

Abstract
An understanding of the process of peptide fragmentation and what parameters are best to obtain the most useful information is important. This is especially true for large-scale proteomics where data collection and data analysis are most often automated and manual interpretation of spectra is rare due to the vast amounts of data generated. We show herein that collisional cell peptide fragmentation, in this case HCD in the Q Exactive, is significantly affected by the normalized energy applied. Both peptide sequence and energy applied determine what ion fragments are observed. However by applying a stepped normalized collisional energy scheme and combining ions from low, medium and high collision energies we are able to increase the diversity of fragmentation ions generated. Application of stepped collision energy to HEK293T lysate demonstrated a minimal effect on peptide and protein identification in a large-scale proteomics dataset, but improved phospho site localization through increased sequence coverage. Stepped HCD is also beneficial for TMT experiments, increasing intensity of TMT reporters used for quantitation without adversely effecting peptide identification.
Introduction
Peptide identification using sequence database centers around the predictability of peptide fragmentation during the collisional dissociation process. These methods are now capable of identifying peptides and proteins on a very large scale, often in a cell lysate where thousands of proteins are identified in a single run. Not only are protein identities determined but additional information can be gained, such as protein isoforms and post-translational modifications (PTMs). Additionally, relative abundances of proteins within a single cell type or between two or more cell types may be determined. Regardless of the application, it is advantageous if a high percentage of the spectra generated in a proteomics experiment be identified. The success of spectra identification can be affected by various parameters such as the data analysis algorithms, databases used, prefractionation, separation conditions, and peptide fragmentation methods. In this study we examine the effect of peptide fragmentation on peptide and protein identification, including the fragmentation ions and abundances generated during fragmentation reactions.
Due to the prevalence of tandem mass spectrometers using ion trap technology, many search engines have been optimized for CID data produced in these devices.[1,2] Ion traps fragment peptides in a helium bath gas environment by applying an auxiliary excitation frequency to increase the motion of a precursor ion. This motion causes more energetic collisions with the bath gas increasing the vibrational excitation of the precursor ion. Once bonds break, the fragment ions are no longer excited by the auxiliary frequency. Ion fragmentation in quadrupole or octopole collision cells is distinctly different in that the precursor ion can undergo multiple fragmentations.[3,4,5]The Q-Exactive mass spectrometer features HCD (higher collisional dissociation) using an octopole collision cell that operates at higher collisional energies than CID.[6,7] This contrasts with excited state fragmentation which rarely produces ions that result from fragmentation at multiple points.[8] As a result, the two different methods of ion activation produce diverse patterns of fragmentations and in particular, collisional cell fragmentation can produce ion types that result from multiple fragmentations.[9] The prevalence of ion types is dependent on both the sequence of the peptide and on the collisional energy applied.[10,11,12] Some studies have suggested that CID is superior to HCD for large scale proteomics. [13,14,15,16] Thus we will explore the effects of differential collisional energies for the elucidation of protein sequences and modifications.
In this study we explore the effects of collisional energy on a select set of peptides and observe the ion types that are produced as a function of collisional energy. As the collisional energy increases, the abundance of ions that are produced from multiple fragmentations increases. Likewise, the amount of precursor that remains unfragmented is inversely correlated to collision energy. Ideally, all of the precursor ions would be fragmented into usable ion types (but not over-fragmented) to produce the highest possible signal of fragment ions. For excited state collisional energy this process is straightforward; the ion is fragmented a single time or not at all. In the case of collisional cell activation (HCD) the ion could remain intact, fragmented at a single site (the ideal case), or fragmented at multiple sites, producing either an informative ion type or producing an ion that is “unusable” in terms of database searching. In order to optimize the percentage of “usable” ion types we examined the use of stepped collisional energy on the proteomics scale. Stepped collisional energy has been most often used for small molecules where identification of the species is difficult and requires a variety of ions which are often not produced with a single collisional energy.[17] During stepped collisional energy the precursor ion is fragmented at a low, medium, and high collisional energy, the fragments from these three energies are combined and are detected simultaneously. The concept of combining spectra from different fragmentation events is not new; MSE (Water’s version of data independent acquisition) utilizes a similar strategy of collecting a high and low energy spectrum, but in this case it is used to deconvolute multiplex spectra.[18] Dayon et al, among others, has used CID (ion trap) spectra for peptide identification combined with HCD for detection of reporter ions for quantitation.[19]
The use of stepped collisional energy is of particular benefit in the case of mass spectrometers where detection is the time limiting factor of the experiment. For the data shown here from a Q Exactive, only a single high resolution detector is available for ion detection. Therefore there is no effective time loss for performing the stepped collisional energy in the Q Exactive relative to fragmentation and detection at a single energy. We show that while there is no significant difference in the number of peptides identified through the use of stepped collisional energy over that of a single energy, stepped HCD is beneficial for phosphorylation site analysis and TMT quantitation. The use of multiple collisional energies increases sequence coverage, improving localization of phosphorylation sites. TMT quantitation is dependent on observation of TMT reporter ions; intensity of reporter ions can be enhanced by use of stepped HCD without sacrificing peptide identification.
