

Abstract
The development of gene targeting approaches has had a tremendous impact on the functional analysis of the mouse genome. A specific application of this technique has been the adaptation of the bacteriophage P1 Cre/loxP site-specific recombinase system which allows for the precise recombination between two loxP sites, resulting in deletion or inversion of the intervening sequences. Because of the efficiency of this system, it can be applied to conditional deletions of relatively short coding sequences or regulatory elements but also to more extensive chromosomal rearrangement strategies. Both mechanistic and functional studies of genomic imprinting have benefited from the development of the Cre/loxP technology. Since imprinted genes within large chromosomal regions are regulated by the action of cis-acting sequences known as imprinting centres, chromosomal engineering approaches are particularly well suited to the elucidation of long-range mechanisms controlling the imprinting of autosomal genes. Here we review the applications of the Cre/loxP technology to the study of genomic imprinting, highlight important insights gained from these studies and discuss future directions in the field.
Keywords: genomic imprinting, Cre-loxP system, chromosome engineering
INTRODUCTION
Site-specific recombination and functional analysis of the mouse genome
The analysis of gene function in the mouse has been revolutionized by the successive development of two key technologies, namely gene targeting by homologous recombination in embryonic stem cells and the application of site-specific recombination systems. Gene targeting and its most common application in the generation of knock-out mice allows for precise modifications of endogenous gene sequences. This technology, which requires arms of homology to the desired target, has greatly benefitted from the availability of an essentially complete genome sequence for the mouse as well as libraries of mapped genomic clones which can be used to construct the appropriate targeting vectors. Whereas gene targeting has been useful in generating modifications such as insertions and deletions at the desired genomic location, by itself it is not suited for applications requiring the conditional control of the inserted mutation or implicating rearrangements between sequences located distantly on the same chromosome or even on different chromosomes. These specific advances have been made possible by introduction of site-specific recombination systems in mammalian cells. A number of recombination and integration systems have been shown to function in mouse cells as well as in vivo, such as Cre-loxP [1], Flp-FRT [2] and ΦC31-att [3–5], but by far the most widely used, and the one which will be discussed here, is the Cre-loxP system.
The Cre-loxP system, originally imported from bacteriophage P1, is a bipartite recombination system. It is composed of the Cre recombinase which acts in trans and its target sites, called loxP, short 34-bp sequences comprised of an asymmetrical core of 8 bp, flanked by 13-bp inverted repeats. Two subunits of the Cre enzyme specifically recognize and bind to the inverted repeats of a single loxP site. When two loxP sites are present in the same nucleus, Cre can catalyze the recombination between these two sites. This basic reaction, and the finding that Cre can act both intramolecularly and intermolecularly, has lead to a number of important applications for the functional analysis of the mouse genome. Here we review some of these key applications and how they could or have been exploited in the study of genomic imprinting in the mouse.
Genomic imprinting
In both human and mouse, well over eighty genes are regulated by the epigenetic phenomenon of genomic imprinting [6]. Imprinting occurs as a consequence of differential epigenetic marks carried into the zygote by the mature gametes at fertilization. As one of the main consequences of these parental marks, imprinted genes are monoallelically expressed only from the paternal or maternal allele during development [7]. Perhaps the most critical epigenetic mark that dictates genomic imprinting is differential DNA methylation of specific cis-acting sequences associated with imprinted genes. For some differentially methylated regions (DMRs) the origin of the differences between paternal and maternal homologues can be traced back to the mature gametes. These regions are referred to as gametic (or primary) DMRs and if they are shown to regulate the imprinted expression of several linked loci, can act as imprinting centres (ICs), also known as imprinting control regions (ICRs) [8]. Since most imprinted genes have been found to play roles in embryonic growth and development as well as placental development [9, 10], aberrations in DNA methylation at ICs will often cause loss of imprinting of genes in cis which can ultimately lead to a range of pre- and post-natal disorders such as Beckwith-Wiedemann syndrome (BWS) in humans.
