

Abstract
Background
Many neurobiological factors may initiate and sustain alcoholism. Recently, dysregulation of the neuroimmune-system by chronic-ethanol (CE) has implicated toll-like receptor-4 (TLR4)-activation. Even though TLR4s are linked to CE-initiation of brain cytokine-mRNAs, the means by which CE influences neuroimmune signaling in the sterile environment of brain remains uncertain. Therefore, the hypothesis is tested that release of an endogenous TLR4 agonist, high-mobility group box 1 (HMGB1) and/or CRF during CE-withdrawal are responsible for CE-protocols increasing cytokine-mRNAs.
Methods
Acute-ethanol 2.75g/kg) and acute-LPS (lipopolysaccharide)(250μg/kg) dosing on cytokine-mRNAs are first compared. Then, the effects of chronic-LPS exposure (250 μg/kg for 10-days) on cytokine-mRNAs are compared to changes induced by CE-protocols [15-days of continuous 7% ethanol-diet (CE-protocol) or three-intermittent 5-day cycles of 7%-ethanol-diet (CIE-protocol)]. Additionally, TLR4-, HMGB1- and down-stream effector mRNAs are assessed after CE, CIE, and chronic-LPS. To test whether HMGB1 and/or CRF support the CE-withdrawal increase in cytokine-mRNAs, the HMGB1-antagonists, glycyrrhizin and ethyl-pyruvate, and a CRF1-receptor-antagonist (CRF1RA) are administered during 24-hours of CE-withdrawal.
Results
While cytokine-mRNAs were not increased following acute-ethanol, acute-LPS increased all cytokine-mRNAs 4-hours after injection. CE produced no change in cytokine-mRNAs prior to CE-removal; however, the CE- and CIE-protocols increased cytokine-mRNAs by 24-hours after withdrawal. In contrast, chronic-LPS produced no cytokine-mRNA changes 24-hours after LPS-dosing. TLR4-mRNA was elevated 24-hours following both CE-protocols and chronic-LPS exposure. While chronic-LPS had no effect on HMGB1-mRNA, withdrawal from CE-protocols significantly elevated HMGB1-mRNA. Systemic administration of HMGB1-antagonists or a CRF1RA significantly reduced the cytokine-mRNA increase following CE-withdrawal. The CRF1RA and the HMGB1-antagonist, ethyl-pyruvate, also reduced the HMGB1-mRNA increase that followed CE-withdrawal.
Conclusion
By blocking HMGB1 or CRF action during CE-withdrawal, evidence is provided that HMGB1- and CRF-release are critical for the CE-withdrawal-induction of selected brain cytokine-mRNAs. Consequently, these results clarify a means by which withdrawal from CE-exposure activates neuroimmune-function in the sterile-environment of brain.
Keywords: Chronic-Ethanol Withdrawal, LPS, Cytokines, HMGB1-Antagonists and CRF1-Receptor Antagonist
Introduction
Although alcohol abuse influences many organs throughout the body, its effect on the brain represents the most significant component in the maintenance of alcoholism. In spite of numerous factors having previously been implicated in initiating and sustaining alcohol abuse, dysregulation of the neuroimmune system has recently come to light as playing a significant role in the consequences of chronic-ethanol-(CE) exposure (Alfonso-Loeches et al. 2010; Breese et al., 2008; Crews et al., 2011, 2012; He and Crews 2008; Knapp et al., 2011; Pascual et al., 2007; 2011; Qin et al., 2008; Valles et al., 2003). Further, recent experimental studies have demonstrated a key link between ethanol and activation of the neuroimmune system via toll-like receptor-4 (TLR4) signaling (Akira and Takeda, 2004; Alfonso-Loeches et al. 2010; et al., 2005; Fernandez-Lizarbe et al., 2008; 2009; Wu et al., 2012; Zou and Crews, 2010).
Crews et al. (2012) found increased expression of TLR4 in post-mortem human alcoholic frontal cortex as well as in brain of mice following CE-exposure. Several additional studies in TLR4-KO mice have supported a role for TLR4 signaling in ethanol-induced increases in cytokine production—evidence which includes preventing ethanol-induced liver injury (Uesugi et al., 2001), ethanol activation of TLR4 signaling in cells (Blanco et al., 2005;), the inflammatory gene expression induced by ethanol in mice (Alfonso-Loeches et al., 2010; Fernandez-Lizabre et al., 2008, 2009), neurodegeneration induced by CE-intake (Alfonso-Loeches et al., 2010), involvement in seizure sensitivity (Maroso et al., 2010), antagonism of the sedation and motor impairment associated with acute-ethanol administration (Wu et al., 2012), and antagonism of the behavioral impairment induced by CE-exposure (Pascual et al., 2011). Collectively, these studies provided convincing evidence that TLR4s contribute to the CE-mediated increase in brain cytokines that contribute to behavioral and pathological changes associated with CE-exposure.
Fernandez-Lizarbe et al. (2008) provided evidence that low to moderate concentrations of ethanol facilitate recruitment of TLR4s into lipid rafts that results in TLR4 activation, similar to lipopolysaccharide (LPS) (Triantafilou et al., 2002). While the LPS-agonist-action on TLR4s (Okun et al., 2009) is used to model bacterial infections, ethanol is unlikely to induce brain cytokine-mRNAs by directly activating TLR4-signaling. Rather, ethanol, unlike LPS (Pålsson-McDermott and O’Neill, 2004), most likely influences TLR4 in the sterile environment of brain by either releasing an endogenous agonist for TLR4s (Crews et al, 2012) or by CRF release during withdrawal from CE-exposure (Breese et al., 2011). Therefore, to determine if ethanol and LPS have similar properties, the present investigation first compares the acute- and chronic actions of LPS with acute-ethanol and CE-protocols on mRNAS for cytokines, TLR4, HMGB1 [i.e., High-mobility group box-1; Andersson and Tracey, 2011), an endogenous TLR4 agonist. Subsequently, a CRF-receptor antagonist (Knapp et al, 2004) and two HMGB1-antagonists (Mollica et al., 2007; Ohnishi et al., 2011; Su et al.,2011) are used to test the hypothesis that in the sterile-environment of brain (Andersson and Tracey, 2011) release of CRF and/or HMGB1 during CE-withdrawal is responsible for increasing brain cytokine-mRNAs (Breese et al., 2011; Fernendez-Lizarbe et al., 2008; Yang et al., 2005). 502
Materials and Methods
Animals
Adult male Sprague-Dawley rats obtained from Charles-River (Raleigh, NC) weighed 180-200g upon arrival. Subsequently, rats were group housed and fed RMH3000 rat-chow (TestDiets, Richmond, IN) for 2-3-days to acclimate them to the new environment (temperatures 70-72° F; humidity 40-60%; and a 12hr-light/12hr-dark cycle) prior to study initiation. Then, all animals were singly housed for acute and chronic-ethanol and LPS-exposures. All methods used were approved by the University of North Carolina—Chapel Hill’s Institutional Animal Care and Use Committee.
TREATMENT PROTOCOLS
Liquid Diet
A nutritionally-complete and calorically-balanced liquid diet for control (CD) and the chronic-ethanol diet (CE)-protocol were used with adjustments made in the amount of dextrose added to each diet type, as previously described (e.g., Frye et al., 1983; Knapp et al., 2004; Overstreet et al., 2002; Wills et al. 2008). Rats were fed either control diet or 7% thanol (wt/vol) in a modified pair-feeding system to balance diet intake.
