

Abstract
Lactams and imides have been shown to consistently provide enantioselectivities substantially higher than other substrate classes previously investigated in the palladium-catalyzed asymmetric decarboxylative allylic alkylation. We have designed several new substrates to probe the contributions of electronic, steric, and stereoelectronic factors that distinguish the lactam/imide series as superior alkylation substrates. These studies culminated in marked improvements on carbocyclic allylic alkylation substrates.
Keywords: asymmetric catalysis, palladium, allylation, heterocyclic compd., high-throughput screen
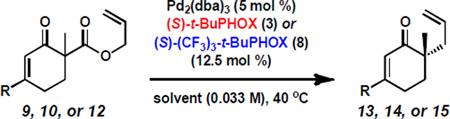
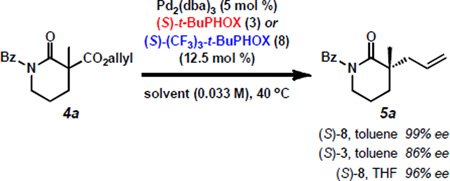
The asymmetric construction of quaternary stereocenters is a topic of great interest in the organic chemistry community. Among the available methods that afford this motif,[1] palladium-catalyzed decarboxylative allylic alkylation[2, 3] has proven particularly effective and, over the last decade, our group has pursued this strategy employing chiral phosphinooxazoline (PHOX) ligands.[4,5] Our initial efforts in this area led to the preparation of enantioenriched α-quaternary ketones (e.g. 2a) in good yields and enantioselectivities using (S)-t-BuPHOX (3)[5a–b] as a chiral ligand (Scheme 1A).[6] Since these early results, we have considerably expanded the scope,[7] demonstrated multiple applications,[8] and performed mechanistic investigations[9] of this powerful transformation. Recently, we discovered that the allylic alkylation of lactams (4a to 5a) and imides (6a to 7a) with (S)-(CF3)3-t-BuPHOX (8)[5c] consistently proceeds with enantioselectivities substantially higher than any other substrate class previously examined in this system (Scheme 1B and C).[10] This observation prompted us to investigate which characteristics distinguish these molecules as superior alkylation substrates. The basic distinctions between these ketone and N-heterocyclic molecules are the deviation in electronic nature of the enolate and the identity of the α'-functionality (i.e. the group flanking the carbonyl at the site opposite of alkylation). Thus, we have designed several new alkylation substrates to examine the relative contribution of each effect. We have found that the exceptional enantioselectivities observed in the lactam/imide series are likely not a result of a purely electronic effect, but a combination of stereoelectronic and steric factors associated with the α'-substituent.
Scheme 1.

Comparison of allylic alkylation of ketones, lactams, and imides.
Initially, we hypothesized that the divergence in enolate electronics between the substrate classes depicted in Scheme 1 could be the major determining factor of the observed enantioselectivities. Insight from our previous work[9,10] suggested that differences in selectivity in the alkylation of electron-poor and -rich molecules could be considerable. To investigate the electronic effect of the nitrogen atom on alkylation selectivity without the influence of α'-functionality, we examined variably functionalized enaminones (12, i.e. vinylogous amides) as electronic analogs of lactams (4, Figure 1). We were particularly drawn to this new class of compounds as our past experience with vinylogous esters (10)[11] and thioesters (11)[11b,12] would provide a foundation and comparison point.[13]
Figure 1.

Ketone and lactam enantioselectivity divergence as inspiration for investigation of enolate electronics using vinylogous systems.
We consequently prepared a number of racemic alkylation precursors and screened these under a series of palladium-catalyzed decarboxylative allylic alkylation conditions (Table 1). This study was performed in a manner similar to our previous investigation of lactams,[10, 14, 15] employing two electronically differentiated chiral ligands, (S)-3 and (S)-8, and four solvents of differing polarity: tetrahydrofuran (THF), methyl tert-butyl ether (MTBE), toluene, and 2:1 hexane–toluene.
Table 1.
