

Abstract
Bicalutamide (Casodex®) is a non-steroidal pure anti-androgen used in the treatment of localized prostate cancer. It is a racemate drug and its activity resides in the (R)-enantiomer, with little in the (S)-enantiomer. A major metabolic pathway for bicalutamide is glucuronidation catalyzed by UDP-glucuronosyltransferase (UGT) enzymes. While (S)bicalutamide is directly glucuronidated, (R)bicalutamide requires hydroxylation prior to glucuronidation. The contribution of human tissues and UGT isoforms in the metabolism of these enantiomers has not been extensively investigated. In this study, both (R) and/or (S)bicalutamide were converted into glucuronide (-G) derivatives following incubation of pure and racemic solutions with microsomal extracts from human liver and kidney. Intestinal microsomes exhibited only low reactivity with these substrates. Km values of liver and kidney samples for (S)bicalutamide glucuronidation were similar, and lower than values obtained with the (R)-enantiomer. Among the 16 human UGTs tested, UGT1A8 and UGT1A9 were able to form both (S) and (R)bicalutamide-G from pure or racemic substrates. UGT2B7 was also able to form (R)bicalutamide-G. Kinetic parameters of the recombinant UGT2B7, UGT1A8 and UGT1A9 enzymes support a predominant role of the UGT1A9 isoform in bicalutamide metabolism. Accordingly, (S)bicalutamide inhibited the ability of human liver and kidney microsomes to glucuronidate the UGT1A9 probe substrate, propofol. In conclusion, the present study provides the first comprehensive analysis of in vitro bicalutamide glucuronidation by human tissues and UGTs, and identifies UGT1A9 as a major contributor for (R) and (S) glucuronidation in the human liver and kidney.
INTRODUCTION
Bicalutamide (Casodex®, Figure 1) is a non-steroidal pure anti-androgen used for the treatment of early localized or locally advanced non-metastatic prostate cancer (reviewed in [1]). The drug is given once daily at a dosage of 150 mg as monotherapy or of 50 mg in combination with chemical or surgical castration. This pure anti-androgen competes with the active hormone, 5α-dihydrotestosterone, for androgen receptor binding and activation, and thus blocks the prostate tumor growth-stimulating effects of androgens [2]. The drug used in clinic corresponds to a racemate of (R) and (S) enantiomers (Figure 1), and its anti-androgenic activity resides almost exclusively in the (R)-enantiomer, with little, if any activity in the (S)-enantiomer [1, 2].
Figure 1. Chemical structure and representative chromatograms of (R) and (S) bicalutamide-glucuronide.

(A and B) Bicalutamide (A) is a pure non-steroidal anti-androgen used in clinic as a racemate of R and S enantiomers. Both enantiomers can be glucuronidated (B).
(C and D) Representative chromatograms of (R)bicalutamide-glucuronide (C) and (S)-glucuronide (D) as obtained from incubated samples. Retention times were 3.1 minutes (min) for (R)bicalutamide-glucuronide and 3.9 minutes for (S)bicalutamide-glucuronide and deuterated internal standard (S)bicalutamide-G, d4.
G: glucuronide; min: minute; cps: count per second
Bicalutamide is cleared almost exclusively by metabolism. In humans, the drug is excreted to similar extents in urine and feces [3]. Two polar metabolites found in urines were identified as the glucuronide (-G) conjugates of (S)bicalutamide and hydroxy(R)bicalutamide [3]. Urinary concentrations of (S)bicalutamide-G is maximal 24 hours after administration of a single dose and represent up to 76% of the urinary metabolite. However, this percentage decreases to 14%, 9 days after dosage [3]. Urinary levels of the hydroxy(R)bicalutamide-G follows an inverse variation, and its relative abundance increases from 14 to 61% of the urinary metabolites during the 9 days after treatment [3]. Similar trends were also observed in feces [3]. These observations indicate that direct glucuronidation is the main metabolic pathway for the rapidly cleared (S)bicalutamide, whereas hydroxylation followed by glucuronidation is a major metabolic pathway for the slowly cleared (R)bicalutamide [1, 3]. The liver is considered the primary organ for bicalutamide clearance in man [1, 3].
Glucuronidation is a major phase II drug-metabolizing reaction in humans [4]. The UDP-glucuronosyltransferase (UGT) enzymes catalyze this enzymatic process, which corresponds to the transfer of glucuronic acid from UDP-glucuronic acid (UDPGA) to hydrophobic molecules [4]. The resulting glucuronide conjugates have low activity, high solubility in water and are easily eliminated from the human body into bile or urine [4]. Human UGTs are classified into 4 families: UGT1, UGT2, UGT3 and UGT8. The most important drug conjugating UGTs belong to the UGT1 and UGT2 families [5]. While the UGT1 family is composed of only 1 subfamily, i.e. UGT1A, the 12 members of the UGT2 are further divided into UGT2A (3 enzymes) and UGT2B (9 enzymes) subfamilies [5–7]. The 9 active human UGT1As are produced from a single gene locus located on chromosome 2q37 [6, 8]. In humans, the liver, kidney and intestine are considered the main sites for drug glucuronidation [9, 10].
Although glucuronidation is the main conjugating reaction for bicalutamide in humans [1, 3], the UGT enzyme(s) involved in this reaction ha(s)ve never been identified. However, such information may be critical for the use of this anti-androgen in a clinical setting, because UGT gene polymorphisms usually cause inter-individual variations in drug metabolism, and so can drastically impact drug efficiency and toxicity [6, 11, 12]. Bicalutamide pharmacokinetics also sustains a large inter-individual variability which can reflect different phenotypes in bicalutamide-conjugating UGTs [1, 13, 14]. The primary objectives of the present study were to identify the human UGT(s) involved in (R) and (S)bicalutamide glucuronidation. Additional experiments were also performed to evaluate whether bicalutamide glucuronidation is restricted to the liver, and if this catabolic reaction is racemate-sensitive in in vitro assays. These observations will improve our understanding of the bicalutamide pharmacology. In the context of polypharmacotherapy of prostate cancer patients, an improved knowledge of bicalutamide metabolism will be helpful in preventing potential drug-drug interactions with drug used in combination and eventually sharing the same glucuronidating pathways.