Acute-LPS and Acute-Ethanol Protocols
Either a single 250 μg/kg dose of LPS (E.coli 0111;B4) (Calbiochem & EMD Chem Inc, San Diego, CA) or saline-vehicle was injected intraperitoneally (IP) to rats that were sacrificed 4-hours later (Fig 1B). For comparison with LPS, oral gavage of either a single 2.75 g/kg ethanol-dose or tap water to controls was utilized (Fig. 1A). For the oral-ethanol dose, peak blood alcohol levels of 185-200 mg% were observed, as found previously (Overstreet et al., 2002; Wills et al., 2008). Four-hours following either acute-ethanol or acute-LPS dosing, rats and corresponding controls were sacrificed and brains collected for assessment of cytokine and TLR4-mRNAs and selected cytokine-peptides.
Figure 1.

Protocols for the experimental treatments described in this paper.
Chronic-LPS Protocol
Chronic-LPS (E.coli 0111;B4) effects on cytokine function were determined after 10-daily injections of LPS (250 μg/kg, IP) (Fig 1D). Controls received saline-injections daily for 10-days. Animals were sacrificed and brains harvested 24-hours following final injection for analysis of mRNAs for cytokines, TLR4, HMGB1, MyD88 (myeloid differentiation factor-88; Janssens and Beyaert, 2002) or NF-Kb-(nuclear factor-κ-light-chain-enhancer of activated B cells; Baeuerle and Henkel, 1994; Fitzgerald et al., 2003).
Continuous (CE) and Intermittent Chronic-Ethanol (CIE) Protocols
The CE-protocol involved 15-continuous days of ethanol liquid diet (7% wt/vol) (Frye et al., 1983) (Fig 1C) and the CIE-protocol involved three 5-day bouts of ethanol-diet separated by 2-days of control diet (Overstreet et al., 2002; Fig 1F). To determine if a difference between the CE- and CIE-exposures occurred, rats were sacrificed 24-hours after withdrawal from each protocol. As before, brains from these groups were collected and processed for mRNAs for cytokines, TLR4, and HMGB1 and other neuroimmune elements. To determine the peak brain cytokine, TLR4 and HMGB1-mRNA production and duration of change for the CE-protocols, animals were sacrificed 24-, 48-, 72-hours and 7-days following withdrawal (Fig. 1E). These brains were collected and processed for cytokine, TLR4, HMGB, myD88 (-mRNAs.
Brain Tissue Collection
All brains were collected following rapid decapitation. Whole brains were extracted and immediately frozen in isopentane at −25° C prior to storage at −80° C for later microdissection of the cortex. Cortical tissue was divided into two halves to provide tissue for qPCR as well as for selected ELISA determinations.
Administration of a CRF1-Receptor antagonist (CRF1RA) and HMGB1-Antagonists During CE-Withdrawal
To test whether CRF and/or HMGB1 were involved in increasing cytokine-mRNAs during CE-withdrawal, antagonists for both were administered IP 4- and 12-hours during the 24-hour period of withdrawal from CE-exposure. The CRF1RA (CP-154,526; 10mg/kg) suspended in 0.5%-carboxymethylcellulose was used to block CRF action (Knapp et al., 2004). To test for a role of HMGB1 in cytokine-mRNA increases after the CE-protocol, the HMGB1-antagonists, [either glycyrrhizin ammonium salt (50 mg/kg) (Sigma, St.Louis, Mo) or ethyl-pyruvate (75 mg/kg) (Sigma, St. Louis, Mo)] were also administered IP 4- and 12-hours during CE-withdrawal. All control subjects received injections of sterile saline IP. To block HMGB1 function, glycyrrhizin acts to prevent released HMGB1 from acting on TLR4 (Girard, 2007; Mollica et al., 2007; Ohnishi et al., 2011), whereas ethyl pyruvate interferes with HMGB1 function by minimizing its release (Dave et al., 2009; Su et al., 2011).
Real-Time RT-PCR Analysis for mRNA
Total RNA was extracted from homogenized micro-dissected cortex from control and CE-ethanol-treated groups and LPS-exposed rats using Trizol (Invitrogen, Carlsbad, CA). RNA was converted to cDNA utilizing RT/PCR as described elsewhere (Qin et al., 2008). SYBR green PCR master mix (Applied Biosystems, Foster City, CA) was used for real-time PCR analysis. The cycle time (CT) values were normalized with β-actin to assess relative differences of mRNA expression between groups. Calculated values were expressed as relative change compared to controls set as 100%. Primer sequences for all mRNA determinations are shown in Table 1.
Table 1.
Primer sequences
| CCL2 | 5′-TCACGCTTCTGGGCCTGTTG-3′ (forward) 5′-CAGCCGACTCATTGGGATCATC-3′ (reverse) |
| IL1-β | 5′-GAAACAGCAATGGTCGGGAC-3′ (forward) 5′-AAGACACGGGTTCCATGGTG-3′ (reverse) |
| TNFα | 5′-ATGTGGAACTGGCAGAGGAG-3′ (forward) 5′-ACGAGCAGGAATGAGAAGAGG-3′ (reverse) |
| TLR4 | 5′-GCCGGAAAGTTATTGTGGTGGT-3′ (forward) 5′-ATGGGTTTTAGGCGCAGAGTTT-3′ (reverse) |
| NF-κB | 5′-GGCAGCACTCCTTATCAA-3′ (forward) 5′-GGTGTCGTCCCATCGTAG -3′ (reverse) |
| MyD88 | 5′-GGCAGGCTGCTAGAGTTGCT -3′ (forward) 5′-TGTGGGACACTGCTCTCCAC-3′ (reverse) |
| HMGB1 | 5′-GAGATCCTAAGAAGCCGAGA-3′ (forward) 5′-CTTCCTCATCCTCTTCATCC-3′ (reverse) |
| β-actin | 5′-ATGGTGGGTATGGGTCAGAAGG-3′ (forward) 5′-GCTCATTGTAGAAAGTGTGGTGCC-3′ (reverse) |
ELISA Determination of Cytokine-Peptides
Cortical samples were collected as described in the Brain-Tissue Collection section 4-hours after acute-ethanol, acute-LPS or 24-hours after CE-withdrawal. These tissues were homogenized in Iscove’s Modified Dubecco-Medium (Invitrogen, Carlsbad, CA) containing a Complete-protease-inhibitor cocktail (Roche Diagnostics, Indianapolis, IN). Homogenized specimens were centrifuged 10 min at 12,000g (4°C). Supernatants collected were stored at −80°C until cytokine-peptides were determined. ELISA kits for the determinations were purchased for IL-1β and TNF-α from R&D Systems (Minneapolis, MN), for CCL2 from BD BioSciences (San Jose, CA), and for HMGB-1 from IBL-International (Hamburg, Germany). CCL2 standard was diluted according to kit instructions, but standards for IL-1β and TNF-α were serially diluted to a specificity of 1.95 pg/ml and 0.78 pg/ml, respectively. Other ELISA procedures were performed according to manufacturer’s directions. All cytokine-levels were corrected by protein concentration calculated using Pierce® BCA Protein assay (Thermo Scientific, Rockford, IL).
Statistical analysis
Data (expressed as mean ± SEM) were evaluated for statistical significance with either the student’s t-test or ANOVA with Fishers Least Significant Difference (LSD) tests for individual comparisons of group pairs as appropriate. Individual data points that were three standard deviations from their respective group means were removed from the group prior to analysis. P-values<0.05 were considered statistically significant.
