

Abstract
The majority of prostate cancers (PCa) express high levels of androgen receptor (AR) and are dependent for their growth on testosterone produced by the testes, which is reduced in the prostate to the higher affinity ligand 5α-dihydrotestosterone (DHT). PCa growth can be suppressed by androgen deprivation therapy, which involves removal of testicular androgens (surgical or medical castration) or treatment with an AR antagonist (or a combination of both), but patients invariably relapse with tumors that have been termed castration recurrent/resistant PCa (CRPC). Importantly, AR transcriptional activity becomes reactivated at this CRPC stage of the disease and remains essential for tumor growth. The objective of this review is to outline one clinically important mechanism contributing to this AR reactivation, which is increased intratumoral synthesis of testosterone and DHT from weak androgens produced by the adrenal glands and possibly de novo from cholesterol. Early studies showed that a substantial fraction of CRPC patients responded to adrenalectomy or medical suppression of adrenal androgen synthesis using agents such as ketoconazole (CYP17A1 inhibitor), and a recent phase III study of a more potent and selective CYP17A1 inhibitor (abiraterone) has demonstrated an improvement in survival. With the pending FDA approval of abiraterone for CRPC, defining the molecular mechanisms contributing to CYP17A1 inhibitor resistance/relapse and AR reactivation is now critical to build on these advances.
Androgen synthesis and metabolism in normal prostate
The androgen receptor (AR) can be weakly stimulated by high concentrations of multiple steroids including weak androgens produced by the adrenal gland such as androst-4-ene-3,17-dione (androstenedione) and dehydroepiandrosterone (DHEA; Fig. 1). However, AR has a much higher affinity for testosterone and 5α-dihydrotestosterone (DHT), and these are the major physiological AR ligands. Most of the circulating testosterone in healthy adult males is synthesized by the testes, with only 5–10% being derived from the adrenal glands. In contrast, human adrenal glands (but not rodent adrenals that lack the enzyme CYP17A1; van Weerden et al. 1992) are a major source of weak androgens, with DHEA-sulfate (DHEA-S) circulating at levels that are ~100-fold higher than testosterone. These weak adrenal androgens can be reduced to testosterone in some peripheral tissues including prostate, although this does not appear to be a significant source of testosterone in men with intact testicular function. DHEA-S taken up by prostate cells is converted to DHEA, which can then be reduced to androstenedione by the enzyme 3β-hydroxysteroid dehydrogenase 1 (HSD3B1) that is expressed in prostate basal epithelial cells (Labrie et al. 2000). Androstenedione in prostate can then be converted to testosterone by the enzyme aldo-keto reductase family 1, member C3 (AKR1C3; Lin et al. 1997, 2004, El-Alfy et al. 1999), which is also called 17β-hydroxysteroid dehydrogenase type 5 (17β-HSD5 or HSD17B5) or 3α-hydroxysteroid dehydrogenase type 2. In contrast, testosterone synthesis by Leydig cells in the testes is mediated by a distinct enzyme, HSD17B3. AKR1C3 in normal prostate has been localized to stromal, endothelial, and perineural cells (Lin et al. 2004). In men with intact testes, the significance of prostatic AKR1C3 as a source of testosterone is not clear. However, the enzyme has other functions such as reduction of estrone to 17β-estradiol and reduction of prostaglandin D2 to prostaglandin F2, which may be significant in normal prostate (Matsuura et al. 1998). In particular, through the latter reaction AKR1C3 can decrease prostaglandin J2 ligands for PPARγ, as these are normally derived from prostaglandin D2 (Desmond et al. 2003).
Figure 1.
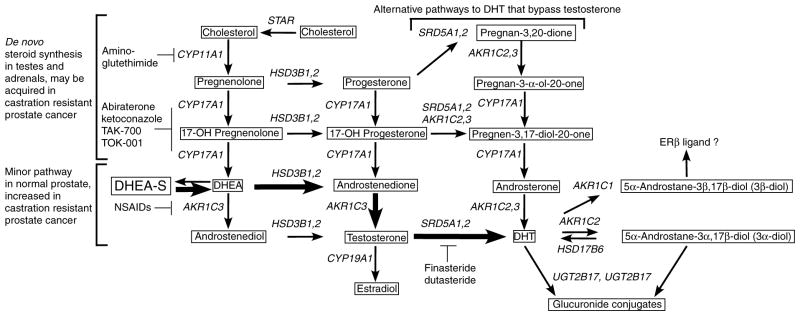
Outline of steps mediating androgen synthesis and metabolism in prostate cancer cells. Pathways that may contribute to androgen synthesis in prostate are outlined. Bold arrows and brackets outline what appears to be the major pathway for generation of testosterone and DHT in PCa cells after ADT when adrenal synthesis of weak androgens is intact and there are high levels of circulating DHEA-S. PCa cells can also synthesize at least trace amounts of testosterone directly from cholesterol through the sequential actions of CYP11A1, CYP17A1, HSD3B1 or 2, and AKR1C3 through the pathway that is normally active only in the adrenal glands and testes (also shown in brackets). It should be noted that testes uses 17β-hydroxysteroid dehydrogenase type 3 rather than AKR1C3 to generate testosterone (not shown). A possible alternative ‘backdoor’ pathway is also shown that may bypass testosterone and generate DHT from androsterone. In some cases more than one enzyme can carry out a particular step (HSD3B1 or HSD3B2; AKR1C2 or AKR1C3; and SRD5A1 or SRD5A2). Inhibitors of enzymes in the pathways are also indicated.