MATERIALS AND METHODS
Materials
(R/S)Bicalutamide, propofol and 4-methylumbelliferone (4-MU) were from Sigma (St. Louis, MO). (R) and (S)bicalutamide, as well as the deuterated [2H4]-(R/S)bicalutamide were from Toronto Research Chemical (Downsview, ON, Canada). The internal standards [2H4]-(S)bicalutamide-G ((S)bicalutamide-G, d4) and 4-MU-G were obtained through in vitro enzymatic assays with [2H4]-(R/S)bicalutamide or 4-MU and human liver microsomes following the reported procedure [15]. All chemicals and reagents were of the highest grade commercially available. HPLC-grade reagents were from VWR Canada (Mississauga, Canada), and Laboratoire MAT (Québec, QC, Canada). UDP-glucuronic acid (UDPGA) and all aglycones were obtained from Sigma and ICN Pharmaceuticals, Inc. (Québec, QC, Canada). Commercially available microsomes from human liver (#452161; pool of microsomes from 19 donors) and intestine (#452210; pool of microsomes from jejunum and duodenum sections of 6 donors), and UGT bacculosomes (#456411, 456413; 456414; 456416; 456407; 456418; 456424; 456427; 456419) were from BD Gentest (Mississauga, ON, Canada). Human kidney microsomes (#S00676; pool of microsomes from 5 donors) were from Celsis-InVitro Technologies (Baltimore, MD). Human embryonic kidney 293 (HEK293) cells were obtained from the American Type Culture Collection (Rockville, MD) and were grown as reported [16]. HEK293 cells expressing human UGTs (UGT-HEK293) were engineered as described [16, 17]. Protein assay reagents were obtained from Bio-Rad Laboratories Inc. (Marnes-la-Coquette, France). Antibiotics and cell culture reagents were from Invitrogen (Burlington, ON, Canada).
Microsome isolation and glucuronidation assays
Microsomal protein extracts from UGT1A- and UGT2B-overexpressing HEK293 cells were prepared by differential centrifugation, and resuspended at a 5 μg/μL concentration. The UGT content in these preparations was previously evaluated through Western blot analyses [16–18].
All glucuronidation assays were performed in the presence of 10 μg microsomes or bacculosomes using the previously reported glucuronidation assay buffer [16], complemented with 1 mM UDPGA and 0.025 μg/ml alamethicin. Assays were ended by adding 100 μl of methanol with 0.02% Butylated Hydroxytoluene [16, 19].
The initial screening of human tissues was performed for 18 hours with 100 μM (R/S), (R) or (S)bicalutamide at 37°C in the presence human liver, kidney, and intestine microsomes (10 μg). Time course analyses were performed in the presence of 100 μM bicalutamide enantiomer solutions for duration varying from 15 minutes to 48 hours. Recombinant human UGTs were also screened with 100 μM (R/S), (R) or (S)bicalutamide through 2-hour long assays with both bacculosomes (UGT2B4, 2B7, 2B15, 2B17, 1A1, 1A3, 1A4, 1A6, 1A7, 1A8, 1A9 and 1A10) and UGT-HEK293 cell microsomes (UGT2B4, 2B7, 2B10, 2B11, 2B15, 2B17, 2B28, 1A1, 1A3, 1A4, 1A5, 1A6, 1A7, 1A8, 1A9 and 1A10) [16, 17].
Kinetic parameters were assessed in liver and kidney samples, as well as with bacculosome preparations of reactive UGTs, namely UGT2B7, UGT1A8 and UGT1A9. Assays were performed using substrate concentrations ranging from 1 to 1000 μM for 2 hours. The enzyme kinetic model was selected as recommended [18, 20], using the Sigma Plot 11.2 assisted by Enzyme Kinetics 1.3 programs (SSI, San Jose, CA). Inhibition constants (Ki) were assessed as above except that propofol and (S)bicalutamide were used at concentrations ranging from 1 to 250 μM and 5 to 500 μM, respectively. The Ki values were estimated using Dixon plots (Sigma Plot 11.2) as reported [21]. Values are expressed as mean ± S.D. of at least two independent experiments performed in triplicate.
Determination of glucuronide levels by liquid chromatography coupled to mass spectrometry: LC-MS/MS
Detection of bicalutamide glucuronides was performed by liquid chromatography (HPLC) coupled to a triple-quadrupole mass spectrometer (LC-MS/MS) (API 3200 Applied Biosystems, Concord, ON, Canada) operating with a turbo ion-spray source using multiple ions monitoring mode. The HPLC system consisted of the Agilent 1200 LC (Agilent Technology, Ville St-Laurent, QC, Canada), coupled to an ACE C18 HL column 100 X 4.6 mm, 3 μm from Canadian Life Science, (Peterborough, ON, Canada). The mobile phase was composed of binary solution, 0.001% formic acid (solvent A), and methanol with 0.001% formic acid (solvent B). Analytes were eluted at 0.9 ml/min flow rate with 70% B. Electro-spray was set to negative-ion mode with ionization, declustering and collision energy potential of −4200 V, −30 V and −30 V, respectively. The ion source temperature was set at 450°C. The following mass ion transitions (m/z) were used for detection: 605.0 → 411.1 for (R)- and (S) bicalutamide-G, and 609.0 → 415.0 for (S)bicalutamide-G, d4 (internal standard). Data are expressed as ratios of peak areas for (R) or (S)bicalutamide-G over (S)bicalutamide-G, d4 (Figure 1).
For propofol-G quantification, 4-MU-G (internal standard) was added to glucuronidation assays for subsequent analyses using the described procedure [22], with the following modifications: the analytical system consisted of a high-performance liquid chromatography module UFLC Prominence (Shimadzu Scientific Instrument Inc., Columbia, MD, USA) and an API 3000 mass spectrometer with a electrospray ion source (Applied Biosystems, Concord, Canada). Samples were injected on to a Gemini C18 100 X 4.6 mm, 3 μm column, (Phenomenex, Torrance, CA). The mobile phase consisted of 85% acetonitrile and 15% water-0.1% formic acid. The flow rate was 0.9 ml/min for a total run time of 3.5 minutes. Ionization was performed at 400°C with an ionization voltage of −4200V, a declustering potential of −10 V, and a collision energy of −30 V. The multiple reaction monitoring (MRM) detection was achieved on a negative mode to monitor ion pairs of propofol-G (m/z 353.1→177.4) and 4-MU-G (m/z 350.9→174.9). Results are expressed as ratio of the area under the curve (AUC) for propofol-G versus AUC for 4-MU-G.
RESULTS
Bicalutamide glucuronidation in human tissues
The liver, kidney and intestine are considered the primary site for drug glucuronidation in humans [9]. While previous studies established the liver as the primary organ for bicalutamide clearance in man [1, 3], how kidney and intestine are able to form glucuronide conjugates of both racemic forms of the drug has never been investigated. Enzymatic assays performed with microsomes from human liver, kidney and intestine showed that bicalutamide glucuronidation is performed mainly by liver and kidney extracts and to a lesser extent by intestinal proteins (Figure 2). Furthermore, the formation of both (R) and (S)bicalutamide-G exhibited a similar pattern of tissue distribution, and the use of either pure or racemic solutions did not influenced this pattern (Figure 2). Nevertheless, the conversion of (R)bicalutamide into its corresponding glucuronide derivatives was more efficient in the presence of the pure enantiomer than with the racemate (Figure 2A and C). (S)Bicalutamide-G was produced at similar levels in the presence of both forms (pure and racemic) (Figure 2B and D).