Experimental
HEK293T cells were grown in Dulbecco’s Modified Eagle’s Medium (D-MEM) with 10% fetal bovine serum (FBS) supplemented with Penicillin Streptomycin. Cells were lysed in PBS by sonication. Lysate samples were precipitated by a methanol/chloroform extraction. Protein pellets were solubilized in 8M urea and 100mM Tris, reduced with 5mM TCEP for 15min, and then alkylated with 10mM iodoacetamide for 20min at 37C. Samples were diluted 1:4 with 100mM Tris buffer and digested with trypsin (1:100 ratio) overnight at 37C.
Phosphopeptide enrichment of HEK lysate was performed using TiO2-coated magnetic beads (Pierce), according to the manufacturer’s protocol. Samples were desalted in a 250μm ID undeactivated silica capillary blocked with a Kasil frit and packed with 3.5cm reverse phase particles (5μm, ODS-AQ, YMC). Peptides were eluted with 250μL 80% ACN/2% formic acid directly on 10μL TiO2 magnetic beads. Beads were placed on a magnetic plate separator and unbound supernatant was removed. Beads were washed three times with 200μL Binding Buffer and once with 200μL Washing Buffer. Phosphopeptides were eluted from magnetic beads by incubation for 10 minutes with 35μL Elution Buffer. Eluted phosphopeptides were dried prior to LC/MS-MS analysis. The equivalent of 50μg of enriched lysate was used per injection.
For quantitative analysis, CFBE (cystic fibrosis bronchial epithelial) cell lysates were labeled with TMTsixplex (Thermo Scientific) according to manufacturer’s instructions. Lysates were solubilized in 0.1M triethyl ammonium bicarbonate (TEAB) and 0.1% Rapigest, reduced and alkylated as above, and digested with trypsin (1:100 ratio) overnight at 37C. For each TMT channel, 100μg of lysate were labeled with 0.2mg of TMTsixplex reagent in 41μL anhydrous ACN for 1h at room temperature. Reactions were quenched by addition of 8μL hydroxylamine solution (5% w/v) and incubated for 15 minutes at room temperature. Samples were mixed 1:1:1:1:1:1 and dried prior to LC/MS-MS analysis. The equivalent of 5μg of lysate per channel was analyzed per replicate.
Peptide mixtures were analyzed by nanoflow liquid chromatography mass spectrometry using an Easy NanoLC II and Q Exactive mass spectrometer (Thermo Scientific, Bremen, Germany). MudPIT columns were prepared by blocking one end of 250μm ID undeactivated silica capillary with a Kasil frit and successively packing 1.5cm of reversed phase (5μm, ODS-AQ C18, YMC) particles, 1.5cm of strong cation exchange resin (5μm Partisphere, Phenomenex) and an additional 1.5cm of reversed phase particles. Samples were pressure loaded onto the MudPIT trapping column using the autosampler of the Easy NanoLC and subsequently washed with buffer A to remove any salts while venting to waste. The MudPIT trapping column was connected via a zero dead volume union to an analytical reverse phase capillary column. The 100μm ID analytical column was generated by pulling the tip to 5μm ID and pressure packing the column (5μm ODS-AQ C18, YMC) until 20cm in length. Electrospray was performed directly from the tip of the analytical column. Six step MudPIT separations were performed with salt pulses of 5ul of 0%, 10%, 20%, 30%, 60%, and 100% of 500mM ammonium acetate prior to the beginning of the reverse phase gradient. Peptides were separated with a linear gradient from 1% to 5%B over the first 5 minutes followed by an increase to 45%B over 120min and then an increase to 98%B over an additional 30 min. The column was held at 98% B for 5 min, reduced to 1% B and re-equilibrated prior to the next salt step. Buffer A was 5% acetonitrile and 0.1% formic acid and buffer B was 80% acetonitrile and 0.1% formic acid. Flow rate was 400nl/min. TMT samples were separated with a 13 step MudPIT method using salt pulses of 0%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90% and 100% of 500mM ammonium acetate followed by two steps containing 90% ammonium acetate and 10%B. Reverse phase gradient from 5% to 45%B was shortened to 90min and the increase to 80%B took place over an additional 20min.
Phosphopeptide enrichment samples were separated over a two hour reverse phase gradient. Peptides were separated with a linear gradient from 1% to 5%B over the first 5 minutes followed by an increase to 55%B over 100min. The column was returned to 1% B over 5 min and re-equilibrated for 10min.