Enaminone Allylic Alkylation Screen.[a]
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Enantiomeric Excess (% ee)[b] | ||||||||
| entry | Substrate | R | Product | ligand | THF | MTBE | Toluene | 2:1 Hex–Tol |
| 1 | 9a | H | 13a | 3 | 87 | 88 | 87 | 87 |
| 2 | 8 | 85 | 86 | 88 | 85 | |||
| 3 | 10a | Oi-Bu | 14a | 3 | 85 | 85 | 86 | 87 |
| 4 | 8 | 86 | 86 | 86 | 88 | |||
| 5 | 12a | NMe(Bn) | 15a | 3 | 61 | 60 | 55 | 52 |
| 6 | 8 | 79 | 78 | 84 | 83 | |||
| 7 | 12b | NPh(Bn) | 15b | 3 | 81 | 87 | 85 | 83 |
| 8 | 8 | 76 | 74 | 82 | 83 | |||
| 9 | 12c | NAc(Bn) | 15c | 3 | 89 | 90 | 88 | 88 |
| 10 | 8 | 83 | 85 | 88 | 86 | |||
| 11 | 12d | NBz(Bn) | 15d | 3 | 86 | 87 | 88 | 87 |
| 12 | 8 | 80 | 83 | 82 | 83 | |||
| 13 | 12e | NBoc(Bn) | 15e | 3 | 87 | 86 | 87 | 82 |
| 14 | 8 | 84 | 84 | 81 | 83 | |||
| 15 | 12f | NTs(Bn) | 15f | 3 | 84 | 83 | 83 | 82 |
| 16 | 8 | 82 | 83 | 83 | 83 | |||
Conditions: enone 9a, vinylogous ester 10a, or enaminone 12a–f (1.0 equiv), Pd2(dba)3 (5 mol %), and (S)-t-BuPHOX (3) or (S)-(CF3)3-t-BuPHOX (8) (12.5 mol %) in solvent (0.033 M) at 40 °C.
Determined by GC, HPLC, or SFC analysis with chiral stationary phase.
Red = with (S)-3 as ligand and blue = with (S)-8 as ligand.
We made several observations upon analysis of the ligand, solvent, and substrate trends that distinguished this substrate class considerably from the previously examined lactams. First, enaminones are obtained in modestly better selectivity with (S)-3 as a ligand in most solvents (entries 7–16), with the exception of methyl enaminone 15a, which significantly favors (S)-8 (entries 5–6). Second, no significant solvent trend is observed for enaminones overall. Third, electron-rich enaminones (i.e. 15a–b) display decreased enantioselectivities and also require extended reaction times for completion,[16] while enaminones bearing electron-withdrawing substituents (15c–f) are all generated in 80–90% ee. By comparison, lactam substrates perform with much higher enantioselectivity with ligand (S)-8 in non-polar solvents.[10, 17] Furthermore, the modest differences in enantioselectivity for the electron-withdrawing enaminones sharply contrasts with the corresponding lactam series, where the N-substituent plays a considerable role, producing significant variation in selectivity.[10,18] These differences suggest that the N-functional group does not contribute to enantioselectivity solely through a perturbation of enolate electronics.
Beyond these considerations, the most striking feature of the screen is that enantioselectivities observed for enaminones 15c–f are approximately equivalent to results obtained for enone 13a,[19] vinylogous ester 14a, and even more general ketone substrates. Of all the enaminones screened, acetyl variant 15c provides the highest selectivity at 90% ee in MTBE, which is only marginally better than the optimal values for the related vinylogous systems. The modest differences between these vinylogous molecules (i.e. 13–15) further suggests that the electronic nature of the enolate is not likely the predominant factor in providing high enantioselectivity for lactams and imides.
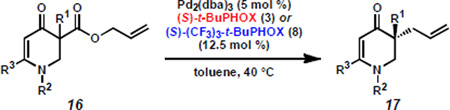
While pursuing enaminones, we also briefly examined the related 2,3-dihydropyridin-4-ones, which possess the nitrogen within the ring (Table 2). N-Carboxybenzyl substituted product 17a is formed with enantioselectivities similar to electronically related enaminones, and electron-rich 2,3-dihydropyridin-4-ones 17b–c are curiously also generated in the same range. Even 2,3-dihydropyridin-4-one 17d is produced in excellent enantioselectivity, despite the highly electron-rich nature of the enolate. These results again allude to other factors beyond enolate electronics that direct alkylation selectivity.