Figure 2. Glucuronide conjugation of bicalutamide enantiomers by microsomes from human liver, kidney and intestine.
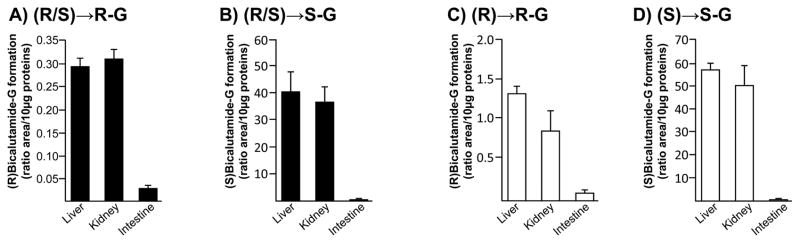
Human liver, kidney or intestine microsomes (10 μg) were incubated in the presence of 100 μM of the racemic (R/S) mixture of bicalutamide (R/S, A and B), or of the pure (R)- (R, C) or (S)-enantiomers (S, D), and UDPGA (1 mM) for 18 hours at 37°C. The formation of (R)bicalutamide-glucuronide (R-G, A and C) and (S)bicalutamide-glucuronide (S-G, B and D) was analyzed by LC-MS/MS. Data represent the mean ± S.D. of two independent experiments performed in triplicate.
G: glucuronide.
Subsequent time-course (Figure 3) and dose-response (Figure 4) experiments were performed to fully establish the enzymatic parameters of bicalutamide glucuronidation by human liver and kidney extracts. Time course experiments performed for up to 48 hours showed that all glucuronidation reactions were linear for 4 hours independently of the tissue (liver or kidney) or substrate (racemic mix or pure enantiomer solutions) (Figure 3). Therefore, subsequent glucuronidation assays were performed for 2 hours. Dose response analyses revealed that liver extracts reached a maximal production of (R)bicalutamide-G in the presence of 300 μM of both (R/S) and (R)bicalutamide and remained stable in the presence of higher drug concentrations (Figure 4A and C). In these assays, the formation of (S)bicalutamide-G was optimal in the presence of 125 μM ((S)bicalutamide, Figure 4D) or 100 μM ((R/S)bicalutamide, Figure 4B) of substrate and again remained stable in the presence of higher concentrations. When incubated in the presence of human kidney microsomes, both (R/S) and (R)bicalutamide provided the highest production levels of (R)-glucuronide at 300 μM (Figure 4E and G). The conversion of (R/S)bicalutamide into (R)bicalutamide-G remained stable when the substrate was used at concentrations varying from 300 to 1 000 μM (Figure 4E), while in the presence of the same amount of (R)bicalutamide, glucuronidation rates tended to decrease (Figure 4G). As for liver extracts, the production of (S)bicalutamide-G reached a maximal level at lower substrate concentrations, i.e. 175 and 200 μM for (S) and (R/S)bicalutamide, respectively, when assayed with microsomal proteins from kidney (Figure 4F and H).
Figure 3. Time course analyses of the conversion of bicalutamide enantiomers into their respective glucuronide conjugates when incubated in the presence of microsomes from human liver and kidney.
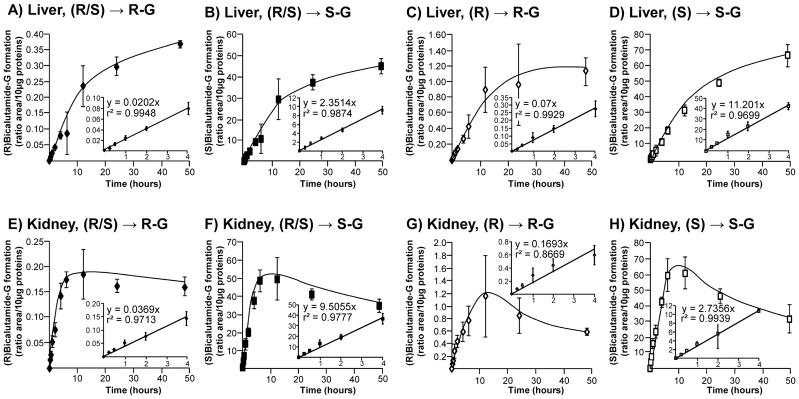
Human liver (A–D) and kidney (E–H) microsomes (10 μg) were incubated in the presence of 100 μM of the racemic (R/S) mixture of bicalutamide (R/S, A, B, E and F), or of the pure (R) (R, C and G) or (S) enantiomers (S, D and H) and UDPGA (1 mM) for increasing durations (15 minutes to 48 hours) at 37°C. The formation of (R)bicalutamide-glucuronide (R-G, A, C, E and G) and (S)bicalutamide-glucuronide (S-G, B, D, F and H) was analyzed by LC-MS/MS. Data represent the mean ± S.D. of two independent experiments performed in triplicate.
G: glucuronide.
Figure 4. Dose-response and kinetic analyses of bicalutamide enantiomers conversion into their respective glucuronide conjugates by microsomes from human liver and kidney.

Human liver (A–D) and kidney (E–H) microsomes (10 μg) were incubated in the presence of increasing concentrations (1 to 1000 μM) of the racemic (R/S) mixture of bicalutamide (R/S, A, B, E and F), or of the pure (R) (R, C and G) or (S) enantiomers (S, D and H) and UDPGA (1mM) for 2 hours at 37°C. The formation of (R)bicalutamide-glucuronide (R-G, A, C, E and G) and (S)bicalutamide-glucuronide (S-G, B, D, F and H) was analyzed by LC-MS/MS. For each panel, large graphs represent the rate of product formation (Y-axis) versus substrate concentration (X-axis) of 2 experiments performed in triplicates (Experiments 1: ·········· and 2: - - - - -) and their mean ( —— ); while small graphs correspond to the mean Eadie-Hofstee plots (rate of product formation (ratio area/min/mg proteins) versus rate of product formation/substrate concentration (ratio area/min/mg proteins/mM)).
G: glucuronide.