While testosterone produced by the testes is the physiological high-affinity ligand for AR in many tissues, in some tissues including prostate it is reduced to the higher affinity ligand DHT (approximately eightfold higher affinity) by type 1 or 2 5α-reductases, with the type 2 5α-reductase (SRD5A2) being the major enzyme in prostate (Fig. 1). This production of DHT presumably allows the prostate to maintain constitutive high levels of AR activity in luminal epithelium. Indeed, treatment with a 5α-reductase inhibitor (finasteride or dutasteride) reduces prostate volume and decreases serum levels of androgen-regulated prostate-specific antigen by ~50%. DHT in prostate is inactivated by the enzyme AKR1C2, which is also termed 3α-hydroxysteroid dehydrogenase type 3 (3α-HSD type 3). Although closely related to AKR1C3 and capable of reducing androstenedione to testosterone, it appears to primarily reduce DHT to 5α-androstane-3α,17β-diol (3α-androstanediol; Ji et al. 2003, Rizner et al. 2003), which is then glucuronidated to form 3α-androstanediol glucuronide by the enzymes UDP glycosyltransferase 2, B15 (UGT2B15) or UGT2B17 (Belanger et al. 2003).
Alternatively, 3α-androstanediol may be oxidized back to DHT by enzymes including retinol dehydrogenase like 3α-hydroxysteroid dehydrogenase (HSD17B6), which is expressed primarily in prostatic stromal cells and appears to be the major oxidase carrying out this function (Bauman et al. 2006b). A third AKR1C family enzyme, AKR1C1, is also expressed in prostate. However, in contrast to AKR1C2, it converts DHT primarily to 5α-androstane-3β,17β-diol (3β-androstanediol; Steckelbroeck et al. 2004), which is a potential endogenous ligand for the estrogen receptor β (Weihua et al. 2002). Genetic differences in several of these enzymes mediating androgen biosynthesis and metabolism have been identified, but their contribution to prostate cancer (PCa) development remains to be firmly established (Lindstrom et al. 2007).
Androgen synthesis and metabolism in PCa before androgen deprivation therapy
Studies using paired PCa and benign prostate tissues have found reductions in AKR1C1 and AKR1C2 mRNA, as well as reduced metabolism of 3H-DHT in malignant versus benign tissues (Ji et al. 2003, 2007). In contrast, significant changes in AKR1C2, AKR1C1, or AKR1C3 transcript levels were not found in studies using epithelial cells cultured from benign prostate versus PCa, although expression levels in the cultured cells could differ from those in vivo (Bauman et al. 2006a). Significantly, immunohistochemical studies with an AKR1C3 specific monoclonal antibody indicate that AKR1C3 protein is absent in benign luminal epithelial cells and expressed in primary PCa cells (Lin et al. 2004). Overall, these results suggest that decreased DHT catabolism (due to lower AKR1C2 expression) and increased tumor cell synthesis of testosterone (mediated by AKR1C3), may contribute to increased testosterone in primary PCa, although the importance of intraprostatic or intratumoral synthesis as a source of testosterone before androgen deprivation therapy (ADT) remains unclear.
Type 2 5α-reductase (SRD5A2) is the major form expressed in normal prostate. However, work from several groups indicates that the type 1 5α-reductase (SRD5A1) is increased in primary PCa based on transcript and protein levels (Titus et al. 2005a). In contrast, levels of SRD5A2 in PCa are not increased and were decreased in some studies. Increased expression of SRD5A1 may have clinical implications as the commonly used 5α-reductase inhibitor (finasteride) is relatively specific for the type 2 enzyme, while dutasteride inhibits both enzymes. Significantly, two large clinical trials showed that both finasteride and dutasteride could decrease the incidence of detectable PCa, although use of these drugs for PCa prevention is not yet recommended as it remains controversial whether they are primarily preventing the detection of low-grade indolent tumors (Gleason score 6) while possibly slightly increasing the incidence of higher grade PCa (Thompson et al. 2003, Andriole et al. 2010). Results from another large clinical trial in men with low-grade PCa who are candidates for surveillance, which is assessing whether dutasteride can prevent tumor progression, should soon be available (Fleshner et al. 2007). Given the prevalence of PCa, and in particular low-grade PCa that may not need to be treated aggressively with radical prostatectomy or radiation, it will be important to address whether all or particular subsets of healthy men or men with low-grade PCa can benefit from treatment with a 5α-reductase inhibitor. Interestingly, a recent study that assessed gene expression in PCa patients who were treated with dutasteride before prostatectomy found highly variable effects on the expression of AR and androgen-regulated genes, despite comparable marked declines in DHT levels, and suggested that AR levels may be a predictor of efficacy (Mostaghel et al. 2010).