Pierce retention time calibration mixture containing 15 tryptic yeast peptides (including ELGQAGVDTYLQTK, LTILEELR, and GILFVGSGVSGGEEGAR) was used as a standard peptide mixture to test the effect of HCD energy on fragmentation. Phosphopeptide TRDIYETDyYRK was purchased from AnaSpec. Peptide standards were analyzed by reverse phase separation on a 20cm analytical column, as described above. 10fmol of peptide mixture was injected through the autosampler and separated with a linear gradient from 0% to 45%B over 25 minutes followed by an increase to 100%B over 10min. The column was held at 98% B for 5 min, reduced to 0% B and re-equilibrated prior to the next injection.
The Q Exactive was operated in the data dependent mode, collecting a full MS scan from 300–1650m/z at 70K resolution and an AGC target of 1e6. The 10 most abundant ions per scan were selected for MS/MS at 17.5K resolution and AGC target of 1e5 and intensity threshold of 1K. Maximum fill times were 10msec and 100msec for MS and MS/MS scans respectively with a dynamic exclusion of 60sec. Normalized collision energy was varied from 12–34 for the peptide standard experiments. HEK samples were analyzed at 20, 25, 30NCE (normalized collisional energy) and at 25 with 20% stepped energy which effectively combines fragmentation at energies of 20, 25 and 30. Fragmentation of peptide standards were manually identified and relative intensities were extracted by Fragmentor v.2.4.1 (http://www.faculty.ucr.edu/~ryanj/fragmentor.html). HEK and CFBE spectra were extracted using RawXtract [20] and protein identification was done with Integrated Proteomics Pipeline (IP2, http://integratedproteomics.com) and ProLuCID by searching against UniProt Human database. Half tryptic searches were performed with carbamidomethylation on cysteine and oxidation on methione as static and variable modifications, respectively. TMT data was searched with static modifications of 229.1629 on lysine side chain and on the N terminus. Data was searched with 6ppm precursor tolerance and 20ppm tolerance for fragment ions. Proteins required a minimum of two peptides present and were filtered to less than 1% FDR using DTASelect. [21]
Results and Discussion
Shown in Figure 1a is the peptide ELGQAGVDTYLQTK, which was fragmented by CID in an LTQ-Orbitrap. The same peptide was fragmented at increasing collisional energy in the HCD cell of the Q Exactive; spectra resulting from normalized energies of 18, 22, 26, 30, and 34 are shown in Figure 1b–f. The CID spectrum displays a predominant series of y ions with lower intensity b series, which is largely independent of the fragmentation energy applied. At the lower HCD energies, 18 and below, the precursor ion dominates the spectrum. As the collisional energy is ramped, the precursor ion is increasingly fragmented into product ions; the abundance of a series of y ions is seen inversely correlated to precursor intensity. As higher energies are applied, ions fragment multiple times causing a shift to lower m/z. At an NCE of 34 the spectrum is largely dominated by ions below 400Da.[22] Although many of the ion types are similar between HCD and CID, there is not a single collisional energy in HCD that can duplicate the pattern and fragmentation by CID.
Figure 1.
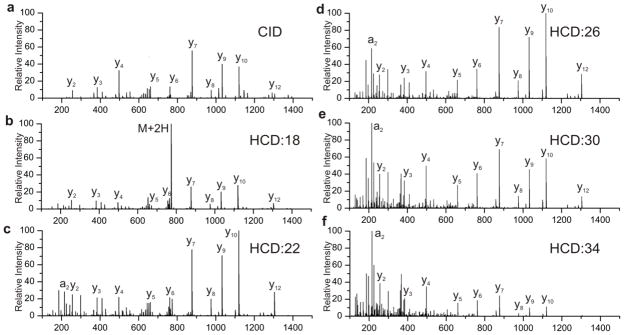
Spectra of the peptide ELGQAGVDTYLQTK by a) CID and at b–f) HCD normalized energies of 18, 22, 26, 30 and 34.
We utilized NoDupe software to provide a quantitative measure of the similarity between the spectra; results shown in Table 1. NoDupe was designed to analyze spectral similarity by generating a spectral contrast angle for two given spectra. [23] If the spectra are identical the reported angle is zero; completely dissimilar spectra will produce a right angle (π/2 rad). Spectral pairs returning a value above the set threshold of 1.1 rad are considered to be non-significant by the software and consequently no value is reported. Not surprisingly the largest degree of spectral similarity is seen between neighboring HCD energies. As the gap between HCD energies increase, the degree of dissimilarity increases. The CID spectrum does not show a high degree of similarity to any of the HCD spectra, qualitatively above, or quantitatively as shown here.
Table 1.