Results from dose-response experiments were also used to determine kinetic parameters of bicalutamide glucuronidation in liver and kidney (Figure 4 and Table 1). For this purpose, Eadie-Hoftee plots (Figure 4) were drawn to identify the kinetic model providing the best estimation [18, 20] for the kinetic constants of affinity (Km), velocity (VmaxApp.) and intrinsic clearance (CLint., here defined as the ratio VmaxApp./Km) (Table 1). While most profiles corresponded to Michaelis-Menten kinetics, (R)bicalutamide-G produced by liver enzymes fitted an intermediate profile between sigmoidal and substrate inhibition, with Hill coefficient values of 1.35 ± 0.15 and 1.81 ± 0.23 for conjugates formed from the racemic mixtures and the pure (R) solution, respectively (Figure 4A and C and Table 1). With renal UGTs, identification of the accurate kinetics was even more complex since Eadie-Hoftee analyses suggested a Michaelis-Menten profile for (R)bicalutamide-G formed from the (R/S) mix (Figure 4E) and a sigmoidal+substrate kinetics for those produced with the pure (R) enantiomer (Figure 4G). In such a context kinetic parameters of both substrates were estimated based on Michaelis-Menten profile (Table 1). Beyond these differences, both liver and kidney extracts led to similar observations (Table 1): their enzymatic affinity was higher for the (S)-enantiomer than for the (R) one with Km values in the 10–20 μM and 50–100 μM ranges, respectively. Km values obtained with the pure (R) and (S) enantiomers were respectively lower and higher than those obtained with the (R/S) mix as substrate (Table 1). Interestingly, the VmaxApp. value of (S)bicalutamide-G formation was not influenced by the type of substrate (pure or mix), while (R)bicalutamide-G was produced at higher velocity with the (R)-enantiomer when compared to the racemic mix (Table 1). Nevertheless, the VmaxApp./Km values for glucuronidation of the 2 enantiomers were higher when both kidney and liver microsomes were incubated with the pure substrate than with the racemic mixture (Table 1).
Table 1.
Kinetic parameters for the glucuronidation of bicalutamide by human tissues.
| Substrate | Formation of (R)bicalutamide-glucuronide | Formation of (S)bicalutamide-glucuronide | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Km | VmaxApp. | n1 | VmaxApp./Km | Km | VmaxApp. | n1 | CLint., VmaxApp./Km | ||
| μM | Ratio area/min/mg | μl/min/mg | μM | Ratio area/min/mg | μl/min/mg | ||||
| Liver | (R/S)bicalutamide | 62.1±5.6 | 0.032±0.003 | 1.35±0.15 | 0.0005±0.00006 | 18.3±2.0 | 3.35±0.45 | / | 0.18±0.03 |
| (R)bicalutamide | 103.5±7.2 | 0.20±0.04 | 1.81±0.23 | 0.0019±0.0005 | |||||
| (S)bicalutamide | 10.6±1.7 | 3.80±0.88 | / | 0.35±0.06 | |||||
| Kidney | (R/S)bicalutamide | 51.4±4.9 | 0.026±0.003 | / | 0.0005±0.00005 | 23.5±1.9 | 5.64±0.57 | / | 0.24±0.01 |
| (R)bicalutamide | 108.9±16.2 | 0.27±0.02 | / | 0.002±0.0003 | |||||
| (S)bicalutamide | 11.6±0.9 | 6.15±0.53 | / | 0.53±0.02 | |||||
Results are expressed as mean ± S.D. of two independent experiments performed in triplicate.
n: Hill coefficient
These observations demonstrate that bicalutamide glucuronidation can occur in the human liver and kidney, and that both tissues exhibit a greater affinity for (S)bicalutamide glucuronidation.
Bicalutamide glucuronidation by recombinant human UGT enzymes
A screening assay realized with both commercial bacculosomes and home-made microsomes from UGT-HEK293 cells demonstrated that UGT1A8 and UGT1A9 exert a predominant role in the formation of bilirubin glucuronide conjugates (Figure 5). However, when microsomes and bacculosomes were assayed with the (R/S) solution, (R)bicalutamide-G was found to be more abundantly produced by UGT2B7 than by UGT1A8 and UGT1A9 (Figure 5A). When the racemic mixture was replaced by the pure (R)-enantiomer, UGT1A9 appeared as the predominant isoform for (R)bicalutamide glucuronidation (Figure 5C). Such discrepancy was not observed in the case of (S)-glucuronide production, which appeared almost exclusively formed by UGT1A8 and UGT1A9 (Figure 5B and D). However, thanks to the sensitivity of the LC-MS/MS method, we were able to detect the production of (S)bicalutamide-G with the UGT2B7 enzyme, but at a much lower rate than with UGT1A9 (Figure 5B and D). Similarly, both (R)- and (S)-G were also produced in the presence of bacculosomes and/or microsomes containing the UGT1A1 (Figure 5A, C and D) and UGT1A3 (Figure 5A–D) proteins. It is however, remarkable that the UGT1A1- and UGT1A3-dependent formation of these conjugates was very low when compared to the ability of UGT1A8, UGT1A9 and UGT2B7 to convert bicalutamide into glucuronide derivatives. As reported [16], UGT1A1, UGT1A3, UGT1A8 and UGT1A9 bacculosome preparations contains a comparable amount of their respective UGT protein, thus indicating that the differences in (R)- and (S)bicalutamide-G production reflect the differential activity of these isoforms.
Figure 5. Glucuronide conjugation of bicalutamide enantiomers by recombinant human UGT enzymes.

Microsomes from UGT-HEK293 cells or commercially available bacculosomes (10 μg) were incubated in the presence of 100 μM of the racemic mixture of (R/S)bicalutamide (R/S, A and B), or of the pure (R) (R, C) or (S) enantiomers (S, D) and UDPGA (1mM) for 2 hours at 37°C. The formation of (R)bicalutamide-glucuronide (R-G, A and C) and (S)bicalutamide-glucuronide (S-G, B and D) was analyzed by LC-MS/MS. Data represent the mean ± S.D. of two independent experiments performed in triplicate.
G: glucuronide.