Androgen synthesis and metabolism in primary PCa after ADT
Although testes are the major source of testosterone in normal men, the intratumoral synthesis of testosterone from weak adrenal androgens appears to be a substantial source of prostatic testosterone after ADT. Several studies have shown that intraprostatic testosterone and DHT levels do not decline as markedly as serum levels after ADT. Prostatic DHT in an early study was found to decline by only 75% after castration versus 86% after ketoconazole (CYP17A1 inhibitor) to suppress both testicular and adrenal androgen production (Geller & Albert 1987). Additional studies have similarly found that prostate tissue levels of DHT in PCa patients treated with ADT therapy before prostatectomy declined by only ~75% versus declines of ~95% in serum levels (Belanger et al. 1989, Mizokami et al. 2004, Nishiyama et al. 2004). In a recent study in healthy men, treatment for 1 month with a GnRH antagonist to suppress testicular androgen synthesis caused a 94% decline in serum testosterone, but only a 70–80% decline in prostate tissue testosterone and DHT (Page et al. 2006). Taken together, these data demonstrate that intraprostatic testosterone synthesis, which presumably serves to maintain high testosterone levels in normal prostate, can be a significant source of testosterone and may buffer primary PCa against the acute effects of ADT. Indeed, an analysis of AR-regulated gene expression in PCa cells from radial prostatectomy specimens in patients receiving neoadjuvant ADT (treatment before prostatectomy) found that while several androgen-regulated genes (NDRG1, FKBP5, and TMPRSS2) were reduced, many others were not suppressed after short- or long-term neoadjuvant ADT (Mostaghel et al. 2007). Based on these findings, current clinical trials are assessing the effects of more aggressive neoadjuvant ADT using a GnRH agonist in combination with CYP17A1 inhibition.
Increased intratumoral conversion of weak adrenal androgens to testosterone and DHT in local and metastatic castration recurrent/resistant PCa
Early studies in men with castration recurrent/resistant PCa (CRPC) and intact prostates found that intraprostatic DHT levels in a small subset were increased relative to those in men immediately after castration, although these findings were not interpreted as providing evidence for increased androgen synthesis (Geller et al. 1984, Geller 1985). A subsequent comprehensive and seminal study by more reliable mass spectrometry methods examined prostatic testosterone and DHT levels in a series of castrated men with CRPC who had tissue obtained from transurethral resections performed to relieve obstruction. Significantly, intraprostatic testosterone levels were not substantially reduced relative to controls with normal serum androgen levels, although DHT levels were reduced to 18% of controls (Mohler et al. 2004, Titus et al. 2005b). In a second critical study, androgen levels were assessed in metastatic CRPC samples obtained through a ‘warm autopsy’ program. Remarkably, testosterone levels in many of the CRPC samples were actually increased relative to control tissues (Montgomery et al. 2008). While DHT levels were less markedly increased, this may have reflected DHT catabolism in the postmortem tissues or other artifacts. Indeed, the analysis of steroid levels in tissues by mass spectrometry remains technically challenging due to issues including steroid metabolism, controlling for losses during extraction and processing, background produced by various biological matrices, poor ionization, and analytical challenges in the separation, detection, and definitive identification of particular steroids amongst highly related compounds. In any case, these data indicated that the progression to CRPC was associated with increased intratumoral accumulation or synthesis of testosterone.
In parallel with these assessments of androgen levels, gene expression studies have identified increased expression of enzymes mediating testosterone and DHT synthesis from weak adrenal androgens as a molecular mechanism mediating the restoration of testosterone (and possibly DHT) levels in CRPC. The initial study used Affymetrix oligonucleotide microarrays to compare global gene expression in a relatively large series of CRPC bone marrow metastases versus laser microdissected primary PCa, and found striking and consistent increases in the levels of AKR1C3 mRNA (approximately fivefold median increase; Stanbrough et al. 2006). The increased expression of AKR1C3 protein in local and metastatic CRPC was further confirmed by immunohistochemistry. This study also found significant increases in type 1 5α-reductase (SRD5A1) and in HSD3B2, which converts DHEA to androstenedione (Stanbrough et al. 2006; Fig. 1). Subsequent studies of CRPC have similarly observed increased expression of enzymes mediating testosterone and DHT synthesis, including AKR1C3 and SRD5A1 (Montgomery et al. 2008, Hofland et al. 2010). Finally, there is a report of a novel 5α-reductase (SRD5A3), which is structurally distinct from SRD5A1 and 2 and not blocked by finasteride or dutasteride, being increased in CRPC (Uemura et al. 2008). However, the extent to which SRD5A3 functions in vivo as a 5α-reductase and its contribution to CRPC remain to be established.