Spectral similarity of ELGQAGVDTYLQTK
| HCD12 | HCD14 | HCD16 | HCD18 | HCD20 | HCD22 | HCD24 | HCD26 | HCD28 | HCD30 | HCD32 | HCD34 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CID | - | - | - | 0.96 | 0.79 | 0.77 | 0.80 | 0.91 | 0.97 | 1.04 | - | - |
| HCD12 | 0.25 | 0.49 | 0.78 | 1.05 | - | - | - | - | - | - | - | |
| HCD14 | 0.31 | 0.62 | 0.90 | - | - | - | - | - | - | - | ||
| HCD16 | 0.38 | 0.68 | 0.93 | - | - | - | - | - | - | |||
| HCD18 | 0.36 | 0.63 | 0.84 | 0.98 | 1.03 | - | - | - | ||||
| HCD20 | 0.35 | 0.57 | 0.73 | 0.81 | 0.92 | 1.03 | - | |||||
| HCD22 | 0.30 | 0.50 | 0.60 | 0.74 | 0.87 | 1.00 | ||||||
| HCD24 | 0.31 | 0.45 | 0.61 | 0.76 | 0.91 | |||||||
| HCD26 | 0.28 | 0.46 | 0.62 | 0.78 | ||||||||
| HCD28 | 0.28 | 0.45 | 0.62 | |||||||||
| HCD30 | 0.26 | 0.45 | ||||||||||
| HCD32 | 0.28 |
To further analyze the source of the dissimilarity between spectra, we examined the fragment ions present. The HCD spectra shown in Figure 1, along with additional collisional energies shown in Table 1 were manually identified and the relative ion abundance of each fragment is plotted as a function of collisional energy in Figure 2. Only a, b, and y ions are plotted for simplicity sake. Other ion types specific to HCD, such as internal ions, are present at higher energies. Plotted in this form, it is easy to see how the fragmentation of the peptide is controlled by the amount of energy applied. As the collisional energy is increased some of the ion types (such as y7 and y9) grow and peak around 20 NCE. With further increases in collisional energy these ions start to undergo additional fragmentation and give way to smaller peptide fragments. This can be seen as y6, y5, y4, y3, and y2 grow at higher collisional energies and the intensities of the larger y ions start to diminish. Thus it can be theorized that the larger ion fragments give way to smaller ion types as the energy is increased.[24] Interestingly there is an initial maximum of fragment ion’s intensities at NCE of 20 which is inversely correlated with the depletion of precursor. A second maximum of the smaller fragment ions is seen around 30, which is inversely correlated with the larger y and b ions. There are a few ions, such as a2 and b2, which are likely produced by degradation of multiple ions as they grow at higher energy and eventually plateau and maintain high ion abundance at higher energies. This could either be due to stability of these ions or due to production from multiple pathways. [25]
Figure 2.

Fragment ions plotted as a function of HCD energy for the peptides a) and b) ELGQAGVDTYLQTK c) and d) LTILEELR
Shown in Figure 2c and d is the same type of plot for the peptide LTILEELR. This peptide, although lower in mass and number of amino acids, and thus number of potential fragment ions, still demonstrates similar trends in fragmentation. Larger y ions reach a maximum around 16 to 18 NCE as the precursor abundance decreases. As higher energies are applied y4, y5, and y6 decrease in intensity and y1 and y2 increase. The larger b ions are only observed at NCE of 20 and lower as they further fragment to yield smaller a and b ions, particularly a2 and b2 in this case. At higher energies the immonium ion for leucine and isoleucine ion quickly dominates the spectrum. In this case the a1 ion and the immonium ion are indistinguishable. Immonium ions are often observed in HCD spectra and provide additional sequence information that is not available with standard ion trap data. [26] Use of immonium ions has been shown to improve database searching by reducing the database search space. [27] By comparing Figures a through d it can be seen that the maximum of informative ions occurs at slightly lower collisional energy for this peptide than for the peptide ELGQAGVDTYLQTK shown in Figure 2a and b. Thus choosing a single collisional energy for an entire experiment could potentially bias the identifiable species.
The fragmentation of the peptide TRDIYETDyYRK (with y as phosphotyrosine) is shown in Figure 3. Y ions are plotted in panel a, b ions in panel b and a ions in panel c. It can be seen that the a ions increase in intensity at higher collisional energy than the b and y ions, likely because they are produced from further fragmentation of the b ions. [28] This is a trend that we have observed for the peptides that we have examined. The peptide shown is of particular interest because it contains a phosphotyrosine. Phosphotyrosine is not highly abundant in cell populations, but it has been shown to be important in regulation of signaling pathways. [29] Furthermore, it has been shown that phosphotyrosine containing peptides can easily be identified by the presence of the phosphotyrosine immonium ion at 216.043 m/z. [30,31] The abundance of the immonium ion (noted pY*) is depicted in Figure 3c as a function of collisional energy. While the phosphotyrosine immonium ion is useful in identification of the phosphopeptide, this informative ion is only present once higher collisional energies are used. If the single collisional energy of 25 is used (which seems to be the best single collisional energy for tryptic peptides based on experimental data not shown) the immonium ion will never be observed for this peptide. However a combination of fragmentations at a high energy and a low energy is much more likely to capture points that are highly informative across a greater species of peptides, and would allow the capture of sequence information as well as immonium ion information. From these few randomly chosen peptides it can easily be seen that selecting low and high energy fragmentations increases the informative fragments in the spectra.
Figure 3.