The mechanism by which genes undergo genomic imprinting is not always obvious. Often differential DNA methylation at ICs leads to differences in gene expression through the interaction of insulators which can alter the positioning of imprinted genes and their access to shared enhancers. One well-studied example of this mechanism lies in the proximal domain of the imprinted distal mouse chromosome 7 (Chr 7) region, which is orthologous to the BWS region on human chromosome 11p15.5. In this region, imprinting centre 1 (IC1) is located upstream of the well-known H19 gene and has been found to regulate the reciprocally imprinted genes H19 and Insulin-like growth factor 2 (Igf2) (Figure 1). IC1 is unmethylated on the maternal allele, such that the insulator CTCF can bind here in an allele-specific manner [11]. This interaction allows the maternal H19 promoter to gain access to shared downstream enhancers, but brings Igf2 into a repressive chromatin domain which blocks it from being expressed maternally. On the other hand, IC1 is methylated on the paternal allele and is known to be bound by methyl-CpG binding protein 3 (MBD3) [12] and probably other unidentified factors. This association allows paternal Igf2 access to enhancers while H19 expression is prevented by the acquisition of promoter DNA methylation on the paternal allele.
Figure 1. Imprinted region on distal mouse chromosome 7.
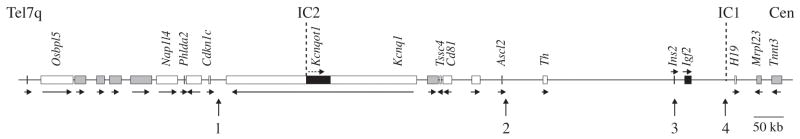
Gene arrangement on distal mouse Chr 7 shown to scale (coordinates from NCBI m37 build), oriented from the telomere (Tel7q, left) to the centromere (Cen, right). Genes showing biallelic, paternal and maternal expressions are represented by shaded, black, and white rectangles, respectively. Horizontal arrows show transcriptional orientation of each gene. The position of the imprinting centres IC1 and IC2 are shown above the diagram, while numbered vertical arrows below the diagram show the positions of loxP site insertions discussed in the text.
In other cases, differential methylation at ICs is thought to regulate imprinting via the production of large noncoding RNAs (ncRNAs) that establish silenced chromatin in cis. One classic example of this lies in the second imprinted domain on distal Chr 7. This distal imprinted domain contains imprinting centre 2 (IC2) which is located within intron 10 of the Kcnq1 gene and is unmethylated on the paternal allele, acting as a promoter for the antisense ncRNA Kcnq1ot1 (Figure 1). Although how this precisely occurs is unclear, the production and elongation of Kcnq1ot1 silences all of the protein-coding genes bidirectionally on the paternal chromosome over a span of over 800 kb [13, 14]. These genes are subsequently only expressed from the maternal allele since Kcnq1ot1 is not transcribed from the methylated maternal IC2. The long-range action of both insulators and ncRNAs in the regulation of imprinted genes points to the observation that most imprinted genes are found in clusters throughout the genome. The mechanisms governing genomic imprinting within these large clusters, including the analysis of cis- and trans-acting factors implicated, as well as the analysis of the function of imprinted genes themselves all constitute important experimental questions which can be address via applications of the Cre-loxP system.
CONDITIONAL MUTAGENESIS: MECHANISTIC AND FUNCTIONAL STUDIES
One of the most powerful and widely used applications of the Cre/loxP system is in conditional mutagenesis [15]. As its name implies, conditional mutagenesis offers a more precise control over the generation of mutant phenotypes by allowing the formation of mosaic embryos or animals in which homozygous mutant cells are confined to a specific cell lineage, in an otherwise wild-type (or heterozygous) environment. Essentially conditional mutagenesis allows the study of phenotypic consequences of the loss of function of a predetermined gene in only a subset of the cells expressing it. Therefore conditional knock-outs can bypass the early embryonic lethality phenotypes often associated with traditional gene knock-outs [16] while offering the possibility to study the function of essential genes in specific lineages. This is achieved via a binary system composed of a conditional allele of the target gene under study and a Cre-expressing allele, guiding the specificity of the conditional system. The conditional allele itself is usually generated by gene targeting in ES cells and consists of a functional, silent mutation in which specific exons are flanked by loxP sites such that deletion of the intervening sequences leads to the formation of a null allele. Cre-expressing alleles have been generated by two different approaches; (i) the random insertion of transgenes in which the Cre gene is controlled by the promoter and enhancer elements of a given tissue-specific gene; or (ii) the insertion (or knock-in) of the Cre recombinase gene into a tissue-specific locus by homologous recombination.