RESULTS
Comparison of Acute-Ethanol with Acute-LPS on Cytokine-mRNAs and Cytokine-Peptides
Acute-ethanol versus acute-LPS effects on cytokine-mRNAs and corresponding cytokine-peptides were determined four-hours after treatments. The acute-ethanol (2.75 g/kg) induced no significant change in CCL2, IL1-β, and TNFα mRNAs (Fig. 2A). Likewise, when additional rats (n=12) were assessed 24-hours following acute-ethanol, no changes in mRNAs for these cytokine-mRNAs were found (Fig 2 legend). Further, no TLR4-mRNA change was observed at either 4-(Fig 2C) or a24-hours (Fig. 2 legend) after acute-ethanol exposure. When cytokine-peptides were assessed 4-hours after acute-ethanol, levels of CCL2 and IL1-β-peptides were not changed, but the TNFα-peptide was significantly increased (Table 2). In contrast to acute-ethanol, an acute-dose of LPS (Fig 1B) caused a several-fold increase in the mRNAs for CCL2 (432%), IL1-β (337%), and TNFα (920%) 4-hours after dosing (Fig 2B). However, the acute-LPS dose did not significantly alter TLR4-mRNA (Fig. 2D). Whereas acute-LPS increased CCL2 and IL1-β-peptides 4-hours after exposure (Table 2), TNFα-peptide was not increase (Table 2). In a second investigation, LPS alternatively induced a significant increase in the TNFα-peptide (Table 2-legend). An explanation for these varying responses for the TNFα-peptide after acute-LPS was not apparent.
Figure 2. Cytokine-mRNA and TLR4-mRNA expression following acute treatment with ethanol (2.75 g/kg) and LPS (250 μg/kg).

(A) The acute single dose of 2.75 g/kg of ethanol I.P resulted in no significant increase in cytokine-mRNAs for CCL2, IL1-β, or TNFα (all p>0.05). (B) The acute single 250 μg/kg dose of LPS IP increased cytokine-mRNAs for CCL2, IL1-β, and TNFα four hours following injection [(CCL2: t(17)=6.07, p<0.0001; IL1-β: t(16)=6.31, p<0.0001; TNFα: t(13)=4.97, p=0.0003). Twenty-four hours after the acute dose of ethanol the levels of cytokine-mRNAs were not significantly different from control P>0.05. Acute-ethanol (C) and Acute LPS (D) treatments did not alter TLR4 mRNA expression either at four hours (Fig C and D) or 24-hrs after their acute dosing (p>0.05). Thus, the acute ethanol exposure did not recapitulate the increase in cytokine-mRNAs induced by acute LPS administration. * Significantly different from control (p<0.05).
Table 2.
Acute-Ethanol or Acute-LPS on CCL2, IL1β, and TNFα-Peptides.
| Treatment | CCL2 | IL1β | TNFα |
|---|---|---|---|
| Water (n) | 29.03 +/− 1.11 | 1.12 +/− 0.04 | 0.15 +/− 0.04 |
| Ethanol (n) | 27.01 +/− 0.78 | 1.15 +/− 0.03 | 0.37 +/− 0.04* |
| Saline (n) | 34.49 +/− 1.94 | 0.81 +/− 0.06 | 0.30 +/− 0.01 |
| LPS (n) | 196.96 +/− 20.55** | 9.33 +/− 1.38*** | 0.28 +/− 0.03 |
Data from cerebral cortex are mean +/− SEM of cytokine-peptides/mg total protein. Acute-LPS (lipopolysaccharide) (250 ug/kg; n = 8) on CCL2 = Chemokine (C-C motif) ligand 2; IL1β = Interleukin-1β; TNFα = Tumor Necrosis Factor-α. Water and ethanol (2.75 g/kg) were administered intragastrically (n = 4 and 8, respectively). Saline and LPS were administered IP (n = 8 for both). TNFα:
t(10) = 3.88, p<0.005 vs water;
CCL2: t(14) = 7.87, p< 0.0001 vs saline;
IL1β: t(14) = 6.14, p<0.0001 vs Saline.
Collectively, these results for acute LPS and acute-ethanol on cytokine-mRNAs and tissue cytokine levels provides a clear distinction between acute-ethanol and acute-LPS administration on neuroimmune responsiveness. Regardless, because a comparable concordance in cytokine-peptide change to that for cytokine-mRNAs was not observed for either of the acute treatments, this lack of agreement is a clear alert that more must be learned about the biological control by which brain cytokine-peptides are synthesized from their corresponding mRNAs.
Comparison of a Chronic-Ethanol Protocol with Chronic-LPS on Cytokine-mRNAs
To assess for potential cytokine-mRNA differences between the CE-protocol and chronic-LPS exposure (see Figs IC & 1D-schematics), measures of cytokine-mRNAs were determined 24-hours following withdrawal from each. After CE-withdrawal, mRNAs for all cytokines showed significant increases compared to control (CCL2=144.9%; IL1-β=97.3%; TNFα=125%; Fig. 3A). In contrast to these increases induced by the CE-exposure, no significant increases in CCL2-, IL1-β-, and TNFα-mRNAs were observed after the last chronic-LPS dose (Fig. 3B). Thus, this comparison of CE- and chronic-LPS-exposure on brain cytokine-mRNAs provided further evidence for distinct influences being present for LPS and ethanol.
Figure 3. Cytokine-mRNA expression following a chronic-ethanol-protocol (Fig 1.C) and a chronic-LPS protocol (Fig 1D).

Twenty-four-hours after withdrawal from 15 days of 7% ethanol-diet (Chronic ethanol), cytokine-mRNAs for CCL2, IL1-β, and TNFα are significantly increased compared to non-treated controls (CD) (t(15)=4.53, p=0.0004; t(20)=3.42, p=0.0027; t(21)=5.443, p<0.0001, respectively. In contrast, 24-hours after the chronic-LPS exposure cytokine-mRNAs for CCL2, IL1-β, and TNFα were not significant increased (all cytokines = p>0.05). Again, the CE- and chronic-LPS protocols differed in their expression of cytokine-mRNAs. * Significantly different from Control (CD) (p<0.05).
Comparison of CE-Exposure Versus Chronic-LPS Treatment on TLR4, HMGB1, myD88, and NF-κB mRNAs
After finding increased cytokine-mRNAs following CE-exposure and the chronic-LPS treatments, the effect of CE and chronic-LPS on mRNAs for TLR4, HMGB1 and other links to TLR4 function were ascertained (Fig 4). Interestingly, a significant increase in TLR4-mRNA was found after 24-hours of withdrawal for both the CE-exposure (63.3% increase) and the exposure to chronic-LPS (79.2% increase) (Fig 4A & 4B), without an observed differences between these groups (p>0.05). Thus, this lack of difference between the CE- and chronic-LPS protocols in the TLR4-mRNA increase provided no further evidence for a distinction between ethanol and LPS action.
Figure 4. Effect of the chronic-ethanol protocol and chronic-LPS protocol on mRNAs for TLR4, HMGB1, My88, and NFκB.

(A). Chronic-ethanol significantly increased TLR4 mRNA expression [t(23)=2.669, p=0.0137]. (B) Chronic-LPS (250 μg/kg/day for 10 days; Fig 1D) also significantly increased TLR4 [t(18)=2.83, p=0.0112]. (C) Chronic-Ethanol significantly increased HMGB1 mRNA exposure [t(16)=3.618, p=0.0023]. (D) Chronic-LPS was without effect on mRNA for HMGB1 (p>0.05). (E-H) Chronic-ethanol and chronic-LPS did not significantly affect either My88 (EF) or NF-κB (GH) mRNA levels (p>0.05). * Significantly different from Control (p<0.05).
Subsequently, a test of whether a change in HMGB1-mRNA would be observe for the CE and chronic-LPS treatments was undertaken. Whereas CE-exposure significantly elevated HMGB1-mRNA by 102.8%, the chronic-LPS did not alter HMGB1-mRNA (Figs 4C & 4D). Further, neither the CE-protocol nor the chronic-LPS protocol induced a significant change in the MyD88- or NF-Kb-mRNAs (Fig 4G & 4H), both of which are linked to TLR4 function (Fig 4E & 4F). Consequently, these data on the induction of HMGB1-mRNA by CE-exposure and chronic-LPS provided another distinction between the action of ethanol and LPS.
