

Summary
DNase hypersensitivity (DHS) analysis coupled with high throughput DNA sequencing (DNase-seq) has emerged as a powerful tool to analyze chromatin accessibility and identify regulatory sequences in genomic DNA on a global scale. In this method, intact nuclei are isolated from fresh tissue or cultured cells and then subjected to limited digestion using DNase I. The resulting short DNA fragments released by DNase digestion, which correspond to regions of open chromatin structure, are subsequently purified and identified by high throughput next generation DNA sequencing. This chapter describes methods used to isolate intact nuclei from mouse liver suitable for DNase-seq studies. The following chapter presents a detailed protocol for DNase I digestion of liver nuclei followed by the isolation of DNase-released fragments for sequencing and genome-wide mapping of DHS sites.
Keywords: DNase I hypersensitivity assay, next generation DNA sequencing, DNase-seq, chromatin structure
1. Introduction
In eukaryotic cells, genomic DNA is wrapped around a core of histone proteins, forming nucleosomes, which are further packaged into chromatin. Chromatin controls the accessibility of regulatory proteins to DNA and plays an important role in many critical nuclear processes, including gene regulation, DNA repair and replication (1). DHS assays using the enzyme DNase I have long been used to identify chromatin regions that are open (i.e., accessible to cleavage by DNase I) (Figure 1). These open chromatin regions are frequently associated with DNA regulatory elements, including gene promoters, distal enhancer sequences, silencers, insulators and locus control regions (2), and thus encompass genomic sequences of substantial biological importance. Recent advances have enabled traditional DHS assays to be coupled with high throughput mapping methods, such as tiling microarrays (DNase-chip) and, more recently, DNA sequencing (DNase-seq), making it possible to obtain high resolution genome-wide maps of DHS sites upon mapping released DNA fragments back to the genome (Fig. 2) (3–6).
Figure 1.
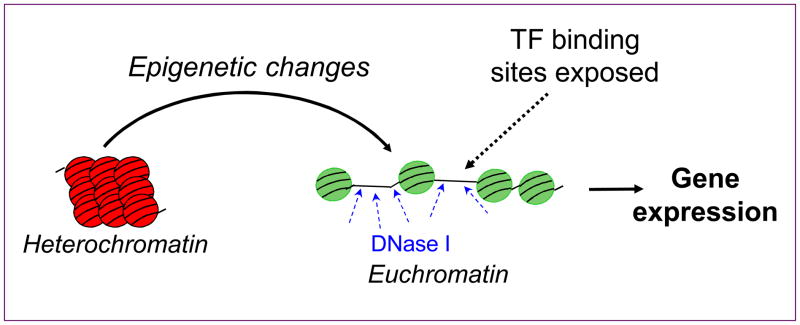
DNase hypersensitivity identifies open genomic regions in chromatin that facilitate transcription factor (TF) binding to chromatin and induce gene expression. Nucleosomes are compacted in closed, inactive heterochromatin but are more open, exposing sites of DNase hypersensitivity in the euchromatin state.
Figure 2.

DNase-seq analysis. Sites of hypersensitivity (HS) are susceptible to cutting by DNase I, which releases many fragments of variable length from each hypersensitive region. The released fragments are then purified, sequenced on one end, and the resultant sequence tags then mapped back to the genome. A peak detection algorithm is used to identify DHS peaks, two of which are shown.
DNase-seq can be carried out with nuclei isolated from intact mammalian tissues, as exemplified by studies from this laboratory on sex differences in mouse liver chromatin structure (7). DNase-seq studies using intact nuclei isolated from fresh tissue, such as mouse liver, have the important advantage of providing detailed information about the regulation of chromatin structure under physiological conditions in vivo. However, care must be taken to ensure that the procedure yields high quality nuclei with minimal disturbance of chromatin structure. In the protocol described here, isolation of fresh, high quality nuclei from tissues is facilitated by sucrose ultracentrifugation. In brief, fresh liver tissue is collected, homogenized, and the nuclei are pelleted by ultracentrifugation through a 2 M sucrose cushion, which helps maintain the integrity of the nuclei and chromatin structure. Only intact nuclei are of sufficiently high density to pass through the sucrose cushion (8). Nuclei isolated and purified by this method give reproducible and reliable DNase-seq results (7). The same protocol can also be used to isolate nuclei from tissue culture cells, alhough in that case a simpler, detergent-based method may work as well (5). We anticipate that the protocol described here can be applied to other tissues that yield high quality nuclei without major modifications.
2. Materials
Prepare all solutions using ultrapure water and analytical grade reagents. Store all buffers at 4°C unless otherwise noted. Protease inhibitors, spermine, spermidine and DTT should be added fresh to each solution just prior to their use.
2.1. Materials for dissection of liver tissue
Scissors, blades, and forceps for tissue dissection.