Conditional deletion of trans-acting factors
In the context of studies of genomic imprinting, conditional mutagenesis offers unique opportunities to address both mechanistic and functional studies. At the mechanistic level, conditional mutations can be exploited to study both cis-acting elements and trans-acting factors implicated in the developmental process of regulation by genomic imprinting. Since several factors implicated in the epigenetic regulation of imprinted genes also play essential roles in other fundamental cellular processes, the study of these epigenetic regulators can only be fully realized using conditional mutagenesis approaches. One such example is in the elucidation of the roles of the DNA methyltransferase genes Dnmt3a and Dnmt3b in the establishment of germline imprints. Several imprinted loci are associated with DNA sequences which are methylated on only one of the parental alleles in somatic cells. In the germline, these imprints are first erased in the precursors of the germ cell lineage, the primordial germ cells, in preparation for reestablishment of new gametic imprints based on the sex of the developing embryo. During gametogenesis, this re-establishment involves de novo DNA methylation which occurs at different stages in the male and female germlines. The finding that the DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation in ES cells as well as in early embryos raised the possibility that these same enzymes were also responsible for the establishment of germline imprints during gametogenesis [17]. However the early post-natal and embryonic lethality phenotypes caused by the Dnmt3a and Dnmt3b knock-outs, respectively, did not allow an analysis of imprinting defects in the germline or in the next generation [17]. To address the issue, the groups of Hiroyuki Sasaki and En Li turned to a conditional mutagenesis approach in the germ cell lineage [18, 19]. They generated conditional alleles of both Dnmt3a and Dnmt3b and obtained germline-specific loss of function by crossing these mutations to the Tnap-Cre knock-in line, which expresses Cre primarily in primordial germ cells [20]. Their analysis showed that whereas loss of Dnmt3b in the germline did not cause any apparent phenotype, Dnmt3a is essential in the male and female germline, notably in the establishment of most germline imprints [19]. Their results also revealed a striking similarity between the conditional germline knock-out of Dnmt3a and the Dnmt3l traditional knock-out mice, suggesting that Dnmt3l acts as an essential germ cell co-factor of Dnmt3a in the de novo methylation of imprinted regions [21, 22]. In a subsequent study, oocyte-specific conditional knock-out of Dnmt3a and Dnmt3b was achieved using a Zp3-Cre transgene [23] to generate embryos deficient in both maternal and zygotic forms of these enzymes [24]. Analysis of paternally methylated regions showed that the differentially methylated regions at H19 and Dlk1/Gtl2 remain methylated in these Dnmt3-deficient preimplantation embryos. By repeating similar experiments with a conditional allele of Dnmt1 [25], this work showed that maternal and zygotic Dnmt1 are necessary and sufficient for maintenance of both maternally and paternally inherited imprints during preimplantation stages [24]. In a more recent application of the Dnmt3a conditional allele, Kaneda et al. showed that postnatal Dnmt3a expressed in growing oocytes is indispensable for de novo methylation at maternal imprints [26]. By generating females carrying the conditional allele at Dnmt3a together with the Zp3-Cre transgene expressed in growing oocytes, the authors were able to show that loss of Dnmt3a activity in oocytes leads to defects in the acquisition of DNA methylation marks both at imprinted genes and at repetitive elements. However, whereas the hypomethylated state of imprinted regions is stably inherited by the resulting embryos, repetitive elements showed re-acquisition of methylation during development. These results show that the establishment of DNA methylation imprints can only occur at a specific stage of oocyte growth and the Dnmt3a is required for this activity.
