal of gastroenterology. 2002;97:1138–42. doi: 10.1111/j.1572-0241.2002.05677.x. [DOI] [PubMed] [Google Scholar]
- 138.Eaton JE, Thackeray EW, Lindor KD. Likelihood of malignancy in gallbladder polyps and outcomes following cholecystectomy in primary sclerosing cholangitis. The American journal of gastroenterology. 2012;107:431–9. doi: 10.1038/ajg.2011.361. [DOI] [PubMed] [Google Scholar]
- 139.Lindstrom L, Lapidus A, Ost A, Bergquist A. Increased risk of colorectal cancer and dysplasia in patients with Crohn’s colitis and primary sclerosing cholangitis. Diseases of the colon and rectum. 2011;54:1392–7. doi: 10.1097/DCR.0b013e31822bbcc1. [DOI] [PubMed] [Google Scholar]
- 140.Thackeray EW, Charatcharoenwitthaya P, Elfaki D, Sinakos E, Lindor KD. Colon neoplasms develop early in the course of inflammatory bowel disease and primary sclerosing cholangitis. Clinical gastroenterology and hepatology: the official clinical practice journal of the American Gastroenterological Association. 2011;9:52–6. doi: 10.1016/j.cgh.2010.09.020. [DOI] [PubMed] [Google Scholar]
- 141.Walker M, Radley S. Adenocarcinoma in an ileoanal pouch formed for ulcerative colitis in a patient with primary sclerosing cholangitis and a liver transplant: report of a case and review of the literature. Diseases of the colon and rectum. 2006;49:909–12. doi: 10.1007/s10350-006-0517-2. [DOI] [PubMed] [Google Scholar]
- 142.Cairns SR, Scholefield JH, Steele RJ, Dunlop MG, Thomas HJ, Evans GD, Eaden JA, Rutter MD, Atkin WP, Saunders BP, Lucassen A, Jenkins P, Fairclough PD, Woodhouse CR. Guidelines for colorectal cancer screening and surveillance in moderate and high risk groups (update from 2002) Gut. 2010;59:666–89. doi: 10.1136/gut.2009.179804. [DOI] [PubMed] [Google Scholar]
- 143.Farraye FA, Odze RD, Eaden J, Itzkowitz SH. AGA technical review on the diagnosis and management of colorectal neoplasia in inflammatory bowel disease. Gastroenterology. 2010;138:746–74. 774e1–4. doi: 10.1053/j.gastro.2009.12.035. quiz e12-3. [DOI] [PubMed] [Google Scholar]
- 144.Vieth M, Behrens H, Stolte M. Sporadic adenoma in ulcerative colitis: endoscopic resection is an adequate treatment. Gut. 2006;55:1151–5. doi: 10.1136/gut.2005.075531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Eaton JE, Smyrk TC, Imam M, Pardi DS, Loftus EV, Jr, Owens VL, Talwalkar JT. Fate of indefinite and low grade dysplasia in ulcerative colitis and primary sclerosing cholangitis before and after liver transplantation. doi: 10.1111/apt.12469. Submitted 2012. Publication pending. [DOI] [PubMed] [Google Scholar]
- 146.Tung BY, Emond MJ, Haggitt RC, Bronner MP, Kimmey MB, Kowdley KV, Brentnall TA. Ursodiol use is associated with lower prevalence of colonic neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Annals of internal medicine. 2001;134:89–95. doi: 10.7326/0003-4819-134-2-200101160-00008. [DOI] [PubMed] [Google Scholar]
- 147.Pardi DS, Loftus EV, Jr, Kremers WK, Keach J, Lindor KD. Ursodeoxycholic acid as a chemopreventive agent in patients with ulcerative colitis and primary sclerosing cholangitis. Gastroenterology. 2003;124:889–93. doi: 10.1053/gast.2003.50156. [DOI] [PubMed] [Google Scholar]
- 148.Wolf JM, Rybicki LA, Lashner BA. The impact of ursodeoxycholic acid on cancer, dysplasia and mortality in ulcerative colitis patients with primary sclerosing cholangitis. Alimentary pharmacology & therapeutics. 2005;22:783–8. doi: 10.1111/j.1365-2036.2005.02650.x. [DOI] [PubMed] [Google Scholar]