As for liver and kidney, kinetic parameters for bicalutamide glucuronidation by recombinant UGTs (commercial bacculosome preparations) were determined through dose-response experiments (Figure 6 and Table 2). Pure sigmoidal kinetics are clearly suggested by the Eadie-Hoftee analyses for UGT1A8-catalyzed reactions (Figure 6A–D and Table 2). All UGT2B7 reactions, as well as the UGT1A9-dependent conversion of (R)bicalutamide into its glucuronide derivative, fitted a substrate inhibition kinetic model (Figure 6G, I and J), while (S)bicalutamide glucuronidation reactions catalyzed by UGT1A9 corresponded to a Michaelis-Menten profile (Figure 6F and H). As shown in table 2, Ki values for UGT2B7 were lower than for UGT1A9 (597.38 ± 143.94 μM), suggesting that (R)bicalutamide self-inhibits its UGT1A9-dependent glucuronidation only at high concentrations. When assayed with pure solutions, the recombinant UGT1A9 enzyme displayed lower Km values for the (S)-enantiomer than for its (R) counterpart (Table 2). However, in the presence of both enantiomers, as found in the racemic mixture, this enzyme exhibited a similar affinity for the production of (S)bicalutamide-G (8.8 ± 2.3 μM) and (R)-glucuronide (7.9 ± 0.4 μM). Similar observations also apply to the recombinant UGT1A8 isoform (Table 2). By contrast, the Km value for (R)bicalutamide-G production by UGT2B7 was higher when the enzyme was incubated with the racemic mix (268.1 ± 175.4 μM) than in the presence of the pure solution (133.3 ± 63.6 μM) (Table 2). The VmaxApp. values of (R)bicalutamide-G formation by UGT1A9 were higher in the presence of the pure (R)-enantiomer than with the (R/S) mixture, while the UGT1A9-dependent formation of (S)bicalutamide-G was not affected by the type of substrate (Table 2). The opposite was observed for UGT1A8 and UGT2B7 since these enzymes showed similar VmaxApp. values for (R)bicalutamide-G production in the presence of pure or racemic substrates, but UGTA8 exhibited an higher (S)bicalutamide-G production in the presence of the pure (S)-enantiomer than with the (R/S) mixture (Table 2). Consequently, UGT1A9 exhibited higher VmaxApp. values than UGT1A8 for the formation of (S)bicalutamide-G from the pure (S) and (R/S) mixture (Table 2). Another interesting difference between these UGTs relates to their respective CLint. values for (S)bicalutamide-G formation (Table 2). Indeed, UGT1A9 displayed a 2-fold higher value in the presence of the pure solution than with the racemic mixture, while a 4-fold opposite relation was detected in the presence of the UGT1A8 bacculosomes. Nevertheless, UGT1A9 was superior to UGT1A8 and UGT2B7 in terms of intrinsic clearance for all glucuronidation reactions, as performed in the current study.
Figure 6. Dose-response and kinetic analyses of bicalutamide enantiomers conversion into their respective glucuronide conjugates by bacculosomes expressing UGT2B7, UGT1A8 and UGT1A9.

UGT1A8 (A–D), UGT1A9 (E–H) and UGT2B7 (I and J) bacculosomes (10 μg) were incubated in the presence of increasing concentrations (1 to 1000 μM) of the racemic (R/S) mixture of bicalutamide (R/S, A, B, E, F and J), or of the pure (R) (R, C, G and I) or (S) enantiomers (S, D and H) and UDPGA (1 mM) for 2 hours at 37°C. The formation of (R)bicalutamide-glucuronide (R-G, A, C, E and G) and (S)bicalutamide-glucuronide (S-G, B, D, F and H) was analyzed by LC-MS/MS. For each panel, large graphs represent the rate of product formation (Y-axis) versus substrate concentration (X-axis) of 2 experiments performed in triplicates (Experiments 1: ·········· and 2: - - - - -) and their mean ( —— ); while small graphs correspond to the mean Eadie-Hofstee plots (rate of product formation (ratio area/min/mg proteins) versus rate of product formation/substrate concentration (ratio area/min/mg proteins/mM).
G: glucuronide.
Table 2.
Kinetic parameters for the glucuronidation of bicalutamide by human UGT enzymes.
| Substrate | Formation of (R)bicalutamide-glucuronide | Formation of (S)bicalutamide-glucuronide | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Km | VmaxApp. | n1 or Ki2 | VmaxApp./Km | Km | VmaxApp. | n1 or Ki2 | CLint., VmaxApp./Km | ||
| μM | Ratio area/min/mg | μM | μl/min/mg | μM | Ratio area/min/mg | μM | μl/min/mg | ||
| UGT1A8 | (R/S)bicalutamide | 93.1±9.5 | 0.007±0.002 | 1.64±0.29 | 0.00008±0.00002 | 75.4±2.4 | 1.36±0.08 | 2.15±0.14 | 0.18±0.01 |
| (R)bicalutamide | 134.1±8.1 | 0.010±0.001 | 1.87±0.20 | 0.00007±0.00001 | |||||
| (S)bicalutamide | 80.6±2.8 | 3.30±0.26 | 2.02±0.14 | 0.04±0.01 | |||||
| UGT1A9 | (R/S)bicalutamide | 8.8±2.3 | 0.007±0.002 | 1.21±0.38 | 0.0008±0.0001 | 7.9±0.4 | 3.97±0.21 | / | 0.49±0.01 |
| (R)bicalutamide | 173.3±35.8 | 0.14±0.02 | 597.38±143.94 | 0.0008±0.0002 | |||||
| (S)bicalutamide | 4.7±0.3 | 4.3±0.16 | / | 0.91±0.05 | |||||
| UGT2B7 | (R/S)bicalutamide | 268.1±175.4 | 0.011±0.001 | 105.91±67.56 | 0.00004±0.00003 | ||||
| (R)bicalutamide | 133.3±63.6 | 0.019±0.003 | 94.31±43.41 | 0.00014±0.0001 | |||||
Results are expressed as mean ± S.D. of two independent experiments performed in triplicate.
n: Hill coefficient;
Ki: Constant of inhibition
Bicalutamide inhibits propofol glucuronidation
To ensure the predominant contribution of the UGT1A9 enzymes for hepatic and renal (S)bicalutamide glucuronidation, human liver and kidney microsomes, as well as UGT1A9 bacculosomes, were incubated in the presence of increasing concentrations of (S)bicalutamide and of the specific UGT1A9 probe substrate, propofol [23] (Figure 7). A marked decrease in the propofol-G formation was shown to be dose-dependent with all enzymatic preparations. The apparent Ki values estimated from Dixon plots were all in the same range: 21.4 ± 1.4 and 13.4 ± 1.3 μM, for liver and kidney microsomes, and 11.9 ± 1.0 μM for UGT1A9 bacculosomes (Figure 7).
Figure 7. (S)Bicalutamide inhibits propofol-glucuronide production by human liver (A) and kidney (B) microsomes and by the UGT1A9 bacculosomes (C).
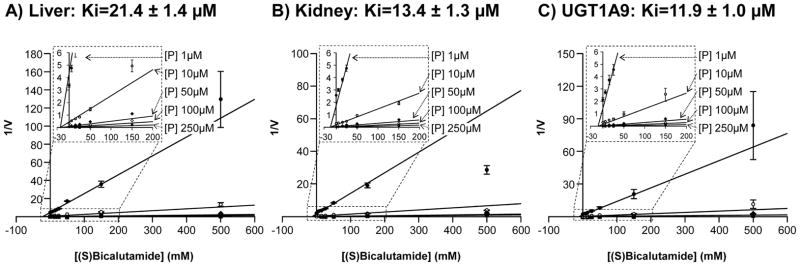
Human liver (A) and kidney (B) microsomes and UGT1A9 (C) bacculosomes were incubated with UDPGA (1 mM), in the presence of increasing concentrations of propofol (P, 1, 10, 50 and 100 μM) and pure (S)bicalutamide (5, 15, 25, 50, 150 and 500μM) for 2 hours at 37°C. Propofol-G formation was analyzed by LC-MS/MS, and Dixon plots analyses were performed to calculate the apparent Ki values. Data represent the mean ± S.D. of two independent experiments performed in triplicate.