Time-Course of Cytokine-mRNAs Following CE-Withdrawal
Upon finding that cytokine-mRNAs increased 24-hours after CE-exposure (Fig 3), mRNA-expression was next determined for cytokine-mRNAs at various times (0, 1, 3, and 7-days) after withdrawal from CE (Fig. 5A). No significant change was observed in cytokine-mRNAs in rats sacrificed while still exposed to alcohol drinking (i.e., T=0). However, in confirmation of previous cytokine-mRNA data after the CE-exposure (Fig 3), mRNA expression significantly increased by 102% for CCL2, by 93.8% for IL1-β, and by 107% for TNFα 24-hours after CE-withdrawal (Fig 5A). Subsequently, by 3-days cytokine-mRNAs gradually declined with changes being approximately 68% for CCL2, 1.0% for IL1-β and 61% for TNFα above control (Fig. 5A). By 7-days, these cytokine-mRNAs returned to control levels. Thus, these findings confirm that the increases in CCL2, IL1-β, and TNFα-mRNAs following 24-hours of abstinence from CE-exposure results in time-dependent changes over the 7-day period.
Figure 5. Time course of changes for cytokine-mRNAs (top) and TLR4- and HMGB1-mRNAs (bottom) while on (o-hr) and 24-hours after withdrawal from the CE-protocol.

For cytokine-mRNAs (top), CCL2, IL1-β, and TNFα mRNAs significantly peaked 24-hours after withdrawal from the CE-protocol compared to control [CCL2-mRNA = Control 100 ± 31.1 vs. CCL2 peak = 202.0 ± 33.3, p=0.025 (F(4,25)= 3.06, p=0.035]. [IL1-β-mRNA = Control 100 ± 11.0 vs. IL1-β peak = 193.8 ± 38.9, p=0.0039) (F(4,27)= 5.05, p=0.036]; TNFα = Control 100 ± 9.3 vs. TNFα-Peak= 207.3 ± 38.7, p=0.0006) (F(4,27)=7.30; p=0.0004]. At time t=0, cytokine-mRNAs were not significantly increased (p>0.05). The peak expression of TLR4-mRNA 24-hours following withdrawal from the CE-protocol (Bottom) was 218.7 ± 19.4 for TLR4 vs 100 ± 13.9 for control (F(4,27)=8.64, p=0.0001) and 164.5 ± 15.6 for HMGB1 vs 100 ± 21.9 for the Control (F(2,26)=3.51, p=0.02). At T=0 (0 hr) TLR4 was not significantly different from control (p>0.05). However, while the HMGB1-mRNA elevation at T=0 (0 hr) is significant, but only to a minor degree (p<0.05), an additional investigation was unable to duplicate this significant increase [Control = 100 ± 16.4% vs HMGB1-mRNA = 120 ± 13.5%; p>0.05]. See text for descriptions of changes for cytokine-mRNAs and the TLR4 and HMGB1-mRNAs 7 days after withdrawal from the CE-protocol.
Time Course of TLR4- and HMGB1 mRNA Changes Following CE-Exposure
Because TLR4 and HMGB1-mRNA expression was elevated after removal from CE-exposure (Fig 4), the time course for TLR4- and HMGB1-mRNAs changes were also determined (Fig 5B). Prior to ethanol withdrawal (T=0), the TLR4-mRNA level was not significantly different from control. However, with 24-hours (1-day) of CE-withdrawal, the TLR4-mRNA had significantly risen by approximately 68% (Fig 5B), consistent with data in Fig 4B. By 72-hours (3-days) after CE-withdrawal, TLR4-mRNA fell to control levels and by 7-days the TLR4-mRNA dropped to 81% below control (p<0.05; Fig 5B).
Prior to ethanol-diet removal (T=0) the level of HMGB1 mRNA was significantly elevated by 35% above control (Fig. 5.B). However, in a second experiment, this HMGB1-mRNA increase was not found to be altered at T=0 (p>0.05; see Fig 5 legend). Regardless, in agreement with Fig. 4B, the HMGB1-mRNA had significantly increased by 119% above control by 24-hours (Fig 5B). Subsequently, similar to TLR-mRNA, HMGB1-mRNA levels dropped to control levels by 72-hours after CE-withdrawal and by 7 days fell to 88% below control (p<0.05; Fig 5B). Thus, a distinct difference was observed in the changes in TLR4 and the HMGB1-mRNAs over time compared to the alterations observed for the cytokine-mRNAs after withdrawal from the CE-exposure.
CE- or Chronic-Intermittent-Ethanol (CIE) Exposures on Cytokine-mRNAs and the CE-Exposure on Cytokine-Peptides
We previously reported that withdrawal from the CIE-protocol (cycled ethanol; Fig 1F) increased seizure-susceptibility and anxiety-like behavior to a greater degree than did a comparable continuous-CE-protocol (McCown and Breese, 1990; Overstreet et al., 2002), a distinction which prompted determining if a divergence in the expression of cytokine-mRNAs would be observed 24-hours after withdrawal from these differing CE-protocols. This assessment (Fig 6) found that cytokine-mRNAs significantly increased after withdrawal from the CE-exposure (i.e., CCL2=79.8%; IL1-β=102.3%; TNFα=96.2%) and the CIE-exposure (i.e., CCL2=70.5%; IL1-β=82.0%; TNFα=108.4%), but a statistical difference between these two exposures was not present. Unlike the increase in cytokine-mRNAs after withdrawal from CE-exposure (Fig 6), cytokine-peptide values determined 24-hours after the CE-protocol exposure did not demonstrate a significant change (Table 3).
Figure 6. Comparison of CE-protocol (Chronic ETOH; see Fig 1C) and Chronic-Intermittent Ethanol (Cycled ETOH; see Fig 1F).

Twenty-four hours after removal from ethanol diet, both the Chronic Ethanol- and Cycled Ethanol-treatment groups showed significant increases in CCL2, IL1-β, and TNFα-mRNA [F(2,18)= 5.211, p=0.018; F(2,24)=9.718, p=0.0009; F(2,25)=6.475, p=0.006, respectively]. However, a statistical difference did not occur between the cytokine-mRNAs for the Chronic ETOH and Cycled ETOH-treatment groups (p>0.05). * Significantly different from Control (p<0.05).
Table 3.
CCL2 , IL1β, and TNFα-Peptides 24-hours after Withdrawal from CE-Exposure.
| Treatment | CCL2 | IL1β | TNFα |
|---|---|---|---|
| Control Diet | 34.5 +/− 1.9 (8) | 1.14 +/− 0.11 (8) | 0.38 +/− 0.07 (8) |
| CE-24-Hr after Withdrawal |
36.7 +/− 1.8 (16) | 1.01 +/− 0.08 (10) | 0.32 +/− 0.04 (9) |
Data from cerebral cortex are mean +/− SEM of cytokine-peptides/mg total protein (n=number). CE = Chronic ethanol exposure. See Methods for Control Diet. CE-24-hr after withdrawal = Rats being removed for 24 hrs from the CE-exposure (see Methods)
Effect of the CE- and CIE-Protocols on mRNAs for TLR4, HMGB1, My88, and NF-κB
Further evaluation of the two-protocols revealed that TLR4-mRNA for the CE-(122.6%) and CIE-(85.2%)-protocols were significantly increased (Fig. 7A), but these increases for these exposures also did not differ (p>0.05). The HMGB1-mRNA for both the CE- and the CIE protocols 24-hours following withdrawal was also significantly increased (CE=64.4%; CIE=57.3%; Fig. 7B), but these increases also were not significantly different (Fig 7B). Finally, analysis of MyD88 and NF-κB-mRNAs determined no significant change for either of these measures in these CE-exposed groups (Fig 7C & 7D). Thus, both the CE- and CIE-groups increased mRNA expression for cytokines, TLR4 and HMGB1 to an equivalent degree without affecting mRNAs for My88 or NF-κB 24-hours after withdrawal.