In addition to increased synthesis, one study found that SLCO1B3, a membrane transporter for steroids including testosterone and DHEA-S, was increased in PCa. Moreover, a common SLCO1B3 polymorphism causing decreased testosterone transport was associated with improved survival, suggesting a significant role for increased transport of steroids in CRPC (Hamada et al. 2008). Finally, expression of enzymes mediating DHT catabolism, including AKR1C2, AKR1C1, and UGT2B15, are also increased in CRPC (Stanbrough et al. 2006). Increased expression of another enzyme mediating androgen inactivation, HSD17B2, was also observed in another study of CRPC obtained from transurethral resections (Fromont et al. 2005). These increases are presumably secondary to the increased intracellular androgen synthesis, although it is possible that AKR1C1 or AKR1C2 is functioning in CRPC as steroid reductases (similarly to AKR1C3) to increase androgen synthesis.
Intratumoral de novo androgen synthesis in CRPC
It now seems clear that PCa cells can adapt to ADT by enhancing their ability to take up and metabolize weak androgens produced by the adrenal glands. Previous studies have also indicated that cholesterol synthesis may be increased in CRPC (Ettinger et al. 2004, Holzbeierlein et al. 2004), but whether CRPC cells can synthesize physiologically significant amounts of androgen de novo from cholesterol has been less clear. Studies conducted primarily in LNCaP cells and xenografts indicate that enzymes required for de novo steroid synthesis, including CYP11A1 and CYP17A1, are expressed and may be increased in castration resistant sublines (Dillard et al. 2008, Locke et al. 2008, 2009). Moreover, these tumors could also synthesize progesterone and detectable levels of DHT, although lower levels of testosterone, ex vivo from labeled precursors. The lower levels of testosterone suggested that these cells may produce DHT through a ‘backdoor’ pathway to generate DHT without going through testosterone (Auchus 2004, Locke et al. 2008; Fig. 1), although the relevance of such a pathway in CRPC is not clear as relatively high testosterone levels are found in clinical CRPC samples. It should also be noted that LNCaP cells express a mutant AR (T877A) that can be strongly activated by progesterone, so may experience less selective pressure for synthesis of testosterone (Veldscholte et al. 1990).
Although PCa cell lines and xenografts can synthesize detectable levels of androgens de novo, it is not yet clear whether this occurs in CRPC patients at levels that are adequate to fully reactivate AR. One study found that CYP17A1 and HSD3B1 mRNA were extremely low in locally recurrent CRPC, which suggested that these tumors may not produce physiologically significant levels of androgens de novo (Hofland et al. 2010). However, mRNA for both enzymes as well as CYP11A1 mRNA were readily detected in metastatic CRPC bone marrow biopsy samples (Stanbrough et al. 2006), and appeared to be increased in some advanced tumors obtained from warm autopsies (Montgomery et al. 2008). These observations suggest that de novo intratumoral androgen synthesis may play a limited or variable role initially as tumor cells adapt to conventional ADT (surgical or medical castration), where high levels of circulating weak androgens from the adrenal glands provide a readily available source of substrate for conversion to testosterone and DHT. In any case, it will be important to determine whether surgical (adrenalectomy or hypophysectomy) or medical (aminoglutethimide, ketoconazole, abiraterone, or other agents in development) suppression of this adrenal synthesis generates selective pressure for tumor cells that are relatively resistant to these agents due to mechanisms including increased expression of CYP17A1, with subsequent increases in de novo androgen synthesis being a potential mechanism for relapse.
Conclusions
Prostate expresses the enzymatic machinery for synthesis of testosterone from weak adrenal androgens, but the importance of this as a source of testosterone for normal prostate epithelium or for PCa development in men with normal testicular function is uncertain. In contrast, the intraprostatic synthesis of testosterone from adrenal-derived precursors likely accounts for the relatively high testosterone levels in prostate after ADT, and may buffer PCa cells from the loss of testicular androgen. The CRPC cells that emerge after ADT upregulate enzymes that convert adrenal androgens to testosterone and DHT (in particular AKR1C3), further enhancing their intratumoral androgen synthesis and reactivating AR transcriptional activity. In addition, AR activity in these cells is likely further enhanced by multiple mechanisms that sensitize AR to low levels of androgens (Yuan & Balk 2009). Secondary hormonal therapies that suppress adrenal gland synthesis of weak androgens are effective in many CRPC patients, and a recent study showing a survival advantage in advanced CRPC patients treated with abiraterone will likely lead to approval of this drug in the near future (Attard et al. 2009, Danila et al. 2010, Reid et al. 2010). While de novo intratumoral androgen synthesis may make a variable contribution to the restoration of intratumoral androgens in patients with intact adrenal gland function, it may become a major source in patients treated with CYP17A1 inhibitors whose adrenal synthesis of weak androgens is blocked. Further studies are clearly needed to determine whether CRPC cells develop relative resistance to CYP17A1 inhibitors (through increased expression of CYP17A1, mutations in CYP17A1, altered drug metabolism, or other mechanisms), resulting in increased de novo androgen synthesis and relapse. Such tumors may then respond to other CYP17A1 inhibitors or to the addition of agents that inhibit other steps in androgen synthesis (Sartor et al. 2009, Taplin et al. 2009).