Table 2.
Allylic alkylation of 2,3-dihydropyridin-4-ones.[a]
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | Substrate (16) |
R1 | R2 | R3 | Product (17) |
ligand | yield (%) | ee (%)[b] |
| 1 | 16a | Me | Cbz | H | 17a | 3 | 80 | 86 |
| 2 | 8 | 98 | 84 | |||||
| 3 | 16b | Me | Bn | H | 17b | 3 | 82 | 88 |
| 4 | 8 | 94 | 86 | |||||
| 5 | 16c | i-Bu | Bn | H | 17c | 3 | 85 | 84 |
| 6 | 8 | 81 | 88 | |||||
| 7 | 16d |  |
17d | 8 | 81 | 91 | ||
Conditions: 2,3-dihydropyridin-4-ones 16, Pd2(dba)3 (5 mol %), and (S)-t-BuPHOX (3) or (S)-(CF3)3-t-BuPHOX (8) (12.5 mol %) in toluene (0.033 M) at 40 °C.
Determined by chiral HPLC or SFC analysis.
Red = with (S)-3 as ligand and blue = with (S)-8 as ligand.
Having investigated the impact of enolate electronics, we diverted our efforts to study the influence of α'-functionality on alkylation selectivity (Scheme 2). Our previous lactam screen identified the benzoyl moiety as the optimal protecting group,[10] providing high to nearly perfect enantioselectivities for a variety of lactams (e.g. 5a). Interestingly, both electron-rich and electron-poor benzoyl lactams (5b–d) as well as naphthoyl lactams (5e–f) also display excellent ee, whereas acetyl lactams (e.g. 5g–h) provide lower selectivity. This prompted us to question whether these results are due to a hybridization or steric effect. Consequently, we synthesized the bulky sp3 hybridized cyclohexoyl lactam 4i and pivaloyl lactam 4j, which proceeded in improved enantioselectivities compared to analogous acetyl lactam 4g, supporting a steric effect.[20]
Scheme 2.

Impact of various sp3 and sp2 acyl protecting groups on allylic alkylation enantioselectivity for lactams.
In conjunction with our investigation of the asymmetric allylic alkylation of lactams, we found that N-benzoyl cyclic imides 7a–b are furnished in excellent ee (Scheme 3). However, we observed that formation of N-methyl imide 7c proceeds with moderate enantioselectivity and is hampered by low reactivity. As this substrate also generates an intermediate imido enolate (as do lactams 5a–j, Scheme 2), this result further supports our conclusion that enolate electronics do not play a major role in the enhanced enantioselectivities observed for N-benzoyl lactams. As such, we examined the influence of alternate N-substituents in the context of the allylic alkylation with imides.
Scheme 3.

Allylic alkylation of cyclic imides.
Gratifyingly, we identified N-benzyloxy imides as excellent substrates for this methodology, generating imides 7d–e in yields and enantioselectivities comparable to their N-benzoyl counterparts. In conjunction with the results obtained for substituted N-acyl lactams 5g–j, we reason that the nature of the α'-substituent leads to the observed enhancements in enantioselectivity, though enolate electronics have been shown to dramatically affect the reaction rate.