Conditional deletion of cis-acting sequence elements
Conditional mutagenesis has also been applied to the study of cis-acting sequences implicated in imprinted expression. This is a particularly relevant application of the Cre technology to the study of imprinting, since cis-acting sequences can act at different levels of the developmental cycle of genomic imprinting. Conditional mutagenesis can therefore be used to dissect the temporal requirement of specific sequence elements in the regulation of imprinted gene expression. One particular example is in the analysis of the H19 DMR (IC1) on distal mouse Chr 7 (Figure 1). This sequence, located approximately −4.0 to −2.0 kb relative to the H19 transcriptional start site is required for the imprinted expression of the flanking H19 and Igf2 genes, as was shown by a traditional knock-out of IC1 [27]. When paternally inherited, the deletion leads to loss of silencing at the paternal H19 gene, whereas upon maternal transmission, the deletion causes reactivation of the normally silent maternal Igf2 allele. Although they clearly indicated a key role for IC1 in the regulation of imprinting at H19 and Igf2, these studies with the full knock-out of IC1 could not address when the cis-acting sequences was important for imprinting, if they are active in the germ line or only in somatic cells, or whether they are continuously required for imprinting of the neighboring genes. The dissection of the temporal and developmental roles of IC1 in imprinting was made possible using a conditional mutagenesis approach [28]. In this study, a conditional allele of IC1 was engineered by gene targeting in ES cells in which upstream H19 sequences from −7.0 to −0.8 kb were flanked by loxP sites (Figure 1, site 4). Animals carrying this conditional allele were obtained and shown to maintain normal imprinting at H19. The authors were then able to compare the effects of deleting IC1 at different developmental stages. Germ line deletion of IC1, using the adenovirus EIIa-Cre transgenic line expressing Cre in early development [29] and assessment of the consequences of the deletion in the subsequent generation, was shown to recapitulate the results previously described for the knock-out allele. Experiments deleting IC1 either in zygotes, by transient production of Cre from an injected expression vector, or in terminally differentiated muscle cells, using the MCK-Cre transgenic line [30], showed that the presence of IC1 is required in early development for acquisition of promoter DNA methylation and silencing at the paternal H19. However, IC1 is dispensable for maintenance of imprinting at H19 in non-dividing cells. Their model proposes that the gametic DNA methylation imprint carried by IC1 spreads to the H19 promoter in early embryos and that this secondary or somatic mark is sufficient to silence H19 in muscle cells. That the secondary imprint at the H19 promoter is in fact also stable on its own through rounds of mitosis was also shown in a subsequent publication using the same conditional mutagenesis approach, coupled with a human CD2-Cre transgene expressed in early T cells [31]. This work on IC1 also confirmed that Igf2 is imprinted via a different mechanism, since deletion of IC1 at any stage of development leads to reactivation of the maternal allele, a result consistent with Igf2 silencing being mediated via an insulator element established on the unmethylated allele of IC1 [28].
Another imprinted domain for which conditional deletion has revealed information about the mechanism of imprinted regulation is the Rasgrf1 domain on mouse chromosome 9. Rasgrf1 is a paternally-expressed imprinted gene with a paternally-methylated DMR located 30 kb 5′ of its promoter and a set of sequence repeats 3′ of the DMR which are required for establishment of proper allele-specific methylation in the male germ line [32]. In a knockout model, the absence of these repeats results in loss of methylation on the paternal allele, which allows CTCF to bind to and silence both alleles of Rasgrf1 [32, 33]. This simple deletion was helpful in assessing the role of the repeats in acquisition of DNA methylation in the germ line, but not their continuing role in the maintenance of paternal-specific DNA methylation during development. To examine this more closely, males carrying a conditional allele with the repeats flanked by loxP sites were mated to females transgenic for Cre recombinase driven by promoters specific to oocytes, E5.5 epiblast, E11 central nervous system, or P11 liver [34]. Of these four tissues, only the deletion of the repeats in oocytes resulted in loss of DNA methylation on the paternal allele, indicating that the repeats are required for maintenance of paternal DNA methylation, but only during a specific time window. In this case, the Cre-loxP system allowed detailed analysis of the requirement for a specific sequence element for the maintenance of imprinting in specific tissues throughout the in utero period [34].
Conditional deletion of imprinted genes
In terms of functional studies, the conditional mutagenesis approach also offers an powerful alternative to traditional knock-out technology in the analysis of the developmental and cellular functions of imprinted genes themselves. Several imprinted genes are expressed during development and play important roles in both embryonic and extra-embryonic functions. It is therefore surprising that so far few of these genes have been studied via conditional mutagenesis. We expect to see more conditional studies of imprinted gene mutations in the future, as more is known about the functions of some of those genes. One gene which has been studied by conditional mutagenesis is the maternally expressed Igf2r gene [35]. Igf2r codes for the mannose 6-phosphate (M6P)/insulin-like growth factor 2 receptor. It is implicated in a number of functions including the lysosomal trafficking of many M6P-glycoproteins as well as the degradative internalization of circulating IGF2. The Igf2r gene is imprinted and maternally expressed. A traditional targeted deletion of Igf2r showed that paternal heterozygotes are viable, but most maternal heterozygotes die perinatally. These IGF2R-deficient mice are 25–30% larger than their wild type litter mates and show major cardiac abnormalities [36]. Because of this early post-natal lethality, the role of IGF2R in post-natal biology has not been studied. Recently, a conditional mutation of Igf2r has been described which opens up new avenues to study the function of this imprinted receptor in adult physiology [35].