Acknowledgments
Funding
This work was supported by grants to S P Balk from the NIH (R01 CA111803 and Prostate SPORE P50 CA090381), DOD Prostate Cancer Research Program (PC060807), and a Challenge Grant from the Prostate Cancer Foundation. C Cai was supported by a postdoctoral award from the DOD Prostate Cancer Research Program (PC08115) and a Career Development Award from the DF/HCC Prostate SPORE (P50 CA090381).
Footnotes
Declaration of interest
The authors declare that they have no conflicts of interest that could be perceived as prejudicing the impartiality of this review.
References
- Andriole GL, Bostwick DG, Brawley OW, Gomella LG, Marberger M, Montorsi F, Pettaway CA, Tammela TL, Teloken C, Tindall DJ, et al. Effect of dutasteride on the risk of prostate cancer. New England Journal of Medicine. 2010;362:1192–1202. doi: 10.1056/NEJMoa0908127. [DOI] [PubMed] [Google Scholar]
- Attard G, Reid AH, Olmos D, de Bono JS. Antitumor activity with CYP17 blockade indicates that castration-resistant prostate cancer frequently remains hormone driven. Cancer Research. 2009;69:4937–4940. doi: 10.1158/0008-5472.CAN-08-4531. [DOI] [PubMed] [Google Scholar]
- Auchus RJ. The backdoor pathway to dihydrotestosterone. Trends in Endocrinology and Metabolism. 2004;15:432–438. doi: 10.1016/S1043-2760(04)00214-0. [DOI] [PubMed] [Google Scholar]
- Bauman DR, Steckelbroeck S, Peehl DM, Penning TM. Transcript profiling of the androgen signal in normal prostate, benign prostatic hyperplasia, and prostate cancer. Endocrinology. 2006a;147:5806–5816. doi: 10.1210/en.2006-0627. 10. 1210/en.2006-0627. [DOI] [PubMed] [Google Scholar]
- Bauman DR, Steckelbroeck S, Williams MV, Peehl DM, Penning TM. Identification of the major oxidative 3alpha-hydroxysteroid dehydrogenase in human prostate that converts 5alpha-androstane-3alpha,17beta-diol to 5alpha-dihydrotestosterone: a potential therapeutic target for androgen-dependent disease. Molecular Endocrinology. 2006b;20:444–458. doi: 10.1210/me.2005-0287. [DOI] [PubMed] [Google Scholar]
- Belanger B, Belanger A, Labrie F, Dupont A, Cusan L, Monfette G. Comparison of residual C-19 steroids in plasma and prostatic tissue of human, rat and guinea pig after castration: unique importance of extratesticular androgens in men. Journal of Steroid Biochemistry. 1989;32:695–698. doi: 10.1016/0022-4731(89)90514-1. 10.1016/0022-4731(89)90514-1. [DOI] [PubMed] [Google Scholar]
- Belanger A, Pelletier G, Labrie F, Barbier O, Chouinard S. Inactivation of androgens by UDP-glucuronosyl-transferase enzymes in humans. Trends in Endocrinology and Metabolism. 2003;14:473–479. doi: 10.1016/j.tem.2003.10.005. 10.1016/j.tem.2003. 10.005. [DOI] [PubMed] [Google Scholar]
- Danila DC, Morris MJ, de Bono JS, Ryan CJ, Denmeade SR, Smith MR, Taplin ME, Bubley GJ, Kheoh T, Haqq C, et al. Phase II multicenter study of abiraterone acetate plus prednisone therapy in patients with docetaxel-treated castration-resistant prostate cancer. Journal of Clinical Oncology. 2010;28:1496–1501. doi: 10.1200/JCO.2009.25.9259. 10. 1200/JCO.2009.25.9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmond JC, Mountford JC, Drayson MT, Walker EA, Hewison M, Ride JP, Luong QT, Hayden RE, Vanin EF, Bunce CM. The aldo-keto reductase AKR1C3 is a novel suppressor of cell differentiation that provides a plausible target for the non-cyclooxygenase-dependent antineoplastic actions of nonsteroidal anti-inflammatory drugs. Cancer Research. 2003;63:505–512. [PubMed] [Google Scholar]
- Dillard PR, Lin MF, Khan SA. Androgen-independent prostate cancer cells acquire the complete steroidogenic potential of synthesizing testosterone from cholesterol. Molecular and Cellular Endocrinology. 2008;295:115–120. doi: 10.1016/j.mce.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Alfy M, Luu-The V, Huang XF, Berger L, Labrie F, Pelletier G. Localization of type 5 17beta-hydroxy-steroid dehydrogenase, 3beta-hydroxysteroid dehydrogenase, and androgen receptor in the human prostate by in situ hybridization and immunocytochemistry. Endocrinology. 1999;140:1481–1491. doi: 10.1210/endo.140.3.6585. 10.1210/en. 140.3.1481. [DOI] [PubMed] [Google Scholar]