CO2 chamber for rodents.
Paper towels, Kimwipe and Petri dishes.
Ice cold 1.15% KCl and 1X Phosphate-buffered saline (PBS).
250 ml beaker, 50 ml conical tubes.
1 ml syringes and 27 G needles (optional).
2.2. Tissue homogenization and isolation of nuclei
Preparative ultracentrifuge and SW28 rotor, or equivalent. Ultra-clear centrifugation tubes (Beckman Coulter, Brea, CA; cat. # 344058).
Potter-Elvehjem Tissue Grinder with Teflon Pestle, 10, 15 or 30 or 55 ml size (Wheaton Science Products, Millville, NJ; see Note 1), and a power drill.
Nuclear homogenization buffer (NEHB): 10 mM HEPES-KOH, pH 7.9, 25 mM KCl, 1 mM EDTA, 2 M sucrose, 10% glycerol, 0.15 mM spermine, 0.5 mM spermidine, 10 mM NaF, 1 mM orthovanadate, 1 mM PMSF, 0.5 mM DTT, and 1X protease inhibitor cocktail (Sigma, St Louis, MO; cat. # P8340) (see Note 2).
Nuclear storage buffer: 20 mM Tris-HCl, pH 8.0, 75 mM NaCl, 0.5 mM EDTA, 50% (v/v) glycerol, 1 mM DTT, and 0.1 mM PMSF.
1 M HEPES-KOH, pH 7.9. Dissolve 238.3 gram HEPES in 700 ml of ultrapure water. Use potassium hydroxide pellets and 1 M KOH solution to adjust pH to 7.9. Bring up to 1000 ml with ultrapure water.
1 M Tris-HCl, pH 8.0. Dissolve 121.1 gram Tris base in 800 ml of ultrapure water. Add concentrated HCl to bring to pH 8.0. Bring up to 1000 ml with ultrapure water.
0.5 M EDTA, pH 8.0 (Sigma, St. Louis, MO).
Dounce homogenizer with B pestle, chilled on ice (Wheaton Science Products, Millville, NJ).
Inverted tissue culture microscope, hemocytometer, and typan blue solution (Sigma, cat. #T8154 or equivalent) for counting nuclei.
1.5-ml microcentrifuge tubes, chilled on ice.
Liquid nitrogen for freezing nuclei.
3. Methods
All steps described here, except animal-related procedures, should be carried out in a 4°C cold room. Samples should be kept at 4°C to minimize non-specific DNA degradation.
3.1. Dissection of liver tissue
Prepare complete NEHB on ice (see Note 2). Precool the ultracentrifuge, rotor, tubes and the tissue grinder before starting the experiments.
Euthanize the mice using approval protocols, e.g., CO2 asphyxiation followed by cervical dislocation. Subsequent steps should be carried out promptly to ensure that the minced liver tissue is submerged in ice-cold NEHB (step 6) as quickly as possible.
If required, remove blood sample from the heart using a 1-ml syringe with 50μl of heparin and a 27 G needle (optional).
Remove each liver and rinse in ice-cold PBS. Snap-freeze a small piece of tissue and store at −80°C, e.g., for RNA isolation at a later time.
Place the liver in a 100 × 15 mm Petri dish on ice containing ice cold 1.15% KCl. Cut the liver into small pieces and wash twice with 1.15% KCl.
Carefully dry off the excess KCl solution with a Kimwipe. Weigh the liver and submerge the minced liver in ice-cold NEHB buffer.
3.2. Tissue homogenization and isolation of nuclei (scale: pool of 3–4 mouse livers) (Note 1)
Homogenize the liver in a 30 ml Potter-Elvehjem Tissue Grinder with 3 to 4 strokes. Use a buffer-to-tissue ratio of ~ 6 (v/w) (see Note 3).
Carefully overlay 25 ml of homogenate on top of 10 ml NEHB in a SW28 tube.
Centrifuge at 25,000 rpm (90,000 g) for 45 min at 2°C. The nuclei should be found in a clear pellet at bottom of tube (see Note 4).
Remove tissue debris floating on the top, and decant the supernatant. Turn the tube upside down in a cold room and wipe the sides of each tube with a Kimwipe soaked with PBS.
Resuspend the pellet in a minimal volume of nuclear storage buffer by applying a few strokes of a Dounce homogenizer (see Note 5).
Dilute a sample of the resuspended nuclei 100-fold with PBS and count the nuclei using a hemocytometer. Trypan Blue can be added to a final concentration of 0.04% to help visualize the nuclei. Nuclei should be counted at least three times to ensure that a reproducible count is obtained.
Snap freeze the suspended nuclei in small aliquots using liquid nitrogen, for example 300 μl aliquots in 1.5 ml micro-centrifugation tubes, and store at −80°C (see Note 6).
Acknowledgments
Supported in part by NIH grant DK33765 (to DJW).