To examine the relative merit of the steric and stereoelectronic effects associated with the α'-group, we prepared enones 9b–c and diosphenol ethers 18a–b and subjected these substrates to the standard conditions employed in the alkylation of lactams and imides (Scheme 4). Enones 13b–c are formed in low enantioselectivity. By contrast, benzyl diosphenol ether 19a, which differs from 13c only in the substitution of oxygen for a methylene group, is generated in 92% yield and 94% ee. This suggests that purely steric or π-stacking interactions are not the sole contributing factors to enantioselectivity. Rather, electronic effects of the α'-substituent exert an important effect on the stereoselectivity of the reaction. However, a certain amount of steric bulk appears critical in obtaining high enantioselectivity as methyl diosphenol ether 19b is produced in 85% ee. In comparison, analogous enone 9a, which bears no α'-functionality, proceeds under the same conditions to afford enone 13a in 88% ee,[19] with vinylogous amides and esters also in the ~80–90% ee range (vide supra). Overall, our studies on the role of the α'-substituent have culminated in the discovery of substrate 18a, which proceeds with the greatest enantioselectivity observed in a Pd(PHOX) catalyst system for a carbocyclic substrate bearing an α-methyl and unsubstituted allyl moiety.
Scheme 4.

Allylic alkylation of α'-functionalized enones.
In summary, we have designed and evaluated a number of novel substrates to probe the influence of enolate electronics and the role of α'-functionality on selectivity in the palladium-catalyzed decarboxylative allylic alkylation. Based on these results, we reason that the high enantioselectivities observed with lactams and imides are a consequence of both electronic and steric effects associated with α'-substituents, and that enolate electronics alone contribute relatively little to the stereochemical outcome of the reaction. Both experimental and theoretical investigations are currently underway in our group to determine the nature and origin of the effect of α'-substitution on this transformation and to use this insight to improve and expand our methods.
Supplementary Material
Acknowledgements
The authors thank NIH-NIGMS (R01GM080269-01), Roche, Abbott Laboratories, Amgen, Boehringer Ingelheim, the Gordon and Betty Moore Foundation, and Caltech for awards and financial support. D.C.D. thanks the National Science Foundation for financial support (Predoctoral Research Fellowship – No. DGE-1144469). W.-B.L. thanks the Shanghai Institute of Organic Chemistry for financial support (SIOC Postdoctoral Fellowship). A.N.M. is grateful for a fellowship by the Deutsche Akademie der Naturforscher Leopoldina. Profs. Sarah Reisman and Theodor Agapie are acknowledged for helpful discussions and suggestions. Drs. David VanderVelde and Scott Ross are acknowledged for NMR assistance. Dr. Mona Shahgoli and Naseem Torian are acknowledged for High-Resolution Mass Spectrometry assistance.
Footnotes
Supporting information for this article is available on the WWW under http://www.chemeurj.org/ or from the author.
References
- 1.For review of approaches for the asymmetric construction of all-carbon quaternary stereocenters, see: Christoffers J, Baro A. Adv. Synth. Catal. 2005;347:1473–1482. Trost BM, Jiang C. Synthesis. 2006:369–396.
- 2.For examples of other efforts in the enantioselective allylic alkylation of enolates catalyzed by palladium, see: Trost BM, Schroeder GM. Chem. — Eur. J. 2005;11:174–184. doi: 10.1002/chem.200400666. Trost BM, Xu J, Schmidt T. J. Am. Chem. Soc. 2009;131:18343–18357. doi: 10.1021/ja9053948. Trost BM, Schäffner B, Osipov M, Wilton DAA. Angew. Chem. Int. Ed. 2011;50:3548–3551. doi: 10.1002/anie.201007803. Nakamura M, Hajra A, Endo K, Nakamura E. Angew. Chem. Int. Ed. 2005;44:7248–7251. doi: 10.1002/anie.200502703. Bélanger É, Cantin K, Messe O, Tremblay M, Paquin J-F. J. Am. Chem. Soc. 2007;129:1034–1035. doi: 10.1021/ja067501q. Bélanger É, Houzé C, Guimond N, Cantin K, Paquin J-F. Chem. Commun. 2008:3251–3253. doi: 10.1039/b803097a. Burger EC, Tunge JA. Org. Lett. 2004;6:4113–4115. doi: 10.1021/ol048149t. Chattopadhyay K, Jana R, Day VW, Douglas JT, Tunge JA. Org. Lett. 2010;12:3042–3045. doi: 10.1021/ol101042x.