CHROMOSOMAL REARRANGEMENTS: MITOTIC EVENTS
One of the powerful applications of the Cre-loxP system involves the design of defined chromosomal rearrangements. Often referred to as chromosome engineering technology, these applications are based on the ability of Cre to catalyze the recombination between targeted loxP sites located at large distances within the genome, on the same (cis configuration) or different (trans configuration) chromosome(s). Furthermore Cre can also recombine efficiently between an integrated loxP site and an extrachromosomal site present on an incoming construct, allowing for the controlled insertion of defined elements at a predetermined chromosomal location. A number of these approaches have been used in the study of genomic imprinting, while others offer new avenues for future work.
Historically, the study of imprinting in the mouse has greatly benefitted from the availability of naturally occurring or randomly generated rearrangements between chromosomes. The first genetic demonstration of functional non-equivalence of paternal and maternal alleles at specific chromosomes was based on the generation of mice carrying uniparental disomy for a chromosome of interest [37]. These studies were based on the availability of Robertsonian translocations between different mouse chromosomes, allowing for the generation of nullisomic or disomic gametes following non-disjunction at meiosis. The refinement of the chromosomal domains responsible for parent-of-origin effects took advantage of reciprocal translocations involving only part of the imprinted chromosome, leading to the breeding of mice with uniparental disomy only for a given chromosomal region [38]. Two different approaches based on the Cre-loxP technology offer new avenues to generate ES cells and embryos carrying defined regions of uniparental disomy.
Cre-induced mitotic recombination
The first strategy is based on the ability to induce Cre-mediated mitotic recombination in ES cells. In 2002, two different studies showed that allelic loxP sites, targeted at the same chromosomal position on the two parental homologues of a given chromosome in ES cells, can be used as targets for a Cre-induced mitotic recombination reaction [39, 40]. Several possible outcomes of such rearrangement must be considered, depending on whether Cre acts in G1 or in G2, as well as the type of segregation (X or Z segregations) observed following G2 recombination (Figure 2). Since these rearrangements occur with loxP sites located in trans, they occur at low frequencies (4.2 × 10−5 to 7.0 × 10−3) and therefore require the reconstitution of a positive selectable marker or a selection for homozygosity in the distal region. In these studies, this selection was provided by the use of a bipartite HPRT1 minigene in Hprt-null ES cells [40], previously developed by others for chromosome engineering approaches [41], or increased resistance to higher doses of the drug G418 conferred by increase dosage of the neo resistant marker [39, 42]. Other systems based on the reconstitution of a drug-selectable marker could easily be adapted for the same purpose [43, 44]. Cre recombination in G2 followed by X segregation leads to loss of heterozygosity (LOH) in the entire chromosome region located distal of the loxP sites (Figure 2). In their experiments, Lui et al. obtained frequencies of LOH ranging from 100% to 23% depending on the chromosome and site of integration of the loxP sites, suggesting a significant position effect on the expected outcome [40]. Nevertheless, the authors were able to recover recombinants showing LOH for most of chromosomes 7 and 11, using loxP sites targeted close to the centromeres. The study based on high-G418 resistance generated cells homozygous for most of chromosome 1 [39]. Since a single parental homologue is duplicated in these LOH cells, they constitute uniparental disomies (UPD) for the recombinant. Although this technology was developed for the purpose of generating mosaics for functional studies [45], in which patches of homozygous mutant cells can be studied in an otherwise heterozygous background, it can also provide a useful source of uniparental material for the study of imprinted genes, imprinted regions, and imprinted phenotypes, both through in vitro differentiation assays and in vivo in chimeras. In fact both the study of Lui et al. [40] and subsequent work based on the same recombination system [46] have used the epigenetic polymorphism at the imprinted gene Snrpn as a marker to follow the segregation of parental chromosomes. This work, supported by the study of targeted genetic polymorphisms, confirmed that the DNA methylation mark at Snrpn on Chr 7 was stable in these recombinant, and lead to the derivation of both maternal and paternal uniparental disomies for most of Chr 7. It was also shown that increased frequencies of recombination observed at specific chromosomal locations were not attributable to the S-phase pairing described at imprinted regions [47, 48], although the possibility that pairing does not occur in ES cells remains to be addressed.