Figure 7. Comparison of CE- and cycled-ethanol treatment groups on mRNAs for TLR4, HMGB1, MyD88 and NF-κB.

(A) TLR4 was significantly elevated in both the chronic and cycled ethanol groups (F(2,23)=8.744, p=0.0017); However, a difference between the chronic and cycled groups was not observed (p>0.05). (B) Both the chronic and cycled ethanol protocol groups significantly elevated HMBG1-mRNA (F(2,24)=3.787, p=0.0466), but no difference between the chronic- and cycled-ethanol protocols was noted (p>0.05). (C&D) Neither the chronic nor the cycled ethanol protocol groups caused a discernible effect on levels of MyD88 or NF-κB p>0.05). * Significantly different from CD (p<0.05).
Effect of a CRF1RA Antagonist and HMGB1-Antagonists on Cytokine- and HMGB1-mRNA Increases Following CE-Withdrawal
To determine if an upstream-influence of CRF-release was contributing to the elevation in cytokine-mRNAs during the 24-hours of CE-withdrawal, a CRF1RA was initially administered twice during the withdrawal from CE-protocol (Fig 1G-schematic). This CRF1RA-treatment significantly reduced the increase in the mRNAs for CCL2, IL1β, and TNFα that followed withdrawal from the CE-exposure (Fig 8). Subsequently, based upon the hypothesis that HMGB1 was involved in the cytokine-mRNA increases, administration of the HMGB1-antagonist, ethyl-pyruvate (Su et al, 2011) twice during the 24-hours of CE-withdrawal (Fig 1G) significantly reduced CCL2, IL1β, and TNFα-mRNA increases induced by the CE-withdrawal (Fig 8). The HMGB1 antagonist, glycyrrhizin (Ohnishi et al. 2011), also significantly reduced the increase in IL1β and TNFα-mRNAs that followed withdrawal from CE-exposure, but did not significantly reduce the withdrawal-induced increase in CCL2-mRNA (Fig 8).
Figure 8. Testing of a CRF1RA [CP154,526 (CP: 10 mg/kg)] or the HMGB1 antagonists, glycyrrhizin (Gly: 50 mg/kg) and ethyl-pyruvate (EP: 75 mg/kg), on the cytokine-mRNA increases induced by 24-hrs of withdrawal (WD) from CE-exposure. Each drug was administered at 3 and 12 hours into the withdrawal period after cessation of ethanol diet exposure.

There were significant group effects for each mRNA [TNFα = F(4, 37) = 6.16, p<0.05; IL1β = F(4,37) = 3.46, p<0.05; and CCL2 = F(4,42) = 4.28, p<0.05]. TNFα, CCL2, & IL1β-mRNAs were significantly elevated 24-hours after withdrawal from the CE-24h-WD protocol (P<0.05), in accord with results in Fig 3. The CRF1 receptor antagonist CP154,526 (CP: 10 mg/kg) and ethyl-pyruvate (EP: 75 mg/kg) significantly reduced the cytokine mRNA increases for TNFα and IL1β 24-hours after removal from the CE-protocol (p<0.05). After the HMGB1 inhibitor, glycyrrhizin (Gly: 50 mg/kg), the suppression of CCL2-mRNA from the CE-24-WE-veh did not reach statistical significance (p=0.059).
*Significantly different from CE-24h-WD (p<0.05).
The effect of the CRF1RA and HMGB1-antagonists on the increase in HMGB1-mRNA increase following CE-withdrawal was also determined (Fig 9). The CRF1RA significantly reduced the HMGB1-mRNA induced by CE-withdrawal. Likewise, ethyl-pyruvate also significantly inhibited the HMGB1-mRNA increase by the CE-withdrawal; however, glycyrrhizin caused only a non-significant reduction (p=0.094) in HMGB1-mRNA.
Figure 9. Testing of the CRF1-receptor antagonist CP154,526 (CP: 10 mg/kg) or the HMGB1-antagonists, glycyrrhizin (Gly: 50 mg/kg) and ethyl-pyruvate (EP: 75 mg/kg), on the HMGB1-mRNA increase induced by withdrawal (WD) from chronic ethanol (CE)-exposure.
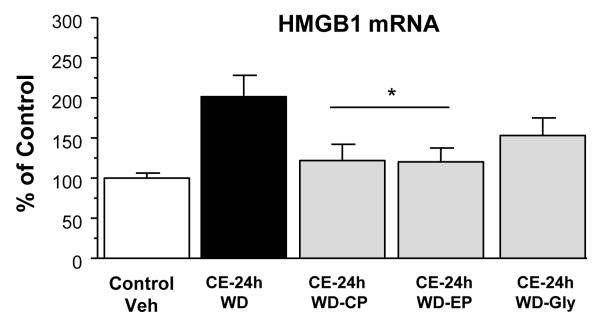
See Figure 8 for further details. Overall effect of groups was significant, [F(4,41) = 3.96, p<0.05]. The CRF1 receptor antagonist [CP154,526 (CP: 10 mg/kg)] and ethyl-pyruvate (EP: 75 mg/kg) significantly reduced the HMGB1-mRNA increase after removal from the CE-24-WD-protocol (p<0.05). The trend toward suppression of HMGB1 with glycyrrhizin after the CE-24-WD protocol did not reach statistical significance (p=0.094).
* Significantly different from CE-24h-WD (p<0.05).
Collectively, these pharmacological strategies supported CRF and HMGB1 being involved in the increased cytokine-mRNA and HMGB1-mRNA expressions observed following withdrawal from the CE-protocol. 2857
DISCUSSION
To account for ethanol induction of cytokines in brain, Fernendez-Lizarbe et al. (2008) suggested that ethanol, similar to LPS, causes a TLR4 accumulation in lipid-rafts to initiate TLR4-signaling (see Introduction). LPS, a membrane component of bacteria (Erridge et al., 2002), increases cytokines in brain, presumably by acting as a TLR4-agonist (Erridge et al., 2002; Okun et al., 2009). However, Qin et al. (2007) reported that the LPS-induced cytokine-mRNA induction in brain was based upon TNFα in blood entering brain to contribute to the LPS-induced cytokine increase—a differing view. Thus, as compelling as the lipid-raft hypothesis is for explaining the basis of TLR4-involvement in ethanol increasing cytokines in brain (Fernendez-Lizarbe et al., 2008), the mechanism by which CE-exposure involves TLR4 in the increase in cytokine-mRNAs in the sterile environment of brain remains unclear, particularly because ethanol is unlikely to directly act as an agonist on TLR4s. Based upon the view that ethanol does not alter TLR4-function directly, this investigation first undertook exploring for potential differences between the agonist action of LPS on TLR4s with the acute and chronic actions of ethanol on mRNA-expression of pro-inflammatory-cytokines and immune signaling proteins.