- 3.For reviews on palladium-catalyzed allylic alkylation, see: Tunge JA, Burger EC. Eur. J. Org. Chem. 2005:1715–1726. Braun M, Meier T. Angew. Chem. Int. Ed. 2006;45:6952–6955. doi: 10.1002/anie.200602169. Mohr JT, Stoltz BM. Chem. Asian J. 2007;2:1476–1491. doi: 10.1002/asia.200700183. Weaver JD, Recio A, III, Grenning AJ, Tunge JA. Chem. Rev. 2011;111:1846–1913. doi: 10.1021/cr1002744.
- 4.For references on the development of oxazoline ligands, see: Schnider P, Koch G, Prétôt R, Wang G, Bohnen FM, Krüger C, Pfaltz A. Chem. Eur. J. 1997;3:887–892. Lightfoot A, Schnider P, Pfaltz A. Angew. Chem. 1998;110:3047–3050. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2897::AID-ANIE2897>3.0.CO;2-8. Angew. Chem. Int. Ed.1998, 37, 2897–2899; Blankenstein J, Pfaltz A. Angew. Chem. 2001;113:4577–4579. Angew. Chem. Int. Ed.2001, 40, 4445–4447; Menges F, Pfaltz A. Adv. Synth. Catal. 2002;344:40–44. Nanchen S, Pfaltz A. Chem. — Eur. J. 2006;12:4550–4558. doi: 10.1002/chem.200501500. Bélanger É, Pouliot M-F, Paquin J-F. Org. Lett. 2009;11:2201–2204. doi: 10.1021/ol9005618. Bélanger É, Pouliot M-F, Courtemanche M-A, Paquin J-F. J. Org. Chem. 2012;77:317–331. doi: 10.1021/jo2019653. Pouliot M-F, Angers L, Hamel J-D, Paquin J-F. Tetrahedron Lett. 2012;53:4121–4123.
- 5.a) Tani K, Behenna DC, McFadden RM, Stoltz BM. Org. Lett. 2007;9:2529–2531. doi: 10.1021/ol070884s. [DOI] [PubMed] [Google Scholar]; b) Krout MR, Mohr JT, Stoltz BM. Org. Synth. 2009;86:181–193. doi: 10.15227/orgsyn.086.0181. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) McDougal NT, Streuff J, Mukherjee H, Virgil SC, Stoltz BM. Tetrahedron Lett. 2010;51:5550–5554. doi: 10.1016/j.tetlet.2010.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Behenna DC, Stoltz BM. J. Am. Chem. Soc. 2004;126:15044–15045. doi: 10.1021/ja044812x. [DOI] [PubMed] [Google Scholar]; b) Mohr JT, Behenna DC, Harned AM, Stoltz BM. Angew. Chem. Int. Ed. 2005;44:6924–6927. doi: 10.1002/anie.200502018. [DOI] [PubMed] [Google Scholar]
- 7.Behenna DC, Mohr JT, Sherden NH, Marinescu SC, Harned AM, Tani K, Seto M, Ma S, Novák Z, Krout MR, McFadden RM, Roizen JL, Enquist JA, Jr, White DE, Levine SR, Petrova KV, Iwashita A, Virgil SC, Stoltz BM. Chem. — Eur. J. 2011;17:14199–14223. doi: 10.1002/chem.201003383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For examples of total syntheses, see: McFadden RM, Stoltz BM. J. Am. Chem. Soc. 2006;128:7738–7739. doi: 10.1021/ja061853f. Enquist JA, Jr, Stoltz BM. Nature. 2008;453:1228–1231. doi: 10.1038/nature07046. Day JJ, McFadden RM, Virgil SC, Kolding H, Alleva JL, Stoltz BM. Angew. Chem. Int. Ed. 2011;50:6814– 6818. doi: 10.1002/anie.201101842. Hong AY, Stoltz BM. Angew. Chem. Int. Ed. 2012;51:9674–9678. doi: 10.1002/anie.201205276.