Fragment ion of the peptide TRDIYETDyYRK plotted as a function of HCD energy
To further extend this idea, fragmentation of a peptide of the sequence GILFVGSGVSGGEEGAR is shown in Figure 4. Fragmentation is shown with NCE of 20, 25, 30, and with a stepped collisional energy combining 20, 25 and 30. Although the peptide was identified through database searching in each of these instances, it can be seen that there are subtle but useful differences. The lower collision energy (20) has not fully fragmented the precursor ion, as the parent ion is observed in fairly high abundance. It is advantageous to have the entire parent ion fragmented in order to produce higher signal, and thus a potentially more sensitive method. At 20 NCE a full y ion series is observed as well as a nearly complete b series. Many of the b ions are in low abundance, particularly the larger b ions. Only a few a ions are observed at this collisional energy, as the energy is not sufficient to fragment the precursor multiple times. At higher collisional energy (30) the parent ion has been completely fragmented. A nearly complete y series is still observed but abundances of the ions change slightly in comparison with the lower energy; doubly charged ions are no longer observed and the y1 ion is much more apparent in the spectrum. Very few b ions are observed at this energy, and only the smaller ions are seen (b4, b3, b2) and they occur at higher abundance than with lower energy, suggesting that the smaller b ions are products of fragmentation of the larger, and now absent, larger b ions. A few a ions are also observed, but just like the b ions, the abundance has shifted to the smaller a ions. The b2 and a2 ions are commonly seen as two of the more abundant ions at higher collisional energy in these experiments.
Figure 4.

Fragment ion abundances observed from the peptide GILFVGSGVSGGEEGAR upon HCD activation at 20, 25, 30 and stepped HCD at 20, 25, 30.
Data from a stepped collisional energy experiment is also shown, with low, medium and high energies of 20, 25 and 30. In most cases the abundance of ions from the stepped experiment is approximately the average between the ion abundance seen at 20, 25 and 30. A complete y series is observed, and a partial b series is seen, the larger b ions are absent in this case, but this is not surprising due to their low abundance at the lower collisional energy. There are also several a and d ions present, providing additional useful information. In this instance the d2 ion is observed, allowing identifiaction of the second amino acid as I. [32,33] Fragmentation at 25NCE is also shown, and has been shown previously to be the overall “best” single collision energy for tryptic peptides. It is interesting to see that the stepped experimental data is distinct from data at 25. Thus, utilizing stepped collisional energy we are able to produce a wider variety and coverage of ions than what is achieved through a single collisional energy.
To extend this concept to a larger scale, HEK lysate was compared at low (20), medium (25) and high (30) energies, and with the stepped energy combining 20, 25 and 30. The results of protein and peptide IDs generated by this comparison of HEK lysate are shown in Table 2. Triplicate experiments were performed at each collisional energy and the resulting data is shown in Table 2 along with the combined and searched data.
Table 2.
Proteomic analysis of HEK lysate
| HCD energy | Proteins | Peptides | Spectra Identified | Unfiltered Spectra | % spectra identified |
|---|---|---|---|---|---|
| HCD20 (1) | 2672 | 18259 | 26879 | 183575 | 14.64% |
| HCD20(2) | 3271 | 31153 | 45331 | 198324 | 22.86% |
| HCD20(3) | 2830 | 24908 | 36416 | 182074 | 20.00% |
| 20 (sum) | 3743 | 39383 | 99849 | 563973 | 17.70% |
| HCD25 (1) | 3550 | 35952 | 51677 | 206399 | 25.04% |
| HCD25 (2) | 3357 | 33029 | 49636 | 204511 | 24.27% |
| HCD25 (3) | 2849 | 26699 | 41183 | 178234 | 23.11% |
| 25 (sum) | 4025 | 49095 | 130621 | 589144 | 22.17% |
| HCD30 (1) | 3548 | 38895 | 55713 | 203659 | 27.36% |
| HCD30 (2) | 3143 | 32685 | 48046 | 181652 | 26.45% |
| HCD30 (3) | 2535 | 22289 | 38796 | 159180 | 24.37% |
| 30 (sum) | 3939 | 49171 | 133589 | 544491 | 24.53% |
| Stepped (1) | 3587 | 37061 | 53342 | 203191 | 26.25% |
| Stepped (2) | 3341 | 35345 | 50381 | 186675 | 26.99% |
| Stepped (3) | 2293 | 17521 | 29961 | 167362 | 17.90% |
| 20, 25, 30 (sum) | 4032 | 49146 | 125321 | 557228 | 22.49% |
Utilizing stepped collisional energy has little to no effect on the number of spectra that are collected per experiment and the additional time to fragment the same precursor at three energies is negligible. Fill times are similar for each of the experiments shown; during the stepped experiment one-third of the fill time is allocated to each of the three energies resulting in a minimal effect on the duty cycle length. One possible disadvantage of the stepped energy method is the dilution of ion signal into more fragmentation pathways, as the total ion capacity remains similar to an experiment performed at a single energy. Shown in Figure 5 is the overlap of the peptides and proteins that are identified by each of the collisional energies. The stepped collisional energy overlaps well with both the single energy experiments but also identifies a distinct group of peptides not identified by the single energy experiments. Although peptide fragmentation is dependent on the HCD energy applied, within this small range of energies the search engine is able to capture relatively the same number of peptides for each experiment. While peptide identification itself does not seem to be adversely or positively affected through the use of stepped collisional energies we next examined a few cases where the use of stepped collisional energy is beneficial.