Footnotes
Nuclei may be isolated from a single adult mouse liver if DNase-seq data are to be collected for individual animals; otherwise, it may be more convenient to pool livers from 3–4 mice and prepare a single preparation of nuclei, using a 30 ml or 55 ml tissue grinder for liver homogenization. If the tissue weight is ~2 gr or less (e.g., when isolating nuclei from a single mouse liver), a 10 or 15 ml tissue grinder can be used and the buffer volumes and sizes of the ultracentrifuge tubes and rotor should be scaled down accordingly.
NEHB is prepared and stored at 4°C as 10 mM HEPES pH 7.9, 25 mM KCl, 1 mM EDTA, 2 M sucrose, and 10% glycerol. The other buffer components: spermine, spermidine, NaF, orthovanadate, PMSF, DTT, and protease inhibitor cocktail are prepared separately as concentrated stock solutions (100 X for NaF, orthovanadate (both in water), PMSF (in isopropanol), and protease inhibitor cocktail (in DMSO); 2000 X for spermine, spermidine, and DTT (all in water)), and stored in aliquots at −20°C. The concentrated components are then added to NEHB just before tissue dissection and homogenization.
A buffer-to-tissue ratio of 6:1 (v/w) is recommended for mammalian tissues. Insufficient buffer during tissue homogenization can lead to impure nuclear preparations. Note that the presence of 2 M sucrose makes it difficult to carry out the homogenization step. As such, gloves and goggles should be worn during homogenization for personal protection in the event of glassware failure. Three to four strokes of the homogenizer are required to disrupt >90% of the liver tissue. Other tissues may require more vigorous conditions for effective homogenization.
A nuclear pellet with a red tinge indicates that the nuclei are contaminated by red blood cells and should be discarded if replalcement material is readily available. Increasing the volume of buffer used for homogenization may help avoid this problem. It may be possible to wash the nuclei by resuspending the nuclear pellet in NEHB and then repeating the centrifugation step.
The nuclei should be resuspended in a minimal volume of nuclear storage buffer prior to counting. For example, nuclei from 3–4 livers should initially be resuspended in approx. 0.5 ml storage buffer. Additional buffer can be added as required once the count is known to adjust the final preparation to 50–100 million nuclei per ml.
Although fresh nuclei may in some cases be preferrable for DHS assays, we have not noticed any difference in results when frozen mouse liver nuclei are used after storage at −80°C for several months.
References
- 1.Bell O, Tiwari VK, Thomä NH, Schübeler D. Determinants and dynamics of genome accessibility. Nat Rev Genet. 2011;12:554–564. doi: 10.1038/nrg3017. [DOI] [PubMed] [Google Scholar]
- 2.Gross DS, Garrard WT. Nuclease hypersensitive sites in chromatin. Annu Rev Biochem. 1988;57:159–197. doi: 10.1146/annurev.bi.57.070188.001111. [DOI] [PubMed] [Google Scholar]
- 3.Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, Furey TS, Crawford GE. High-resolution mapping and characterization of open chromatin across the genome. Cell. 2008;132:311–322. doi: 10.1016/j.cell.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crawford GE, Davis S, Scacheri PC, Renaud G, Halawi MJ, Erdos MR, Green R, Meltzer PS, Wolfsberg TG, Collins FS. DNase-chip: a high-resolution method to identify DNase I hypersensitive sites using tiled microarrays. Nat Methods. 2006;3:503–509. doi: 10.1038/NMETH888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sabo PJ, Kuehn MS, Thurman R, Johnson BE, Johnson EM, Cao H, Yu M, Rosenzweig E, Goldy J, Haydock A, Weaver M, Shafer A, Lee K, Neri F, Humbert R, Singer MA, Richmond TA, Dorschner MO, McArthur M, Hawrylycz M, Green RD, Navas PA, Noble WS, Stamatoyannopoulos JA. Genome-scale mapping of DNase I sensitivity in vivo using tiling DNA microarrays. Nat Methods. 2006;3:511–518. doi: 10.1038/nmeth890. [DOI] [PubMed] [Google Scholar]
- 6.Song L, Crawford GE. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb Protoc. 2010;2010 doi: 10.1101/pdb.prot5384. pdb.prot5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ling G, Sugathan A, Mazor T, Fraenkel E, Waxman DJ. Unbiased, genome-wide in vivo mapping of transcriptional regulatory elements reveals sex differences in chromatin structure associated with sex-specific liver gene expression. Mol Cell Biol. 2010 Dec;30(23):5531–44. doi: 10.1128/MCB.00601-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lichtsteiner S, Wuarin J, Schibler U. The interplay of DNA-binding proteins on the promoter of the mouse albumin gene. Cell. 1987;51:963–973. doi: 10.1016/0092-8674(87)90583-6. [DOI] [PubMed] [Google Scholar]