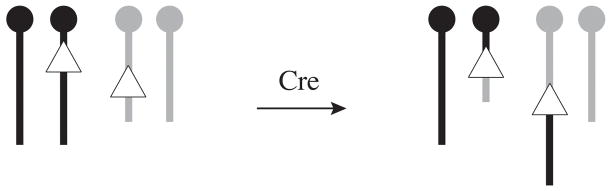
Figure 2. Cre/loxP-induced mitotic recombination: generation of uniparental disomic cells.

Mitotic recombination is induced by transient Cre production in a cell carrying allelic loxP sites (arrowheads) on the paternal (black) and maternal (gray) homologues. Centromeres are represented by the circles at the top of each chromosome. If Cre recombination occurs in G1, two recombinant chromosomes are produced and replicated during S phase (S). At mitosis (M), the two daughter cells produced are identical and balanced, with no loss of heterozygosity. If Cre recombination occurs in G2 between one sister chromatids per homologue, two different segregation patterns can occur. In X segregation (X), the recombinants are segregated to different daughter cells. As a consequence, both cells have lost heterozygosity for all markers distal of the loxP sites. This constitutes regions of paternal and maternal uniparental disomy. As in the case of G1 Cre, Z segregation (Z), in which recombinants co-segregate, does not lead to any loss of heterozygosity.
Despite the low frequency of recombination events noted in the ES cell-based systems, a number of efforts have been made to adapt this LOH approach to mosaic analysis in vivo [49–52]. As in the case of the ES cells system, these studies are based on the targeting of allelic loxP sites on parental homologues for a specific autosome. Where the in vivo strategies offer more flexibility is in the selection of the transgenic Cre line driving the recombination events. By generating animals trans-heterozygous for the loxP site insertions and breeding in a chosen tissue-specific Cre line, mitotic recombination and LOH can be targeted to a specific tissue or organ. The detection of LOH cells, which have undergone G2 Cre recombination followed by X segregation, remains a challenge, and two different approaches have been exploited. In the first strategy, one of the loxP site alleles is tagged with a human CD2 cell surface marker and LOH cells are detected based on the loss of hCD2 expression [50]. Using a Pgk-Cre line to drive recombination, up to 1% of B lymphocytes were shown to be negative for hCD2 expression. This system can only detect one of the two daughter cells produced by X segregation. In the second strategy, complementary bipartite GFP and DsRed2 expression cassettes were engineered to flank intronic loxP sites in the targeting vectors [52]. Upon recombination in G2, both the GFP and DsRed2 expression cassettes are reconstituted on different homologues. As a consequence, the four possible products of segregation can be distinguished on the basis of their fluorescent properties: Z segregation leads to unlabeled and doubly labeled cells, whereas X segregation, which is accompanied by LOH, leads to GFP-positive and DsRed2-positive singly labeled cells.
As discussed above for the ES cell-based approach, these studies therefore show that it is possible to generate and tag cells having undergone G2 recombination followed by X segregation in vivo, resulting in LOH. As these cells represent uniparental disomies for the chromosomal region located distal of the recombination breakpoint defined by the loxP sites, they could offer new avenues for the study of imprinted regions in vivo. Taking advantage of these tools, it may be possible to discover novel imprinted genes in those regions of the mouse genome which are known to show parent-of-origin effects, but contain no known imprinted genes.
Nonhomologous recombination and reciprocal translocations
The second strategy based on Cre-induced chromosomal engineering which offers new opportunities for the analysis of imprinted regions is the generation of specific chromosomal translocations (Figure 3). In this approach, the breakpoints of the translocation are defined by targeted loxP site insertions positioned on nonhomologous chromosomes in the same relative orientation with respect to the centromeres. A trans-recombination reaction catalyzed by Cre will result in the reciprocal exchange of the chromosome arms located distal of the loxP sites and the generation of a balanced translocation. As in the case of homologous recombination discussed above, this system has been demonstrated both in ES cell in cultures using transient Cre expression [53, 54] as well as in vivo with a Cre transgene [55, 56]. Because the breakpoints of such rearrangement can be predetermined through the design of the necessary targeting vectors for loxP site insertions, essentially any translocation can be engineered using this approach. The first application of this strategy to imprinted regions is therefore in the refinement of domains showing imprinted phenotypes in uniparental disomies and in the generation of new material for the analysis of these regions during development.