By comparing acute and chronic-ethanol and LPS-exposures, we gained valuable insight as to the possible mechanism by which ethanol depends upon TLR4-signaling to increase brain cytokines (see Introduction). An acute intoxicating dose of ethanol did not increase cytokine-mRNAs in cortex of Sprague-Dawley rats 4-hours after administration—a finding distinct from the several-fold increase in the cytokine-mRNAs observed 4-hours after acute-LPS administration (Fig 2). Consistent with these findings, Buck et al. (2011) also found no increase in IL-6 or IL1β-mRNAs in Sprague-Dawley rats 12-hours after ethanol (4g/kg). However, Qin et al. (2008) reported in C57Bl/6 mice that a single 5g/kg oral dose of ethanol increased TNFα- and CCL2-mRNAs in whole brain 24-hours after ethanol-dosing, without a change IL1β-mRNA. Other than the differing species, whole-brain versus cortex, and/or the higher dose of ethanol utilized, another possible reason for this distinction between studies could in part relate to Sprague-Dawley rats being less sensitive than mice to challenges which increase cytokine function (see Porterfield et al, 2011). While a significant change in cytokine-mRNAs was lacking after acute-ethanol administration, CE-exposure produced an approximate doubling of cytokine-mRNAs in cortex 24-hours after CE-withdrawal. Distinct from this cytokine-mRNA increase by CE-withdrawal, chronic-LPS produced no elevation in cortical cytokine-RNAs 24-hours after removal from LPS dosing—a presumed reflection of the tachyphylaxis that occurs following repeated LPS-exposure (Vartanian and Stenzel-Poore, 2010; West and Heagy, 2002; Ziegler–Heitbrock, 1995). Thus, the present findings comparing acute- and chronic-ethanol to acute- and chronic-LPS exposures provide clear evidence for a fundamental difference in the ability of LPS and ethanol to influence neuroimmune-function in brain.
Previous studies in our lab established that CIE-exposure is more effective than exposure to the CE-protocol at engendering anxiety-like symptoms during ethanol withdrawal (Breese et al., 2011; Overstreet et al., 2002) or for increasing kindling of seizures (McCown and Breese, 1990). Based on the possibility that CIE might produce a greater induction of cytokine-mRNAs than the CE-exposure, these two CE-protocols were compared for their ability to induce cytokine-mRNAs 24-hours following CE-withdrawal. The increase in CCL2, IL1β, and TNFα cytokine-mRNAs observed after CE-withdrawal did not differ from the cytokine-mRNA increase observed during withdrawal from the CIE-protocol. Thus, unlike the “kindling” responsible for CIE-facilitation of withdrawal anxiety (Breese et al, 2005; 2011; Overstreet et al., 2002) and the kindling of seizures (McCown and Breese, 1990), cytokine-mRNA changes following the two CE-protocols were comparable. One possible explanation for not observing a kindling-mediated difference between these CE-protocols could be from having measured the elevation of cytokine-mRNAs only in cortex—a site not associated with CIE-susceptibility for seizures (McCown and Breese, 1990) or with facilitation of anxiety (Overstreet et al., 2002; Breese et al. 2011). Consequently, the relationship between brain site and neuroimmune function after these differing CE-protocols will need exploration in future investigations to resolve whether cytokine-mRNAs in differing brain sites explain this lack of difference in cytokine-mRNA expression observed by these differing CE-protocols.
Studies in whole-brain of C57Bl/6 mice found that 24-hours of ethanol abstinence from 10-days of ethanol administration (5-gm/kg-IG/day) increased CCL2- and TNFα-mRNAs, but not mRNAs for IL1-β or other cytokines (Qin et al., 2008). Whereas the CE-protocol produced an increase in cytokine-mRNAs in cortex 24-hours after withdrawal, the time-course determination demonstrated that cytokine-mRNAs were not changed while still being exposed to the CE-diet (T=0). Therefore, rather than just exposure to the CE-protocol, the findings were a clear indication that the cytokine-mRNA increase associated with the CE-protocol is not due to the chronic-ethanol exposure alone, but is dependent upon the 24-hour withdrawal-period from the CE-exposure.
Given that the increase in cytokine-mRNAs induced by withdrawal from CE occurs in the sterile environment of brain, a critical issue remained as to how this CE-withdrawal drives this increased expression of cytokine-mRNAs. Therefore, based upon the documented evidence for the TLR4 importance in ethanol’s action (see Introduction), mRNA levels were determined for TLR4 as well for HMGB1, an endogenous TLR4-agonist (Andersson and Tracey, 2011; Lin et al., 2005). Acute-ethanol caused no change in TLR4-mRNA, but the CE- and CIE-protocols (Fig 7A) elevated TLR4-mRNA as well as HMGB1-mRNA without affecting mRNAs for either MyD88- or NF-κB. Even though the HMGB1-mRNA was nearly doubled following withdrawal from the CE-protocols, the HMGB1-mRNA prior to removal from the CE-exposure (T=0) revealed at best only a small change—a time when TLR4 and cytokine-mRNAs levels were not different from control. The TLR4- and HMGB1-mRNAs peaked 24-hours after the withdrawal from the CE-protocol and gradually fell to control level by 3-days and below the control levels by 7-days. The loss of the HMGB1- and TLR4-mRNA temporal relationship with the cytokine-mRNAs by 7 days after the removal from the CE-protocol days was certainly unexpected. Therefore, the biological basis of this difference should be sought. Regardless, the increase in the HMGB1- and TLR4-mRNAs having a temporal relationship with the elevated cytokine-mRNAs 24-hours after CE-withdrawal was considered a critical clue for understanding how expression of cytokine-mRNAs was increased during the CE-withdrawal.
From the demonstration that withdrawal from CE-exposure increased HMGB1-mRNA and chronic-LPS did not, the possibility was considered that HMGB1 may be an endogenous ligand released from neurons in brain (Faraco et al., 2007) during the CE-withdrawal that would activate TLR4s to increase brain cytokine-mRNAs (Andersson and Tracy, 2011; Faraco et al., 2007; Lin et al., 2012; Müller et al., 2001; Yang et al., 2005; Yu et al., 2006). This intriguing presumption that HMGB1 release onto TLR4s contributes to the cytokine-mRNA increases during CE-withdrawal was potentially consistent with various ethanol actions being dependent upon TLR4 function (Alfonso-Loeches et al., 2010; Blanco et al., 2005; Fernandez-Lizarbe et al., 2008; 2009; see Introduction). In support of this view, Crews et al. (2012) observed that HMGB1-neutralizing antibodies added to brain slice cultures blunted ethanol-induction of IL1β, a finding suggestive that this activation occurs through a HMGB1/TLR-signaling mechanism. Nonetheless, because a CRF1RA prevents facilitation of anxiety following CE-withdrawal (Breese et al., 2011; Knapp et al., 2004), the possible involvement of CRF-release was also considered as a potential contributor to the increase in cytokine-mRNAs during withdrawal from CE.
To test this hypothesis that HMGB1 and/or CRF support the cytokine-mRNA increase induced by CE-withdrawal, antagonists for HMGB1 and CRF were administered twice during the 24-hour period of withdrawal from the CE-protocol. The HMGB1-antagonists utilized were glycyrrhizin (Faraco et al., 2007; Mollica et al., 2007; Ohnish et al., 2011) and ethyl pyruvate (Davis et al., 2009; Su et al., 2011). The administration of the CRF1RA, ethyl-pyruvate, and glycyrrhizin during the 24-hour period of CE-withdrawal all significantly reduced the CCL2, IL1β and TNFα-mRNA increases that followed withdrawal. Thus, these seminal findings clearly supported both HMGB1 and CRF being critical for the increased expression of cytokine-mRNAs during the 24-hour withdrawal period that follows exposure to the CE.