- Ettinger SL, Sobel R, Whitmore TG, Akbari M, Bradley DR, Gleave ME, Nelson CC. Dysregulation of sterol response element-binding proteins and downstream effectors in prostate cancer during progression to androgen independence. Cancer Research. 2004;64:2212–2221. doi: 10.1158/0008-5472.CAN-2148-2. [DOI] [PubMed] [Google Scholar]
- Fleshner N, Gomella LG, Cookson MS, Finelli A, Evans A, Taneja SS, Lucia MS, Wolford E, Somerville MC, Rittmaster R. Delay in the progression of low-risk prostate cancer: rationale and design of the Reduction by Dutasteride of Clinical Progression Events in Expectant Management (REDEEM) trial. Contemporary Clinical Trials. 2007;28:763–769. doi: 10.1016/j.cct.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Fromont G, Chene L, Vidaud M, Vallancien G, Mangin P, Fournier G, Validire P, Latil A, Cussenot O. Differential expression of 37 selected genes in hormone-refractory prostate cancer using quantitative taqman real-time RT-PCR. International Journal of Cancer. 2005;114:174–181. doi: 10.1002/ijc.20704. [DOI] [PubMed] [Google Scholar]
- Geller J. Rationale for blockade of adrenal as well as testicular androgens in the treatment of advanced prostate cancer. Seminars in Oncology. 1985;12:28–35. [PubMed] [Google Scholar]
- Geller J, Albert J. Effects of castration compared with total androgen blockade on tissue dihydrotestosterone (DHT) concentration in benign prostatic hyperplasia (BPH) Urological Research. 1987;15:151–153. doi: 10.1007/BF00254427. [DOI] [PubMed] [Google Scholar]
- Geller J, Albert JD, Nachtsheim DA, Loza D. Comparison of prostatic cancer tissue dihydrotestosterone levels at the time of relapse following orchiectomy or estrogen therapy. Journal of Urology. 1984;132:693–696. doi: 10.1016/s0022-5347(17)49829-6. [DOI] [PubMed] [Google Scholar]
- Hamada A, Sissung T, Price DK, Danesi R, Chau CH, Sharifi N, Venzon D, Maeda K, Nagao K, Sparreboom A, et al. Effect of SLCO1B3 haplotype on testosterone transport and clinical outcome in caucasian patients with androgen-independent prostatic cancer. Clinical Cancer Research. 2008;14:3312–3318. doi: 10.1158/1078-0432.CCR-07-4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofland J, van Weerden WM, Dits NF, Steenbergen J, van Leenders GJ, Jenster G, Schroder FH, de Jong FH. Evidence of limited contributions for intratumoral steroidogenesis in prostate cancer. Cancer Research. 2010;70:1256–1264. doi: 10.1158/0008-5472.CAN-09-2092. [DOI] [PubMed] [Google Scholar]
- Holzbeierlein J, Lal P, LaTulippe E, Smith A, Satagopan J, Zhang L, Ryan C, Smith S, Scher H, Scardino P, et al. Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. American Journal of Pathology. 2004;164:217–227. doi: 10.1016/S0002-9440(10)63112-4. 10. 1016/S0002-9440(10)63112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Q, Chang L, VanDenBerg D, Stanczyk FZ, Stolz A. Selective reduction of AKR1C2 in prostate cancer and its role in DHT metabolism. Prostate. 2003;54:275–289. doi: 10.1002/pros.10192. [DOI] [PubMed] [Google Scholar]
- Ji Q, Chang L, Stanczyk FZ, Ookhtens M, Sherrod A, Stolz A. Impaired dihydrotestosterone catabolism in human prostate cancer: critical role of AKR1C2 as a pre-receptor regulator of androgen receptor signaling. Cancer Research. 2007;67:1361–1369. doi: 10.1158/0008-5472.CAN-06-1593. [DOI] [PubMed] [Google Scholar]
- Labrie F, Luu-The V, Lin SX, Simard J, Labrie C. Role of 17 beta-hydroxysteroid dehydrogenases in sex steroid formation in peripheral intracrine tissues. Trends in Endocrinology and Metabolism. 2000;11:421–427. doi: 10.1016/S1043-2760(00)00342-8. [DOI] [PubMed] [Google Scholar]
- Lin HK, Jez JM, Schlegel BP, Peehl DM, Pachter JA, Penning TM. Expression and characterization of recombinant type 2 3 alpha-hydroxysteroid dehydrogenase (HSD) from human prostate: demonstration of bifunctional 3 alpha/17 beta-HSD activity and cellular distribution. Molecular Endocrinology. 1997;11:1971–1984. doi: 10.1210/me.11.13.1971. [DOI] [PubMed] [Google Scholar]