Figure 3. Reciprocal translocations in ES cells.
Cre can catalyze a trans recombination between loxP sites targeted on different chromosomes. The resulting ES cells are balanced for the translocation and can be used to establish new chromosome-specific translocation lines or to study the effect of the disruption caused by the translocation itself. Black and gray chromosomes represent different autosomes.
The second application of site-specific translocations is in the analysis of long-range mechanisms regulating the coordinated imprinted expression of genes clustered into large imprinted domains regulated by a unique imprinting centre. Such a strategy was used to analyze the imprinted regulation of the Cdkn1c gene on distal mouse Chr 7 [57]. In this study, the Cre/loxP system was used to engineer a translocation between the distal imprinted domain on Chr 7 and a telomeric position on Chr 11, using a previously established system in Hprt-deficient ES cells based on the bipartite HPRT1 minigene [58]. In this distal imprinted domain several protein-coding genes are preferentially or exclusively expressed from the maternal chromosome. On the paternal allele, these genes are silenced by a cis mechanism implicating the imprinted non-coding RNA Kcnq1ot1, which is maintained silent on the maternal homologue by a gametic DNA methylation imprint inherited directly from oocytes. In their study, Cleary et al. introduced a loxP site in between the differentially methylated promoter of Kcnq1ot1 and a cluster of distal imprinted genes including Cdkn1c (Figure 1, site 1). A reciprocal translocation between this site on Chr 7 and the loxP site on Chr 11 was obtained by transient expression of Cre recombinase in ES cells and selection for reconstitution of the HPRT1 minigene by growth in HAT medium. The outcome of this rearrangement is the translocation of Chr 7 genes distal of the loxP site to the end of Chr 11. Interestingly, it was shown that the ES cells carrying this balanced translocation cause a cell-autonomous defect in high-contribution chimeras. Since the phenotype observed in these embryos is reminiscent of the developmental abnormalities described for the Cdkn1c knock-out mice, the results suggest that the translocation implicated the maternal allele of Cdkn1c and that the expression of this gene was affected by the translocation. Germline transmission of the translocated chromosomes was eventually obtained in low-contribution chimeras and imprinting analysis showed that whereas all the Chr 7 genes analyzed and located proximal of the breakpoint were unaffected, Cdkn1c and two other distal genes studied showed reduced expression and loss of silencing when paternally inherited from the translocated chromosome [57]. This work established that linkage between Cdkn1c and the proximal part of the domain was essential for epigenetic silencing on the paternal chromosome.
MEIOTIC RECOMBINATION
One of the key limitations of trans recombinations catalyzed by Cre is the low frequencies at which the recombinants are recovered in dividing somatic cells. This limits the applications of these approaches in vivo unless specific cell labeling systems are involved, as described above for the mitotic recombination. The ability to recover recombinants in vivo is also desirable as more loxP site containing alleles are generated, providing a useful mouse resource in itself for further manipulation of the genome. To overcome these limitations an elegant in vivo system based on meiotic Cre recombination was developed [59]. Termed targeted meiotic recombination (TAMERE), this approach is based on the use of the Sycp1-Cre transgenic line which expresses Cre recombinase in primary spermatocytes [60]. The rationale behind TAMERE is that trans recombination between loxP sites targeted at non-allelic positions of homologous chromosomes would be more efficient at meiosis, when chromosome pairing occurs in preparation for meiotic recombination. Coding for a component of the synaptonemal complex, the Sycp1 gene is produced in early meiotic cells and the Sycp1 promoter-driven Cre transgenic line was shown to be expressed during leptotene to zygotene stages of male meiosis [60]. In the TAMERE strategy, two loxP sites inserted in the same orientation on homologous chromosomes are bred into a male hemizygous for the Sycp1-Cre transgene. In the germline of such a “trans-loxer”, Cre will catalyze the unequal recombination between the non-allelic loxP sites, resulting in mature gametes carrying the reciprocal products of this rearrangement, the deletion and duplication of the chromosomal region define by the loxP site breakpoints segregated in different gametes (Figure 4). By simply breeding this male to wild type females, paternal heterozygous progeny carrying the deletion and duplication allele can be recovered and used to establish the new mutant line. This strategy for the in vivo generation of novel deletion and duplication alleles was developed in the laboratory of Denis Duboule. They used TAMERE to generate several new alleles at the HoxD locus, which they recovered at frequencies ranging from 0.1 to 20% from trans-loxing males [59, 61, 62].