G: glucuronide; P: propofol, V: propofol-G formation expressed as the ratio of the area under the curve (AUC) for propofol-G versus AUC of 4-methylumbelliferone-G/min/mg of proteins); Ki: constant of inhibition.
DISCUSSION
This study demonstrates the ability of human liver and kidney to form glucuronide conjugates from bicalutamide enantiomers, and illustrates the predominant role played by the human UGT2B7, UGT1A8 and UGT1A9 isoforms in this catabolic process.
This investigation was enabled by the development of a novel protocol for the simultaneous analysis of the (R) and (S)bicalutamide-glucuronide derivatives. While efficient, sensitive and enantiomer-specific, our approach was limited by the absence of commercially available sources for (R) and (S)bicalutamide-G that could have been used as analytical standards for fully quantitative LC-MS/MS determination. Nevertheless, the established quantification method provides solid estimation of glucuronide production levels, based on LC peak areas. For each conjugate these areas can be compared in different samples, while potential differences in the ionization properties of the 2 enantiomers limit our ability to adequately compare (R) and (S)bicalutamide glucuronidation levels, even within a single sample. Nonetheless, it is remarkable that the huge differences observed between (S)- and (R)bicalutamide-G formation in the presence of liver and kidney extracts is consistent with the previous determination of (S)bicalutamide as a directly glucuronidated molecule in humans [1, 3]. In clinics, the (R)bicalutamide metabolism is slower and requires hydroxylation before glucuronidation, which is in accordance with the low activity level detected in the present study [1, 3]. Similarly, the higher affinity of liver and kidney extracts for (S)bicalutamide glucuronidation when compared to the (R)-G formation is consistent with the stereoselectivity of drug glucuronidation [23–25], and suggests that in the presence of the racemic drug, these tissues will preferentially produce (S)bicalutamide-glucuronide conjugates.
Our data reveal that the liver and kidneys exhibit similar conjugating activities, affinity and even intrinsic clearances for (R) and (S)bicalutamide glucuronidation, thus suggesting that both organs contribute to bicalutamide clearance in vivo. These results also suggest that kidneys may eventually contribute to the formation of urinary glucuronide metabolites of bicalutamide. On the other hand, when compared with results from kidney and liver, microsomes from human intestine sections (jejunum and duodenum) were only feebly reactive with both (R) and (S) molecules. Because, elevated intestinal glucuronidation is closely associated with low oral drug bioavailability [26], this observation is in agreement with the elevated bioavailability of bicalutamide as observed in healthy volunteers receiving oral doses of the racemate drug [1, 3].
The initial screening of human drug-conjugating UGTs for reactivity with pure ((R) and (S)) and racemic (R/S) bicalutamide solutions revealed the elevated capacities of UGT1A8, UGT1A9 and even UGT2B7 to convert these molecules into their respective glucuronide derivatives. However, various observations point out UGT1A9 as a dominant player in these catabolic reactions: first, the high reactivity of UGT2B7 was restricted to the less glucuronidated (R)bicalutamide enantiomer. Second, when compared to UGT1A8, UGT1A9 has higher affinity (Km), velocity (VmaxApp.) and intrinsic clearance (VmaxApp./Km) values for the formation of (S)bicalutamide-G from pure or racemic substrates. Third, (S)bicalutamide efficiently inhibited the hepatic and renal glucuronidation of propofol, a UGT1A9 specific substrate [23]. Last, the pattern of bicalutamide glucuronidation in human liver, kidney and intestine extracts indicate a minor contribution for UGT1A8 and UGT2B7 in (R) and (S)bicalutamide glucuronidation in vivo. Indeed, in contrast to the abundant UGT1A9 and UGT2B7 enzymes, UGT1A8 is not–or barely [27]–expressed in liver and found at low level in the kidney when compared to UGT1A9 [28], and thus can minimally contribute to the elevated formation of bicalutamide-G observed with these organs [7, 9, 29]. Similarly, the low glucuronidation activity detected with microsomal proteins from the intestine (jejunum and duodenum) is consistent with the previous demonstration that UGT1A9 is 2-time less abundant than UGT1A8 [28, 30]. In these tissues, UGT1A8 and UGT2B7 proteins are actually abundant [7, 9, 28–30]. Thus, from the 3 reactive enzymes, only UGT1A9 exhibits a pattern of tissue expression consistent with the tissue distribution of bicalutamide glucuronidation.
UGT1A9 is a polymorphic enzyme, and various single nucleotide polymorphisms identified within the human gene cause alteration of the UGT1A9 protein expression and/or activity [22, 28, 31, 32]. Recent investigations established the large inter-individual variability of hepatic UGT1A9 protein contents, and demonstrated the strong influence of the UGT1A9 phenotype on drug biodisposition as exemplified with the immunosuppressive drug mycophenolic acid [20, 31–33]. It is therefore likely that UGT1A9 polymorphisms also interfere with bicalutamide glucuronidation. Such interference could take part to the great inter-individual variability of bicalutamide pharmacokinetics in clinic [1]. Beyond expression and activity, UGT1A9 polymorphisms also affect the stereoselectivity of this enzyme for racemic drug glucuronidation, as observed with (R/S) racemates of flurbiprofen, a non-steroidal anti-inflammatory drug [24]. Again, it can be envisioned that UGT1A9 polymorphisms may also influence the enzyme ability to differentiate bicalutamide enantiomers. Therefore, additional investigations will have to establish how UGT1A9 polymorphisms influence not only the rate of (S)bicalutamide elimination, but also the differential metabolism of each enantiomer.
From the racemic prescribed mixture, (S)bicalutamide is the primary target for direct glucuronidation, while (R)bicalutamide is the active anti-androgenic component [1]. The (R) enantiomer requires hydroxylation prior to glucuronidation [1, 3], and the absence of available sources for hydroxy(R)bicalutamide precluded any examination of its tissue and UGT-specific glucuronidation in the context of the present study. Thus, future investigations are required to fully evaluate how this enzyme influences bicalutamide’s pharmacological activity. Such investigations are also required to determine whether UGT enzymes unreactive with bicalutamide can glucuronidate its phase I hydroxyl(R)bicalutamide derivative. Nevertheless, (S)bicalutamide may now be considered as a potential inhibitor for the glucuronide conjugation of other UGT1A9 drug substrates [23], as observed in the present study with propofol. The possible metabolic drug-drug interactions resulting from such an inhibition are of major clinical relevance regarding prostate cancer treatment. Indeed, two recent reports identified: 1) UGT1A9 as the main UGT enzyme for hepatic glucuronidation of the anti-angiogenic drug, sorafenib [34]; and 2) the combination of sorafenib plus bicalutamide as a promising therapeutic strategy for castration-resistant prostate cancer patients [35]. Our results therefore highlight a potential risk for drug-drug interactions when combining sorafenib and bicalutamide, since these 2 UGT1A9 substrates can compete with each other for glucuronidation. In the context of a worldwide clinical implementation of the sorafenib plus bicalutamide combination, future investigations will have to ensure that each compound minimally affects its partner’s pharmacokinetics in order to evaluate the risk of drug interactions, and eventually recommend dosage adjustments.