- 9.a) Keith JA, Behenna DC, Mohr JT, Ma S, Marinescu SC, Oxgaard J, Stoltz BM, Goddard WA., III J. Am. Chem. Soc. 2007;129:11876–11877. doi: 10.1021/ja070516j. [DOI] [PubMed] [Google Scholar]; b) Sherden NH, Behenna DC, Virgil SC, Stoltz BM. Angew. Chem. Int. Ed. 2009;48:6840–6843. doi: 10.1002/anie.200902575. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Keith JA, Behenna DC, Sherden N, Mohr JT, Ma S, Marinescu SC, Nielsen RJ, Oxgaard J, Stoltz BM, Goddard WA., III J. Am. Chem. Soc. 2012;134:19050–19060. doi: 10.1021/ja306860n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Behenna DC, Liu Y, Yurino T, Kim J, White DE, Virgil SC, Stoltz BM. Nature Chem. 2012;4:130–133. doi: 10.1038/nchem.1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.For examples of research on vinylogous esters, see: White DE, Stewart IC, Grubbs RH, Stoltz BM. J. Am. Chem. Soc. 2008;130:810–811. doi: 10.1021/ja710294k. Krout MR. Ph.D. Dissertation. CA: California Institute of Technology; 2009. Sep, Progress Toward the Asymmetric Total Synthesis of Variecolin and Gas-Phase Studies of the Twisted Amide 2-Quinuclidone. Hong AY, Krout MR, Jensen T, Bennett NB, Harned AM, Stoltz BM. Angew. Chem. Int. Ed. 2011;50:2756–2760. doi: 10.1002/anie.201007814. Bennett NB, Hong AY, Harned AM, Stoltz BM. Org. Biomol. Chem. 2012;10:56–59. doi: 10.1039/c1ob06189e. Hong AY, Bennett NB, Krout MR, Jensen T, Harned AM, Stoltz BM. Tetrahedron. 2011;67:10234–10248. doi: 10.1016/j.tet.2011.10.031.
- 12.For examples of research on vinylogous thioesters, see: Levine SR, Krout MR, Stoltz BM. Org. Lett. 2009;11:289–292. doi: 10.1021/ol802409h. Petrova KV, Mohr JT, Stoltz BM. Org. Lett. 2009;11:293–295. doi: 10.1021/ol802410t.
- 13.Concurrent with our efforts, Trost has also performed palladium-catalyzed asymmetric allylic alkylations to form enantioenriched α-quaternary vinylogous esters and thioesters. See: Trost BM, Pissot-Soldermann C, Chen I, Schroeder GM. J. Am. Chem. Soc. 2004;126:4480–4481. doi: 10.1021/ja0497025. Trost BM, Schroeder GM. Chem. — Eur. J. 2005;11:174–184. doi: 10.1002/chem.200400666. Trost BM, Pissot-Soldermann C, Chen I. Chem. — Eur. J. 2005;11:951–959. doi: 10.1002/chem.200400558. Trost BM, Bream RN, Xu J. Angew. Chem. Int. Ed. 2006;45:3109–3112. doi: 10.1002/anie.200504421.
- 14.For another example of a screen performed with a Symyx core module, see: McDougal NT, Virgil SC, Stoltz BM. Synlett. 2010:1712–1716. doi: 10.1055/s-0030-1258094.
- 15.See Supporting Information.
- 16.Corresponding electron-rich lactams (N-substituent = Me or Bn) exhibit little to no conversion to the desired products and, as such, are poor alkylation substrates.
-
17.For example, selectivities for lactam 5a vary from 99% ee with (S)-8 to 86% ee with (S)-3 in toluene and to 96% ee with (S)-8 in THF.
-
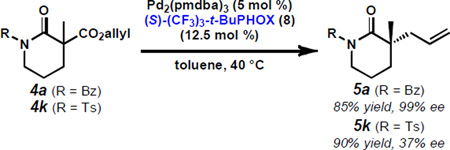
18.For example, with (S)-8 in toluene, observed selectivities for lactams vary from 99% ee (5a, R = Bz) to 37% ee (5b, R = Ts).
- 19.Employing optimized conditions with (S)-3 in Et2O, enone 13a has been isolated in 90% ee. See reference 6b.
- 20.The increase in steric bulk for substrates 4i–j also slows the reaction dramatically. See Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.