- 149.Lindstrom L, Boberg KM, Wikman O, Friis-Liby I, Hultcrantz R, Prytz H, Sandberg-Gertzen H, Sangfelt P, Rydning A, Folvik G, Gangsoy-Kristiansen M, Danielsson A, Bergquist A. High dose ursodeoxycholic acid in primary sclerosing cholangitis does not prevent colorectal neoplasia. Alimentary pharmacology & therapeutics. 2012;35:451–7. doi: 10.1111/j.1365-2036.2011.04966.x. [DOI] [PubMed] [Google Scholar]
- 150.Jorgensen KK, Lindstrom L, Cvancarova M, Castedal M, Friman S, Schrumpf E, Foss A, Isoniemi H, Nordin A, Holte K, Rasmussen A, Bergquist A, Vatn MH, Boberg KM. Colorectal neoplasia in patients with primary sclerosing cholangitis undergoing liver transplantation: a Nordic multicenter study. Scandinavian journal of gastroenterology. 2012;8–9:1021–9. doi: 10.3109/00365521.2012.685754. [DOI] [PubMed] [Google Scholar]
- 151.Epstein MP, Kaplan MM. A pilot study of etanercept in the treatment of primary sclerosing cholangitis. Digestive diseases and sciences. 2004;49:1–4. doi: 10.1023/b:ddas.0000011827.87103.2e. [DOI] [PubMed] [Google Scholar]
- 152.Sandborn WJ, Gasink C, Gao LL, Blank MA, Johanns J, Guzzo C, Sands BE, Hanauer SB, Targan S, Rutgeerts P, Ghosh S, de Villiers WJ, Panaccione R, Greenberg G, Schreiber S, Lichtiger S, Feagan BG. Ustekinumab induction and maintenance therapy in refractory Crohn’s disease. The New England journal of medicine. 2012;367:1519–28. doi: 10.1056/NEJMoa1203572. [DOI] [PubMed] [Google Scholar]
- 153.Parikh A, Leach T, Wyant T, Scholz C, Sankoh S, Mould DR, Ponich T, Fox I, Feagan BG. Vedolizumab for the treatment of active ulcerative colitis: a randomized controlled phase 2 dose-ranging study. Inflammatory bowel diseases. 2012;18:1470–9. doi: 10.1002/ibd.21896. [DOI] [PubMed] [Google Scholar]
- 154.Seavey MM, Dobrzanski P. The many faces of Janus kinase. Biochemical pharmacology. 2012;83:1136–45. doi: 10.1016/j.bcp.2011.12.024. [DOI] [PubMed] [Google Scholar]
- 155.Sandborn WJ, Ghosh S, Panes J, Vranic I, Su C, Rousell S, Niezychowski W. Tofacitinib, an oral Janus kinase inhibitor, in active ulcerative colitis. The New England journal of medicine. 2012;367:616–24. doi: 10.1056/NEJMoa1112168. [DOI] [PubMed] [Google Scholar]
- 156.Ghoreschi K, Jesson MI, Li X, Lee JL, Ghosh S, Alsup JW, Warner JD, Tanaka M, Steward-Tharp SM, Gadina M, Thomas CJ, Minnerly JC, Storer CE, LaBranche TP, Radi ZA, Dowty ME, Head RD, Meyer DM, Kishore N, O’Shea JJ. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550) Journal of immunology. 2011;186:4234–43. doi: 10.4049/jimmunol.1003668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Meyer DM, Jesson MI, Li X, Elrick MM, Funckes-Shippy CL, Warner JD, Gross CJ, Dowty ME, Ramaiah SK, Hirsch JL, Saabye MJ, Barks JL, Kishore N, Morris DL. Anti-inflammatory activity and neutrophil reductions mediated by the JAK1/JAK3 inhibitor, CP-690,550, in rat adjuvant-induced arthritis. Journal of inflammation. 2010;7:41. doi: 10.1186/1476-9255-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Distler JH, Distler O. Tyrosine kinase inhibitors for the treatment of fibrotic diseases such as systemic sclerosis: towards molecular targeted therapies. Annals of the rheumatic diseases. 2010;69 (Suppl 1):i48–51. doi: 10.1136/ard.2009.120196. [DOI] [PubMed] [Google Scholar]