Figure 5.

Overlap of a) peptides and b) proteins identified from Mudpit proteomic analysis of HEK lysate with HCD energies of 20, 25, 30 and stepped HCD at 20,25 and 30.
Phosphorylation site localization is highly dependent on peptide sequence and on sequence coverage. Shown in Figure 6 is the comparison of the number of phosphopeptides identified through the use of a single collisional energy (25) and with the stepped experiment with energies at 20, 25, and 30. Peptides are grouped according to Ascore with higher scores indicating better site localization. Use of multiple collisional energies increases the number of identified phosphorylation sites by ~30%. The use of stepped collisional energy increases not only the number of phosphorylation sites overall but also the confidence of the sites. Percentage of the phosphorylation sites is shown in figure 6b. Stepped collisional energy shows an increase in the Ascore of the peptides, indicating an increased localization of the sites. This increased confidence in site localization is likely due to an increase in sequence coverage which is seen with use of multiple collisional energies. Even in the case of phosphorylated peptides which are often in low abundance the use of stepped collisional energy did not adversely affect the number of peptides identified.
Figure 6.

a) Phosphorylation sites identified by 25NCE and stepped HCD at 20,25 and 30NCE grouped by Ascore. b) Percentage of phosphorylation sites identified by single energy and stepped HCD, stepped HCD provides an increase in Ascores and site localization. c)TMT reporter intensity of identified peptides with 25NCE and with stepped NCE of 30±20%
The effect of stepped HCD on tandem mass tagged (TMT) peptides was also investigated. TMT peptides are labeled with isobaric amine reactive labels which impart quantitative information upon collisional activation. TMT reporter ions are formed through cleavage of the amide bond by collisional energy to create a cluster of ions between 126 and 131m/z. The abundance of reporter ions is proportional to the collisional energy used with higher energy producing an increase in abundance of the reporter ions. Accurate quantitation of channels is dependent on the intensity of the reporter ions, but generation of TMT reporters needs to be balanced with adequate production of peptide sequence ions. Cell lysate labeled with TMT were analyzed with MudPIT LCMS on the Q Exactive either using a single collisional energy at 25 or with the use of stepped collisional energy centered at 30 with + 20% (effectively 24, 30, and 36NCE). The energies used for the stepped experiments were higher than the other experiments previously described to demonstrate the use of the higher collisional energy in the context of the stepped experiment. Use of the stepped HCD not only shows an increase in the TMT reporter ion intensities by approximately an order of magnitude, but also an increase in the number of peptides identified.
Conclusion
Due to the diversity of peptide sequence, “one size fits all” is not always the case when it comes to generating useful spectra. By taking advantage of stepped normalized collision energy we are able to increase the diversity of useful fragment ions. We show applications of stepped HCD for phosphorylation site localization and TMT quantitation. Increased sequence coverage by the use of multiple collisional energies yields an increase in the number of phospho peptides identified as well as increased Ascores, a measurement of the confidence in site localization. Stepped HCD is beneficial for TMT experiments as the reporter ion intensity is increased through the use of a higher HCD energy without sacrificing detection of peptide fragmentation needed for peptide identification.
Acknowledgments
We thank Aaron Aslanian for culture of HEK cells and Claire Delahunty for the critical reading of the manuscript. Financial support was provided by NIH grants P41 RR011823/GM103533, R01 MH067880-09, P01 AG031097-03, and R01 HL079442-07.