The consequences of the CRF1RA and the HMGB1-antagonists were also tested against the HMGB1-mRNA induction during withdrawal from the CE-protocol. Finding that the CRF1RA and ethyl-pyruvate, but not glycyrrhizin, reduced the HMGB1-mRNA increase that follows CE-withdrawal potentially provided further clarification of the potential mechanism by which cytokine-mRNAs increase during withdrawal from the CE-protocol. One interpretation from the outcome with the CRF1RA reduction of the HMGB1-mRNA strongly suggests that the blockade of the action of CRF-released during CE-withdrawal is controlling activation of HMGB1-mRNA—a mechanism which in turn prevents the formation and release of HMGB1 to increase cytokine-mRNAs during the period of CE-withdrawal (see Fig 8). However, in respect to the CRF1RA reduction in the expression of HMGB1-mRNA, a degree of caution should be considered until established that this antagonist does not have an influence on other distinct receptors associated directly with brain neuroimmune-function. Regardless, an explanation for the ethyl-pyruvate preventing HMGB1-release (Dave et al., 2009) to prevent the cytokine-mRNA increase following withdrawal from the CE-protocol could relate to an intracellular action of this HMGB1-antagonist preventing expression of HMGB1-mRNA. Based upon this mechanism proposed for ethyl-pyruvate preventing HMGB1-release by minimizing further expression of HMGB1-mRNA, a future study to critically test this view would seem warranted. On the other hand, glycyrrhizin binding to HMGB1 (Girard, 2007; Mollica et al., 2007), while blocking the increase in the cytokine-mRNAs, did not significantly alter the HMGB1-mRNA increase induced by the withdrawal from the CE-protocol. Consequently, the present data defining the distinct effects of ethyl-pyruvate and glycyrrhizin on HMGB1-mRNA would seem consistent with mechanisms previously proposed for these HMGB1-antagonists preventing HMGB1-action (Dave et al., 2009; Girard et al. 2007; Su et al., 2011). In conclusion, the CRF1RA and HMGB1-antagonists preventing HMGB1 and CRF from increasing cytokine-mRNAs induced by CE-withdrawal should be considered a first step in clarifying the biological means by which neural and neuroimmune-processes can interact to influence cytokine-mRNA expression in the sterile-milieu of brain. 4488
ACKNOWLEDGEMENTS
The authors wish to thank Dr. A. Leslie Morrow for generous contribution of time, space and equipment to this project. We also acknowledge and thank Zachary Zimomra, Bob Angel, and Todd O’Buckley for their excellent technical assistance. Dr. Joyce Besheer provided invaluable help with the oral-gavage procedure.
Funding: This work was supported by the National Institutes of Health, National Institute on Alcoholism and Alcohol Abuse [AA11605, AA17462, AA14949] and the Bowles Center for Alcohol Studies.
REFERENCES
- Akira S, Takeda K. Toll-like receptor signaling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- Alfonso-Loeches S, Pascual-Lucas M, Blanco AM, Sanchez-Vera I, Guerri C. Pivotal role of TLR4 receptors in alcohol-induced neuroinflammation and brain damage. J Neurosci. 2010;30:8285–8295. doi: 10.1523/JNEUROSCI.0976-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29:139–162. doi: 10.1146/annurev-immunol-030409-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeuerle PA, Henkel T. Function and activation of NF-kappa B in the immune system. Annu Rev Immunol. 1994;12:141–79. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- Blanco AM, Vallés SL, Pascual M, Guerri C. Involvement of TLR4/type I IL-1 receptor signaling in the induction of inflammatory mediators and cell death induced by ethanol in cultured astrocytes. J Immunol. 2005;175:6893–6899. doi: 10.4049/jimmunol.175.10.6893. [DOI] [PubMed] [Google Scholar]
- Breese GR, Knapp DJ, Overstreet DH, Navarro M, Wills TA, Angel RA. Repeated lipopolysaccharide (LPS) or cytokine treatments sensitize ethanol withdrawal-induced anxiety-like behavior. Neuropsychopharmacology. 2008;33:867–876. doi: 10.1038/sj.npp.1301468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breese GR, Overstreet DH, Knapp DJ. Conceptual framework for the etiology of alcoholism: a “kindling”/stress hypothesis. Psychopharmacology. 2005;178:367–380. doi: 10.1007/s00213-004-2016-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breese GR, Sinha R, Heilig M. Chronic alcohol neuroadaptation and stress contribute to susceptibility for alcohol craving and relapse. Pharmacol Ther. 2011;129:149–171. doi: 10.1016/j.pharmthera.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck HM, Hueston CM, Bishop C, Deak T. Enhancement of the hypothalamic-pituitary-adrenal axis but not cytokine responses to stress challenges imposed during withdrawal from acute alcohol exposure in Sprague-Dawley rats. Psychopharmacology. 2011;218:203–215. doi: 10.1007/s00213-011-2388-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Zou J, Qin L. Induction of innate immune genes in brain create the neurobiology of addiction. Brain Behav Immun. 2011;25:S4–S12. doi: 10.1016/j.bbi.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews F, Qin L, Sheedy D, Vetreno, Zou J. Increased DANGER Signaling through HMGB1 and TLR Receptors in Alcohol. Biol Psychiatry Resubmission. 2012 doi: 10.1016/j.biopsych.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave SH, Tilstra JS, Matsuyoshi M, Li F, Demarco RA, Beer-Stolz D, Sepulveda AR, Fink MP, Lotze MT, Plevy SE. Ethyl pyruvate decreases HMGB1 release and ameliorates murine colitis. J Leukoc Biol. 2009;86:633–643. doi: 10.1189/jlb.1008662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erridge C, Bennett-Guerrero E, Poxton IR. Structure and function of lipopolysaccharides. Microbes Infect. 2002;4:837–851. doi: 10.1016/s1286-4579(02)01604-0. [DOI] [PubMed] [Google Scholar]
- Faraco G, Fossati S, Bianchi ME, Patrone M, Pedrazzi M, Sparatore B, Moroni F, Chiarugi A. High mobility group box 1 protein is released by neural cells upon different stresses and worsens ischemic neurodegeneration in vitro and in vivo. J Neurochem. 2007;103:590–603. doi: 10.1111/j.1471-4159.2007.04788.x. [DOI] [PubMed] [Google Scholar]
- Fernandez-Lizarbe S, Pascual M, Gascon MS, Blanco A, Guerri C. Lipid rafts regulate ethanol-induced activation of TLR4 signaling in murine macrophages. Mol Immunol. 2008;45:2007–2016. doi: 10.1016/j.molimm.2007.10.025. [DOI] [PubMed] [Google Scholar]
- Fernandez-Lizarbe S, Pascual M, Guerri C. Critical role of TLR4 response in the activation of microglia induced by ethanol. J Immunol. 2009;183:4733–4744. doi: 10.4049/jimmunol.0803590. [DOI] [PubMed] [Google Scholar]
- Fitzgerald KA, Rowe DC, Barnes BJ, Caffrey DR, Visintin A, Latz E, Monks B, Pitha PM, Golenbock DT. LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J Exp Med. 2003;198:1043–1055. doi: 10.1084/jem.20031023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye GD, McCown TJ, Breese GR. Characterization of susceptibility to audiogenic seizures in ethanol-dependent rats after microinjection of gamma-aminobutyric acid (GABA) agonists into the inferior colliculus, substantia nigra or medial septum. J Pharmacol Exp Ther. 1983;227:663–670. [PMC free article] [PubMed] [Google Scholar]
- Girard JP. A direct inhibitor of HMGB1 cytokine. Chem Biol. 2007;14:345–347. doi: 10.1016/j.chembiol.2007.04.001. [DOI] [PubMed] [Google Scholar]