- 159.Yoshiji H, Noguchi R, Kuriyama S, Ikenaka Y, Yoshii J, Yanase K, Namisaki T, Kitade M, Masaki T, Fukui H. Imatinib mesylate (STI-571) attenuates liver fibrosis development in rats. American journal of physiology Gastrointestinal and liver physiology. 2005;288:G907–13. doi: 10.1152/ajpgi.00420.2004. [DOI] [PubMed] [Google Scholar]
- 160.Seidel C, Fenner M, Langer F, Bantel H, Ganser A, Grunwald V. Imatinib-induced liver cirrhosis in a patient with advanced gastrointestinal stroma tumor (GIST) BMC cancer. 2012;12:186. doi: 10.1186/1471-2407-12-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Rockey DC. Current and future anti-fibrotic therapies for chronic liver disease. Clinics in liver disease. 2008;12:939–62. xi. doi: 10.1016/j.cld.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Satapathy SK, Sakhuja P, Malhotra V, Sharma BC, Sarin SK. Beneficial effects of pentoxifylline on hepatic steatosis, fibrosis and necroinflammation in patients with non-alcoholic steatohepatitis. Journal of gastroenterology and hepatology. 2007;22:634–8. doi: 10.1111/j.1440-1746.2006.04756.x. [DOI] [PubMed] [Google Scholar]
- 163.Bharucha AE, Jorgensen R, Lichtman SN, LaRusso NF, Lindor KD. A pilot study of pentoxifylline for the treatment of primary sclerosing cholangitis. The American journal of gastroenterology. 2000;95:2338–42. doi: 10.1111/j.1572-0241.2000.02324.x. [DOI] [PubMed] [Google Scholar]
- 164.Olsson R, Broome U, Danielsson A, Hagerstrand I, Jarnerot G, Loof L, Prytz H, Ryden BO, Wallerstedt S. Colchicine treatment of primary sclerosing cholangitis. Gastroenterology. 1995;108:1199–203. doi: 10.1016/0016-5085(95)90220-1. [DOI] [PubMed] [Google Scholar]
- 165.He H, Mennone A, Boyer JL, Cai SY. Combination of retinoic acid and ursodeoxycholic acid attenuates liver injury in bile duct-ligated rats and human hepatic cells. Hepatology. 2011;53:548–57. doi: 10.1002/hep.24047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lindor KD. Farnesoid X receptor agonists for primary biliary cirrhosis. Current opinion in gastroenterology. 2011;27:285–8. doi: 10.1097/MOG.0b013e32834452c8. [DOI] [PubMed] [Google Scholar]
- 167.Poupon R. Ursodeoxycholic acid and bile-acid mimetics as therapeutic agents for cholestatic liver diseases: An overview of their mechanisms of action. Clinics and research in hepatology and gastroenterology. 2012;36 (Suppl 1):S3–S12. doi: 10.1016/S2210-7401(12)70015-3. [DOI] [PubMed] [Google Scholar]
- 168.Halilbasic E, Fiorotto R, Fickert P, Marschall HU, Moustafa T, Spirli C, Fuchsbichler A, Gumhold J, Silbert D, Zatloukal K, Langner C, Maitra U, Denk H, Hofmann AF, Strazzabosco M, Trauner M. Side chain structure determines unique physiologic and therapeutic properties of norursodeoxycholic acid in Mdr2−/− mice. Hepatology. 2009;49:1972–81. doi: 10.1002/hep.22891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Fickert P, Wagner M, Marschall HU, Fuchsbichler A, Zollner G, Tsybrovskyy O, Zatloukal K, Liu J, Waalkes MP, Cover C, Denk H, Hofmann AF, Jaeschke H, Trauner M. 24-norUrsodeoxycholic acid is superior to ursodeoxycholic acid in the treatment of sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology. 2006;130:465–81. doi: 10.1053/j.gastro.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 170.Al Mamari S, Djordjevic J, Halliday JS, Chapman RW. Improvement of serum alkaline phosphatase to <1.5 upper limit of normal predicts better outcome and reduced risk of cholangiocarcinoma in primary sclerosing cholangitis. Journal of hepatology. 2012 doi: 10.1016/j.jhep.2012.10.013. [DOI] [PubMed] [Google Scholar]