References
- 1.Frank AM. Predicting Intensity Ranks of Peptide Fragment Ions. Journal of Proteome Research. 2009;8(5) doi: 10.1021/pr800677f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yen C-Y, Meyer-Arendt K, Eichelberger B, Sun S, Houel S, Old WM, Knight R, Ahn NG, Hunter LE, Resing KA. A Simulated MS/MS Library for Spectrum-to-spectrum Searching in Large Scale Identification of Proteins. Molecular & Cellular Proteomics. 2009;8(4) doi: 10.1074/mcp.M800384-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Biemann K. SEQUENCING OF PEPTIDES BY TANDEM MASS-SPECTROMETRY AND HIGH-ENERGY COLLISION-INDUCED DISSOCIATION. Methods in Enzymology. 1990;193:455–479. doi: 10.1016/0076-6879(90)93433-l. [DOI] [PubMed] [Google Scholar]
- 4.Hunt DF, Yates JR, Shabanowitz J, Winston S, Hauer CR. PROTEIN SEQUENCING BY TANDEM MASS-SPECTROMETRY. Proceedings of the National Academy of Sciences of the United States of America. 1986;83(17):6233–6237. doi: 10.1073/pnas.83.17.6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Graaf EL, Altelaar AFM, van Breukelen B, Mohammed S, Heck AJR. Improving SRM Assay Development: A Global Comparison between Triple Quadrupole, Ion Trap, and Higher Energy CID Peptide Fragmentation Spectra. Journal of Proteome Research. 2011;10(9) doi: 10.1021/pr200156b. [DOI] [PubMed] [Google Scholar]
- 6.Michalski A, Damoc E, Hauschild J-P, Lange O, Wieghaus A, Makarov A, Nagaraj N, Cox J, Mann M, Horning S. Mass Spectrometry-based Proteomics Using Q Exactive, a High-performance Benchtop Quadrupole Orbitrap Mass Spectrometer. Molecular & Cellular Proteomics. 2011;10(9) doi: 10.1074/mcp.M111.011015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kelstrup CD, Young C, Lavallee R, Nielsen ML, Olsen JV. Optimized Fast and Sensitive Acquisition Methods for Shotgun Proteomics on a Quadrupole Orbitrap Mass Spectrometer. Journal of Proteome Research. 2012;11(6):3487–3497. doi: 10.1021/pr3000249. [DOI] [PubMed] [Google Scholar]
- 8.Mayer PM, Poon C. THE MECHANISMS OF COLLISIONAL ACTIVATION OF IONS IN MASS SPECTROMETRY. Mass Spectrometry Reviews. 2009;28(4) doi: 10.1002/mas.20225. [DOI] [PubMed] [Google Scholar]
- 9.Michalski A, Neuhauser N, Cox J, Mann M. A Systematic Investigation into the Nature of Tryptic HCD Spectra. Journal of proteome research. 2012;11(11):5479–91. doi: 10.1021/pr3007045. [DOI] [PubMed] [Google Scholar]
- 10.van Dongen WD, Ruijters HFM, Luinge HJ, Heerma W, Haverkamp J. Statistical analysis of mass spectral data obtained from singly protonated peptides under high-energy collision-induced dissociation conditions. Journal of Mass Spectrometry. 1996;31(10) doi: 10.1002/(SICI)1096-9888(199610)31:10<1156::AID-JMS407>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 11.Cox KA, Gaskell SJ, Morris M, Whiting A. Role of the site of protonation in the low-energy decompositions of gas-phase peptide ions. Journal of the American Society for Mass Spectrometry. 1996;7(6):522–531. doi: 10.1016/1044-0305(96)00019-0. [DOI] [PubMed] [Google Scholar]
- 12.Dongre AR, Jones JL, Somogyi A, Wysocki VH. Influence of peptide composition, gas-phase basicity, and chemical modification on fragmentation efficiency: Evidence for the mobile proton model. Journal of the American Chemical Society. 1996;118(35):8365–8374. [Google Scholar]
- 13.Jedrychowski MP, Huttlin EL, Haas W, Sowa ME, Rad R, Gygi SP. Evaluation of HCD- and CID-type Fragmentation Within Their Respective Detection Platforms For Murine Phosphoproteomics. Molecular & Cellular Proteomics. 2011;10(12):9. doi: 10.1074/mcp.M111.009910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frese CK, Altelaar AFM, Hennrich ML, Nolting D, Zeller M, Griep-Raming J, Heck AJR, Mohammed S. Improved Peptide Identification by Targeted Fragmentation Using CID, HCD and ETD on an LTQ-Orbitrap Velos. Journal of Proteome Research. 2011;10(5) doi: 10.1021/pr1011729. [DOI] [PubMed] [Google Scholar]
- 15.Shen YF, Tolic N, Purvine SO, Smith RD. Improving Collision Induced Dissociation (CID), High Energy Collision Dissociation (HCD), and Electron Transfer Dissociation (ETD) Fourier Transform MS/MS Degradome-Peptidome Identifications Using High Accuracy Mass Information. Journal of Proteome Research. 2012;11(2):668–677. doi: 10.1021/pr200597j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagaraj N, D’Souza RCJ, Cox J, Olsen JV, Mann M. Feasibility of Large-Scale Phosphoproteomics with Higher Energy Collisional Dissociation Fragmentation. Journal of Proteome Research. 2010;9(12):6786–6794. doi: 10.1021/pr100637q. [DOI] [PubMed] [Google Scholar]