- Lin HK, Steckelbroeck S, Fung KM, Jones AN, Penning TM. Characterization of a monoclonal antibody for human aldo-keto reductase AKR1C3 (type 2 3alpha-hydroxysteroid dehydrogenase/type 5 17beta-hydroxy-steroid dehydrogenase); immunohistochemical detection in breast and prostate. Steroids. 2004;69:795–801. doi: 10.1016/j.steroids.2004.09.014. [DOI] [PubMed] [Google Scholar]
- Lindstrom S, Adami HO, Balter KA, Xu J, Zheng SL, Stattin P, Gronberg H, Wiklund F. Inherited variation in hormone-regulating genes and prostate cancer survival. Clinical Cancer Research. 2007;13:5156–5161. doi: 10.1158/1078-0432.CCR-07-0669. [DOI] [PubMed] [Google Scholar]
- Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC, Wood CA, Ettinger SL, Gleave ME, Nelson CC. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Research. 2008;68:6407–6415. doi: 10.1158/0008-5472.CAN-07-5997. [DOI] [PubMed] [Google Scholar]
- Locke JA, Nelson CC, Adomat HH, Hendy SC, Gleave ME, Guns ES. Steroidogenesis inhibitors alter but do not eliminate androgen synthesis mechanisms during progression to castration-resistance in LNCaP prostate xenografts. Journal of Steroid Biochemistry and Molecular Biology. 2009;115:126–136. doi: 10.1016/j.jsbmb.2009.03.011. 10.1016/j.jsbmb.2009. 03.011. [DOI] [PubMed] [Google Scholar]
- Matsuura K, Shiraishi H, Hara A, Sato K, Deyashiki Y, Ninomiya M, Sakai S. Identification of a principal mRNA species for human 3alpha-hydroxysteroid dehydrogenase isoform (AKR1C3) that exhibits high prostaglandin D2 11-ketoreductase activity. Journal of Biochemistry. 1998;124:940–946. doi: 10.1093/oxfordjournals.jbchem.a022211. [DOI] [PubMed] [Google Scholar]
- Mizokami A, Koh E, Fujita H, Maeda Y, Egawa M, Koshida K, Honma S, Keller ET, Namiki M. The adrenal androgen androstenediol is present in prostate cancer tissue after androgen deprivation therapy and activates mutated androgen receptor. Cancer Research. 2004;64:765–771. doi: 10.1158/0008-5472.CAN-03-0130. [DOI] [PubMed] [Google Scholar]
- Mohler JL, Gregory CW, Ford OH, III, Kim D, Weaver CM, Petrusz P, Wilson EM, French FS. The androgen axis in recurrent prostate cancer. Clinical Cancer Research. 2004;10:440–448. doi: 10.1158/1078-0432.CCR-1146-03. [DOI] [PubMed] [Google Scholar]
- Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, True LD, Nelson PS. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Research. 2008;68:4447–4454. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostaghel EA, Page ST, Lin DW, Fazli L, Coleman IM, True LD, Knudsen B, Hess DL, Nelson CC, Matsumoto AM, et al. Intraprostatic androgens and androgen-regulated gene expression persist after testosterone suppression: therapeutic implications for castration-resistant prostate cancer. Cancer Research. 2007;67:5033–5041. doi: 10.1158/0008-5472.CAN-06-3332. [DOI] [PubMed] [Google Scholar]
- Mostaghel EA, Geng L, Holcomb I, Coleman IM, Lucas J, True LD, Nelson PS. Variability in the androgen response of prostate epithelium to 5alpha-reductase inhibition: implications for prostate cancer chemoprevention. Cancer Research. 2010;70:1286–1295. doi: 10.1158/0008-5472.CAN-09-2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama T, Hashimoto Y, Takahashi K. The influence of androgen deprivation therapy on dihydrotestosterone levels in the prostatic tissue of patients with prostate cancer. Clinical Cancer Research. 2004;10:7121–7126. doi: 10.1158/1078-0432.CCR-04-0913. [DOI] [PubMed] [Google Scholar]
- Page ST, Lin DW, Mostaghel EA, Hess DL, True LD, Amory JK, Nelson PS, Matsumoto AM, Bremner WJ. Persistent intraprostatic androgen concentrations after medical castration in healthy men. Journal of Clinical Endocrinology and Metabolism. 2006;91:3850–3856. doi: 10.1210/jc.2006-0968. [DOI] [PubMed] [Google Scholar]