- He J, Crews FT. Increased MCP-1 and microglia in various regions of the human alcoholic brain. Exp Neurol. 2008;210:349–358. doi: 10.1016/j.expneurol.2007.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens S, Beyaert R. A universal role for MyD88 in TLR/IL-1R-mediated signaling. Trends Biochem Sci. 2002;27:474–482. doi: 10.1016/s0968-0004(02)02145-x. [DOI] [PubMed] [Google Scholar]
- Knapp DJ, Overstreet DH, Moy SS, Breese GR. SB242084, flumazenil, and CRA1000 block ethanol withdrawal anxiety in rats. Alcohol. 2004;32:101–111. doi: 10.1016/j.alcohol.2003.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp DJ, Whitman BA, Wills TA, Angel RA, Overstreet DH, Criswell HE, Ming Z, Breese GR. Cytokine involvement in stress may depend on corticotrophin releasing factor to sensitize ethanol withdrawal anxiety. Brain Behav Immun. 2011;25:S146–154. doi: 10.1016/j.bbi.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q, Yang XP, Fang D, Ren X, Zhou H, Fang J, Liu X, Zhou S, Wen F, Yao X, Wang JM, Su SB. High-mobility group box-1 mediates toll-like receptor 4-dependent angiogenesis. Arterioscler Thromb Vasc Biol. 2011;31:1024–1032. doi: 10.1161/ATVBAHA.111.224048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroso M, Balosso S, Ravizza T, Liu J, Aronica E, Iyer AM, Rossetti C, Molteni M, Casalgrandi M, Manfredi AA, Bianchi ME, Vezzani1 A. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nature Med. 2010;16:413–420. doi: 10.1038/nm.2127. [DOI] [PubMed] [Google Scholar]
- McCown TJ, Breese GR. Multiple withdrawals from chronic ethanol “kindles” inferior-collicular seizure activity: evidence for kindling of seizures associated with alcoholism. Alcohol Clin Exp Res. 1990;14:394–399. doi: 10.1111/j.1530-0277.1990.tb00492.x. [DOI] [PubMed] [Google Scholar]
- Mollica L, De Marchis F, Spitaleri A, Dallacosta C, Pennacchini D, Zamai M, Agresti A, Trisciuoglio L, Musco G, Bianchi ME. Glycyrrhizin binds to high-mobility group box 1 protein and inhibits its cytokine activities. Chem Biol. 2007;14:431–441. doi: 10.1016/j.chembiol.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Müller S, Scaffidi P, Degryse B, Bonaldi T, Ronfani L, Agresti A, Beltrame M, Bianchi ME. New EMBO members’ review: the double life of HMGB1 chromatin protein: architectural factor and extracellular signal. EMBO J. 2001;20:4337–4340. doi: 10.1093/emboj/20.16.4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi M, Katsuki H, Fukutomi C, Takahashi M, Motomura M, Fukunaga M, Matsuoka Y, Isohama Y, Izumi Y, Kume T, Inoue A, Akaike A. HMGB1 inhibitor glycyrrhizin attenuates intracerebral hemorrhage-induced injury in rats. Neuropharmacology. 2011;61(5-6):975–80. doi: 10.1016/j.neuropharm.2011.06.026. [DOI] [PubMed] [Google Scholar]
- Okun E, Griffioen KJ, Lathia JD, Tang SC, Mattson MP, Arumugam TV. Toll-like receptors in neurodegeneration. Brain Res Rev. 2009;59:278–292. doi: 10.1016/j.brainresrev.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overstreet DH, Knapp DJ, Breese GR. Accentuated decrease in social interaction in rats subjected to repeated ethanol withdrawals. Alcohol Clin Exp Res. 2002;26:1259–1268. doi: 10.1097/01.ALC.0000023983.10615.D7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pålsson-McDermott EM, O’Neill LAJ. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology. 2004;113:153–162. doi: 10.1111/j.1365-2567.2004.01976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual M, Baliño P, Alfonso-Loeches S, Aragón CM, Guerri C. Impact of TLR4 on behavioral and cognitive dysfunctions associated with alcohol-induced neuroinflammatory damage. Brain Behav Immun. 2011;25:S80–91. doi: 10.1016/j.bbi.2011.02.012. [DOI] [PubMed] [Google Scholar]
- Pascual M, Blanco AM, Cauli O, Minarro J, Guerri C. Intermittent ethanol exposure induces inflammatory brain damage and causes long-term behavioural alterations in adolescent rats. Eur J Neurosci. 2007;25:541–550. doi: 10.1111/j.1460-9568.2006.05298.x. [DOI] [PubMed] [Google Scholar]
- Porterfield VM, Zimomra ZR, Caldwell EA, Camp RM, Gabella KM, Johnson JD. Rat strain differences in restraint stress-induced brain cytokines. Neurosci. 2011;188:48–54. doi: 10.1016/j.neuroscience.2011.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, He J, Hanes RN, Pluzarev O, Hong JS, Crews FT. Increased systemic and brain cytokine production and neuroinflammation by endotoxin following ethanol treatment. J Neuroinflammation. 2008;5:10. doi: 10.1186/1742-2094-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X, Wang H, Zhao J, Pan H, Mao L. Beneficial effects of ethyl pyruvate through inhibiting high-mobility group box 1 expression and TLR4/NF-κB pathway after traumatic brain injury in the rat. Mediators Inflamm. 2011;2011:807142. doi: 10.1155/2011/807142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triantafilou M, Miyake K, Golenbock DT, Triantafilou K. Mediators of innate immune recognition of bacteria concentrate in lipid rafts and facilitate lipopolysaccharide-induced cell activation. J Cell Sci. 2002;115:2603–2611. doi: 10.1242/jcs.115.12.2603. [DOI] [PubMed] [Google Scholar]
- Uesugi T, Froh M, Arteel GE, Bradford BU, Thurman RG. Toll-like receptor 4 is involved in the mechanism of early alcohol-induced liver injury in mice. Hepatology. 2001;34:101–108. doi: 10.1053/jhep.2001.25350. [DOI] [PubMed] [Google Scholar]
- Valles SL, Blanco AM, Azorin I, Guasch R, Pascual M, Gomez-Lechon MJ, Renau-Piqueras J, Guerri C. Chronic ethanol consumption enhances interleukin-1-mediated signal transduction in rat liver and in cultured hepatocytes. Alcohol Clin Exp Res. 2003;27:1979–1986. doi: 10.1097/01.ALC.0000099261.87880.21. [DOI] [PubMed] [Google Scholar]
- Vartanian K, Stenzel-Poore M. Toll-like receptor tolerance as a mechanism for neuroprotection. Transl Stroke Res. 2010;1:252–260. doi: 10.1007/s12975-010-0033-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MA, Heagy W. Endotoxin tolerance: A review. Crit Care Med. 2002;30:S64–S73. [PubMed] [Google Scholar]
- Wills TA, Knapp DJ, Overstreet DH, Breese GR. Differential dietary ethanol intake and blood ethanol levels in adolescent and adult rats: effects on anxiety-like behavior and seizure thresholds. Alcohol Clin Exp Res. 2008;32:1350–1360. doi: 10.1111/j.1530-0277.2008.00709.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Lousberg EL, Moldenhauer LM, Hayball JD, Coller JK, Rice KC, Watkins LR, Somogyi AA, Hutchinson MR. Inhibiting the TLR4-MyD88 signalling cascade by genetic or pharmacological strategies reduces acute alcohol-induced sedation and motor impairment in mice. Br J Pharmacol. 2012;165:1319–1329. doi: 10.1111/j.1476-5381.2011.01572.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, Czura CJ, Tracey KJ. The cytokine activity of HMGB1. J Leukoc Biol. 2005;78:1–8. doi: 10.1189/jlb.1104648. [DOI] [PubMed] [Google Scholar]
- Yu M, Wang H, Ding A, Golenbock DT, Latz E, Czura C, Fenton M, Matthew J, Tracey KJ, Yang H. Hmgb1 Signals Through Toll-Like Receptor (Tlr) 4 and Tlr2 Shock. 2006;26:174–179. doi: 10.1097/01.shk.0000225404.51320.82. 2006. [DOI] [PubMed] [Google Scholar]
- Ziegler-Heitbrock HW. Molecular mechanism in tolerance to lipopolysaccharide. J Inflamm. 1995;45:13–26. [PubMed] [Google Scholar]
- Zou J, Crews F. Induction of innate immune gene expression cascades in brain slice cultures by ethanol: key role of NFκB and proinflammatory cytokines. Alcohol Clin Exp Res. 2010;34:777–789. doi: 10.1111/j.1530-0277.2010.01150.x. [DOI] [PubMed] [Google Scholar]