- 17.Bushee JL, Argikar UA. An experimental approach to enhance precursor ion fragmentation for metabolite identification studies: application of dual collision cells in an orbital trap. Rapid Communications in Mass Spectrometry. 2011;25(10) doi: 10.1002/rcm.4996. [DOI] [PubMed] [Google Scholar]
- 18.Chakraborty AB, Berger SJ, Gebler JC. Use of an integrated MS-multiplexed MS/MS data acquisition strategy for high-coverage peptide mapping studies. Rapid Communications in Mass Spectrometry. 2007;21(5):730–744. doi: 10.1002/rcm.2888. [DOI] [PubMed] [Google Scholar]
- 19.Dayon L, Pasquarello C, Hoogland C, Sanchez JC, Scherl A. Combining low- and high-energy tandem mass spectra for optimized peptide quantification with isobaric tags. Journal of Proteomics. 2010;73(4):769–777. doi: 10.1016/j.jprot.2009.10.015. [DOI] [PubMed] [Google Scholar]
- 20.McDonald WH, Tabb DL, Sadygov RG, MacCoss MJ, Venable J, Graumann J, Johnson JR, Cociorva D, Yates JR. MS1, MS2, and SQT - three unified, compact, and easily parsed file formats for the storage of shotgun proteomic spectra and identifications. Rapid Communications in Mass Spectrometry. 2004;18(18):2162–2168. doi: 10.1002/rcm.1603. [DOI] [PubMed] [Google Scholar]
- 21.Tabb DL, McDonald WH, Yates JR. DTASelect and contrast: Tools for assembling and comparing protein identifications from shotgun proteomics. Journal of Proteome Research. 2002;1(1):21–26. doi: 10.1021/pr015504q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Falick AM, Hines WM, Medzihradszky KF, Baldwin MA, Gibson BW. LOW-MASS IONS PRODUCED FROM PEPTIDES BY HIGH-ENERGY COLLISION-INDUCED DISSOCIATION IN TANDEM MASS-SPECTROMETRY. Journal of the American Society for Mass Spectrometry. 1993;4(11) doi: 10.1016/1044-0305(93)87006-X. [DOI] [PubMed] [Google Scholar]
- 23.Tabb DL, MacCoss MJ, Wu CC, Anderson SD, Yates JR. Similarity among tandem mass spectra from proteomic experiments: Eetection, significance, and utility. Analytical Chemistry. 2003;75(10) doi: 10.1021/ac026424o. [DOI] [PubMed] [Google Scholar]
- 24.Jue AL, Racine AH, Glish GL. The effect of ion trap temperature on the dissociation of peptide ions in a quadrupole ion trap. International Journal of Mass Spectrometry. 2011;301(1–3) [Google Scholar]
- 25.Yalcin T, Csizmadia IG, Peterson MR, Harrison AG. The structure and fragmentation of B-n (n>=3) ions in peptide spectra. Journal of the American Society for Mass Spectrometry. 1996;7(3) doi: 10.1016/1044-0305(95)00677-X. [DOI] [PubMed] [Google Scholar]
- 26.Papayannopoulos IA. THE INTERPRETATION OF COLLISION-INDUCED DISSOCIATION TANDEM MASS-SPECTRA OF PEPTIDES. Mass Spectrometry Reviews. 1995;14(1) [Google Scholar]
- 27.Eng JK, McCormack AL, Yates JR. AN APPROACH TO CORRELATE TANDEM MASS-SPECTRAL DATA OF PEPTIDES WITH AMINO-ACID-SEQUENCES IN A PROTEIN DATABASE. Journal of the American Society for Mass Spectrometry. 1994;5(11):976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 28.Paizs B, Szlavik Z, Lendvay G, Vekey K, Suhai S. Formation of a(2)(+) ions of protonated peptides. An ab initio study. Rapid Communications in Mass Spectrometry. 2000;14(9) doi: 10.1002/(SICI)1097-0231(20000515)14:9<746::AID-RCM939>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 29.Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127(3):635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 30.Steen H, Kuster B, Fernandez M, Pandey A, Mann M. Detection of tyrosine phosphorylated peptides by precursor ion scanning quadrupole TOF mass spectrometry in positive ion mode. Analytical Chemistry. 2001;73(7) doi: 10.1021/ac001318c. [DOI] [PubMed] [Google Scholar]
- 31.Olsen JV, Macek B, Lange O, Makarov A, Horning S, Mann M. Higher-energy C-trap dissociation for peptide modification analysis. Nature Methods. 2007;4(9):709–712. doi: 10.1038/nmeth1060. [DOI] [PubMed] [Google Scholar]
- 32.Johnson RS, Martin SA, Biemann K, Stults JT, Watson JT. NOVEL FRAGMENTATION PROCESS OF PEPTIDES BY COLLISION-INDUCED DECOMPOSITION IN A TANDEM MASS-SPECTROMETER - DIFFERENTIATION OF LEUCINE AND ISOLEUCINE. Analytical Chemistry. 1987;59(21):2621–2625. doi: 10.1021/ac00148a019. [DOI] [PubMed] [Google Scholar]
- 33.Johnson RS, Martin SA, Biemann K. COLLISION-INDUCED FRAGMENTATION OF (M+H)+IONS OF PEPTIDES - SIDE-CHAIN SPECIFIC SEQUENCE IONS. International Journal of Mass Spectrometry and Ion Processes. 1988;86:137–154. [Google Scholar]