- Reid AH, Attard G, Danila DC, Oommen NB, Olmos D, Fong PC, Molife LR, Hunt J, Messiou C, Parker C, et al. Significant and sustained antitumor activity in post-docetaxel, castration-resistant prostate cancer with the CYP17 inhibitor abiraterone acetate. Journal of Clinical Oncology. 2010;28:1489–1495. doi: 10.1200/JCO.2009.24.6819. 10.1200/JCO.2009.24. 6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizner TL, Lin HK, Peehl DM, Steckelbroeck S, Bauman DR, Penning TM. Human type 3 3alpha-hydroxy-steroid dehydrogenase (aldo-keto reductase 1C2) and androgen metabolism in prostate cells. Endocrinology. 2003;144:2922–2932. doi: 10.1210/en.2002-0032. [DOI] [PubMed] [Google Scholar]
- Sartor O, Nakabayashi M, Taplin ME, Ross RW, Kantoff PW, Balk SP, Oh WK. Activity of dutasteride plus ketoconazole in castration-refractory prostate cancer after progression on ketoconazole alone. Clinical Genitourinary Cancer. 2009;7:E90–E92. doi: 10.3816/CGC.2009.n.030. [DOI] [PubMed] [Google Scholar]
- Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM, Febbo PG, Balk SP. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Research. 2006;66:2815–2825. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- Steckelbroeck S, Jin Y, Gopishetty S, Oyesanmi B, Penning TM. Human cytosolic 3alpha-hydroxysteroid dehydrogenases of the aldo-keto reductase superfamily display significant 3beta-hydroxysteroid dehydrogenase activity: implications for steroid hormone metabolism and action. Journal of Biological Chemistry. 2004;279:10784–10795. doi: 10.1074/jbc.M313308200. [DOI] [PubMed] [Google Scholar]
- Taplin ME, Regan MM, Ko YJ, Bubley GJ, Duggan SE, Werner L, Beer TM, Ryan CW, Mathew P, Tu SM, et al. Phase II study of androgen synthesis inhibition with ketoconazole, hydrocortisone, and dutasteride in asymptomatic castration-resistant prostate cancer. Clinical Cancer Research. 2009;15:7099–7105. doi: 10.1158/1078-0432.CCR-09-1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson IM, Goodman PJ, Tangen CM, Lucia MS, Miller GJ, Ford LG, Lieber MM, Cespedes RD, Atkins JN, Lippman SM, et al. The influence of finasteride on the development of prostate cancer. New England Journal of Medicine. 2003;349:215–224. doi: 10.1056/NEJMoa030660. [DOI] [PubMed] [Google Scholar]
- Titus MA, Gregory CW, Ford OH, III, Schell MJ, Maygarden SJ, Mohler JL. Steroid 5alpha-reductase isozymes I and II in recurrent prostate cancer. Clinical Cancer Research. 2005a;11:4365–4371. doi: 10.1158/1078-0432.CCR-04-0738. [DOI] [PubMed] [Google Scholar]
- Titus MA, Schell MJ, Lih FB, Tomer KB, Mohler JL. Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer. Clinical Cancer Research. 2005b;11:4653–4657. doi: 10.1158/1078-0432.CCR-05-0525. [DOI] [PubMed] [Google Scholar]
- Uemura M, Tamura K, Chung S, Honma S, Okuyama A, Nakamura Y, Nakagawa H. Novel 5 alpha-steroid reductase (SRD5A3, type-3) is overexpressed in hormone-refractory prostate cancer. Cancer Science. 2008;99:81–86. doi: 10.1111/j.1349-7006.2007.00656.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldscholte J, Ris-Stalpers C, Kuiper GG, Jenster G, Berrevoets C, Claassen E, van Rooij HC, Trapman J, Brinkmann AO, Mulder E. A mutation in the ligand binding domain of the androgen receptor of human LNCaP cells affects steroid binding characteristics and response to anti-androgens. Biochemical and Biophysical Research Communications. 1990;173:534–540. doi: 10.1016/S0006-291X(05)80067-1. [DOI] [PubMed] [Google Scholar]
- van Weerden WM, Bierings HG, van Steenbrugge GJ, de Jong FH, Schroder FH. Adrenal glands of mouse and rat do not synthesize androgens. Life Sciences. 1992;50:857–861. doi: 10.1016/0024-3205(92)90204-3. 10.1016/0024-3205 (92)90204-3. [DOI] [PubMed] [Google Scholar]
- Weihua Z, Lathe R, Warner M, Gustafsson JA. An endocrine pathway in the prostate, ERbeta, AR, 5alpha-androstane-3beta,17beta-diol, and CYP7B1, regulates prostate growth. PNAS. 2002;99:13589–13594. doi: 10.1073/pnas.162477299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, Balk SP. Mechanisms mediating androgen receptor reactivation after castration. Urologic Oncology. 2009;27:36–41. doi: 10.1016/j.urolonc.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]