In conclusion, the present study provides the first comprehensive analysis of in vitro bicalutamide glucuronidation by human tissues and UGTs. We identify UGT1A9 as a major contributor for (R) and (S) glucuronidation in the liver and kidney, and our results highlight the need for future studies focusing on UGT1A9 polymorphisms as possible interferences for bicalutamide metabolism. Finally, our observations also point out the need for a thorough evaluation of potential metabolic interactions when combining sorafenib and bicalutamide, 2 drugs metabolized by UGT1A9.
Acknowledgments
This study was supported by grants from the Canadian Institute of Health Research (CIHR), the Canadian Foundation for Innovation (CFI) and the Natural Sciences and Engineering Research Council of Canada (NSERC). O. Barbier is holder of a salary grant from CIHR (New investigator award #MSH95330). S. Caron is supported by a scholarship from the Fonds pour l’Enseignement et la Recherche (FER) from the Faculty of Pharmacy, Laval University (Québec).
References
- 1.Cockshott ID. Bicalutamide: clinical pharmacokinetics and metabolism. Clin Pharmacokinet. 2004;43:855–78. doi: 10.2165/00003088-200443130-00003. [DOI] [PubMed] [Google Scholar]
- 2.Mukherjee A, Kirkovsky L, Yao XT, Yates RC, Miller DD, Dalton JT. Enantioselective binding of Casodex to the androgen receptor. Xenobiotica; the fate of foreign compounds in biological systems. 1996;26:117–22. doi: 10.3109/00498259609046693. [DOI] [PubMed] [Google Scholar]
- 3.McKillop D, Boyle GW, Cockshott ID, Jones DC, Phillips PJ, Yates RA. Metabolism and enantioselective pharmacokinetics of Casodex in man. Xenobiotica; the fate of foreign compounds in biological systems. 1993;23:1241–53. doi: 10.3109/00498259309059435. [DOI] [PubMed] [Google Scholar]
- 4.Dutton GJ. Glucuronidation of drugs and other compounds. Boca Raton, FL: CRC Press; 1980. [Google Scholar]
- 5.Mackenzie PI, Walter Bock K, Burchell B, Guillemette C, Ikushiro S, Iyanagi T, et al. Nomenclature update for the mammalian UDP glycosyltransferase (UGT) gene superfamily. Pharmacogenet Genomics. 2005;15:677–85. doi: 10.1097/01.fpc.0000173483.13689.56. [DOI] [PubMed] [Google Scholar]
- 6.Guillemette C, Lévesque É, Harvey M, Bellemare J, Menard V. UGT genomic diversity: beyond gene duplication. Drug metabolism reviews. 2010;42:24–44. doi: 10.3109/03602530903210682. [DOI] [PubMed] [Google Scholar]
- 7.Knights KM, Miners JO. Renal UDP-glucuronosyltransferases and the glucuronidation of xenobiotics and endogenous mediators. Drug metabolism reviews. 2010;42:63–73. doi: 10.3109/03602530903208561. [DOI] [PubMed] [Google Scholar]
- 8.Gong QH, Cho JW, Huang T, Potter C, Gholami N, Basu NK, et al. Thirteen UDPglucuronosyltransferase genes are encoded at the human UGT1 gene complex locus. Pharmacogenetics. 2001;11:357–68. doi: 10.1097/00008571-200106000-00011. [DOI] [PubMed] [Google Scholar]
- 9.Mackenzie PI, Hu DG, Gardner-Stephen DA. The regulation of UDP-glucuronosyltransferase genes by tissue-specific and ligand-activated transcription factors. Drug metabolism reviews. 2010;42:99–109. doi: 10.3109/03602530903209544. [DOI] [PubMed] [Google Scholar]
- 10.Tukey RH, Strassburg CP. Human UDP-glucuronosyltransferases: metabolism, expression, and disease. Annual review of pharmacology and toxicology. 2000;40:581–616. doi: 10.1146/annurev.pharmtox.40.1.581. [DOI] [PubMed] [Google Scholar]
- 11.Guillemette C. Pharmacogenomics of human UDP-glucuronosyltransferase enzymes. Pharmacogenomics J. 2003;3:136–58. doi: 10.1038/sj.tpj.6500171. [DOI] [PubMed] [Google Scholar]
- 12.Nagar S, Remmel RP. Uridine diphosphoglucuronosyltransferase pharmacogenetics and cancer. Oncogene. 2006;25:1659–72. doi: 10.1038/sj.onc.1209375. [DOI] [PubMed] [Google Scholar]
- 13.Cockshott ID, Cooper KJ, Sweetmore DS, Blacklock NJ, Denis L. The pharmacokinetics of Casodex in prostate cancer patients after single and during multiple dosing. European urology. 1990;18 (Suppl 3):10–7. doi: 10.1159/000463972. [DOI] [PubMed] [Google Scholar]
- 14.Tyrrell CJ, Denis L, Newling D, Soloway M, Channer K, Cockshott ID. Casodex 10–200 mg daily, used as monotherapy for the treatment of patients with advanced prostate cancer. An overview of the efficacy, tolerability and pharmacokinetics from three phase II dose-ranging studies. Casodex Study Group. European urology. 1998;33:39–53. doi: 10.1159/000019526. [DOI] [PubMed] [Google Scholar]
- 15.Caron P, Trottier J, Verreault M, Bélanger J, Kaeding J, Barbier O. Enzymatic production of bile acid glucuronides used as analytical standards for liquid chromatography-mass spectrometry analyses. Mol Pharm. 2006;3:293–302. doi: 10.1021/mp060021l. [DOI] [PubMed] [Google Scholar]
- 16.Trottier J, Verreault M, Grepper S, Monté D, Bélanger J, Kaeding J, et al. Human UDP-glucuronosyltransferase (UGT)1A3 enzyme conjugates chenodeoxycholic acid in the liver. Hepatology. 2006;44:1158–70. doi: 10.1002/hep.21362. [DOI] [PubMed] [Google Scholar]
- 17.Caillier B, Lépine J, Tojcic J, Ménard V, Pérusse L, Bélanger A, et al. A pharmacogenomics study of the human estrogen glucuronosyltransferase UGT1A3. Pharmacogenet Genomics. 2007;17:481–95. doi: 10.1097/FPC.0b013e32806d87a4. [DOI] [PubMed] [Google Scholar]
- 18.Benoit-Biancamano MO, Adam JP, Bernard O, Court MH, Leblanc MH, Caron P, et al. A pharmacogenetics study of the human glucuronosyltransferase UGT1A4. Pharmacogenet Genomics. 2009;19:945–54. doi: 10.1097/FPC.0b013e3283331637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trottier J, El Husseini D, Perreault M, Paquet S, Caron P, Bourassa S, et al. The human UGT1A3 enzyme conjugates norursodeoxycholic acid into a C23-ester glucuronide in the liver. The Journal of biological chemistry. 2010;285:1113–21. doi: 10.1074/jbc.M109.073908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miners JO, Mackenzie PI, Knights KM. The prediction of drug-glucuronidation parameters in humans: UDP-glucuronosyltransferase enzyme-selective substrate and inhibitor probes for reaction phenotyping and in vitro-in vivo extrapolation of drug clearance and drug-drug interaction potential. Drug metabolism reviews. 2010;42:196–208. doi: 10.3109/03602530903210716. [DOI] [PubMed] [Google Scholar]
- 21.Bélanger AS, Caron P, Harvey M, Zimmerman PA, Mehlotra RK, Guillemette C. Glucuronidation of the antiretroviral drug efavirenz by UGT2B7 and an in vitro investigation of drug-drug interaction with zidovudine. Drug metabolism and disposition: the biological fate of chemicals. 2009;37:1793–6. doi: 10.1124/dmd.109.027706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Girard H, Court MH, Bernard O, Fortier LC, Villeneuve L, Hao Q, et al. Identification of common polymorphisms in the promoter of the UGT1A9 gene: evidence that UGT1A9 protein and activity levels are strongly genetically controlled in the liver. Pharmacogenetics. 2004;14:501–15. doi: 10.1097/01.fpc.0000114754.08559.27. [DOI] [PubMed] [Google Scholar]
- 23.Court MH. Isoform-selective probe substrates for in vitro studies of human UDP- glucuronosyltransferases. Methods in enzymology. 2005;400:104–16. doi: 10.1016/S0076-6879(05)00007-8. [DOI] [PubMed] [Google Scholar]
- 24.Wang H, Yuan L, Zeng S. Characterizing the effect of UDP-glucuronosyltransferase (UGT) 2B7 and UGT1A9 genetic polymorphisms on enantioselective glucuronidation of flurbiprofen. Biochemical pharmacology. 2011;82:1757–63. doi: 10.1016/j.bcp.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 25.Court MH, Duan SX, Guillemette C, Journault K, Krishnaswamy S, Von Moltke LL, et al. Stereoselective conjugation of oxazepam by human UDP-glucuronosyltransferases (UGTs): S-oxazepam is glucuronidated by UGT2B15, while R-oxazepam is glucuronidated by UGT2B7 and UGT1A9. Drug metabolism and disposition: the biological fate of chemicals. 2002;30:1257–65. doi: 10.1124/dmd.30.11.1257. [DOI] [PubMed] [Google Scholar]
- 26.Furukawa T, Yamano K, Naritomi Y, Tanaka K, Terashita S, Teramura T. Method for predicting human intestinal first-pass metabolism of UGT substrate compounds. Xenobiotica; the fate of foreign compounds in biological systems. 2012;42:980–8. doi: 10.3109/00498254.2012.680620. [DOI] [PubMed] [Google Scholar]
- 27.Izukawa T, Nakajima M, Fujiwara R, Yamanaka H, Fukami T, Takamiya M, et al. Quantitative analysis of UDP-glucuronosyltransferase (UGT) 1A and UGT2B expression levels in human livers. Drug metabolism and disposition: the biological fate of chemicals. 2009;37:1759–68. doi: 10.1124/dmd.109.027227. [DOI] [PubMed] [Google Scholar]
- 28.Harbourt DE, Fallon JK, Ito S, Baba T, Ritter JK, Glish GL, et al. Quantification of human uridine-diphosphate glucuronosyl transferase 1A isoforms in liver, intestine, and kidney using nanobore liquid chromatography-tandem mass spectrometry. Analytical chemistry. 2012;84:98–105. doi: 10.1021/ac201704a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oda S, Nakajima M, Hatakeyama M, Fukami T, Yokoi T. Preparation of a specific monoclonal antibody against human UDP-glucuronosyltransferase (UGT) 1A9 and evaluation of UGT1A9 protein levels in human tissues. Drug metabolism and disposition: the biological fate of chemicals. 2012;40:1620–7. doi: 10.1124/dmd.112.045625. [DOI] [PubMed] [Google Scholar]
- 30.Ohno S, Nakajin S. Determination of mRNA expression of human UDP-glucuronosyltransferases and application for localization in various human tissues by real-time reverse transcriptase-polymerase chain reaction. Drug metabolism and disposition: the biological fate of chemicals. 2009;37:32–40. doi: 10.1124/dmd.108.023598. [DOI] [PubMed] [Google Scholar]
- 31.Kuypers DR, Naesens M, Vermeire S, Vanrenterghem Y. The impact of uridine diphosphate-glucuronosyltransferase 1A9 (UGT1A9) gene promoter region single-nucleotide polymorphisms T-275A and C-2152T on early mycophenolic acid dose-interval exposure in de novo renal allograft recipients. Clinical pharmacology and therapeutics. 2005;78:351–61. doi: 10.1016/j.clpt.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 32.Lévesque É, Delage R, Benoit-Biancamano MO, Caron P, Bernard O, Couture F, et al. The impact of UGT1A8, UGT1A9, and UGT2B7 genetic polymorphisms on the pharmacokinetic profile of mycophenolic acid after a single oral dose in healthy volunteers. Clinical pharmacology and therapeutics. 2007;81:392–400. doi: 10.1038/sj.clpt.6100073. [DOI] [PubMed] [Google Scholar]
- 33.Bernard O, Guillemette C. The main role of UGT1A9 in the hepatic metabolism of mycophenolic acid and the effects of naturally occurring variants. Drug metabolism and disposition: the biological fate of chemicals. 2004;32:775–8. doi: 10.1124/dmd.32.8.775. [DOI] [PubMed] [Google Scholar]
- 34.Peer CJ, Sissung TM, Kim A, Jain L, Woo S, Gardner ER, et al. Sorafenib is an inhibitor of UGT1A1 but is metabolized by UGT1A9: implications of genetic variants on pharmacokinetics and hyperbilirubinemia. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:2099–107. doi: 10.1158/1078-0432.CCR-11-2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beardsley EK, Hotte SJ, North S, Ellard SL, Winquist E, Kollmannsberger C, et al. A phase II study of sorafenib in combination with bicalutamide in patients with chemotherapy-naive castration resistant prostate cancer. Investigational new drugs. 2012;30:1652–9. doi: 10.1007/s10637-011-9722-5. [DOI] [PubMed] [Google Scholar]