Figure 4. Targeted meiotic recombination (TAMERE).

Recombination between loxP sites located in trans at non-allelic positions on homologous chromosomes can be induced by Cre in vivo. Triple transgenic males carrying the loxP site insertions on paternal (black) and maternal (gray) homologues of a given chromosome and the Sycp1-Cre transgene will undergo a Cre-mediated recombination during chromosome pairing at meiosis I (MI) at high frequencies. Following meiosis II (MII) mature gametes carrying either the deletion in the intervening sequence (Del) or its duplication (Dp) can be recovered and these new mutations can established in a mouse line by direct breeding.
TAMERE has been used to generate reciprocal deletion and duplication alleles within the imprinted domain on distal mouse Chr 7 [44]. Using loxP site insertions immediately upstream of the Ins2 gene [63] and downstream of the Ascl2 gene [44] a ~280 kb region of distal Chr 7 was deleted and duplicated in the Del7AI and Dp7AI alleles (Figure 1, sites 2 and 3). In this case, the recombinants were each recovered in approximately 17% of the progeny from two trans-loxing males [44]. The deletion allele was designed to remove the entire interval between two independently regulated imprinted sub-domains, each controlled by its own imprinting centre: Ins2 is located at the distal end of the proximal IC1 sub-domain regulated by the paternal DNA methylation imprint upstream of H19, whereas Ascl2 is the most proximal imprinted gene regulated by the non-coding RNA Kcnq1ot1 discussed above (IC2 sub-domain; Figure 1). Analysis of heterozygous progeny carrying a maternal or a paternal Del7AI allele showed that imprinting at IC1 and IC2 is unaffected by this large deletion and the juxtaposition of the Ins2 and Ascl2 genes. Both paternal and maternal inheritance of the Del7AI allele are viable although the maternal heterozygotes are growth retarded. These results suggest that although the deleted sequences are dispensable for imprinting in the region, they do contain cis-acting elements required for normal expression of flanking genes.
INTEGRATION OF EXTRA-CHROMOSOMAL ELEMENTS
The observation that Cre can catalyze both intramolecular and intermolecular recombination between loxP sites led to the development of site-specific integration strategies [43, 64]. In this approach, an integrated loxP site is used as a docking site for the site-specific integration of an incoming extra-chromosomal circular molecule (Figure 5A). Upon recombination the entire loxP site containing construct is integrated as a loxP-flanked single copy insertion, or “pop-in”. Because the excision of the integrated construct by an intramolecular reaction is favored over the intermolecular integration, a number of strategies have been used to recover stable integrants in ES cells. Successful integration has been achieved by transient expression of Cre from a transfected expression vector in combination with reconstitution of a positive selectable marker at the site of recombination. Selections based on bipartite neo or tk constructs and the HPRT1 minigene have all been described [43, 53, 64, 65]. The rationale behind these approaches is that although the integrated form can readily be excised by Cre, stable integrants can still be recovered under strong selection since Cre is only transiently produced in these cells. Higher frequencies of integration can be obtained by using heterotypic mutant lox sites which can recombine with each other in the forward integrative reaction, but which yield non-compatible lox sites flanking the insertion [66]. The integrated construct is therefore maintained in a stable non-excisable state, even in the continuous presence of Cre [67].
Figure 5. Site-specific intermolecular reactions.

Cre can catalyze the recombination between an integrated genomic loxP site and a second site present on an incoming molecule transfected in ES cells. Selection for the desired recombination events requires reconstitution of a bipartite selectable marker, the non-functional components of which are present at the integrated loxP site (M 3′) as well as on the incoming DNA construct (M 5′). Cre-mediated intermolecular recombination reconstitutes a functional cassette (M 5′-loxP-M 3′) which provides the phenotypic selection for the recovery of stable recombinants in transient Cre expression experiments. (A) In site-specific integration reactions, the extra-chromosomal loxP site is present on a circular molecule. Cre can catalyze the integration of this entire construct and yield recombinants in which the integrated DNA is flanked by loxP sites. (B) In recombinase-mediated chromosome truncation, the incoming construct is linear and carries a terminal array of telomere repeats. In this case, the Cre-mediated intermolecular reaction catalyzes the exchange between this incoming artificial telomere and the distal end of the chromosome carrying the docking site. The outcome is the generation of a chromosomal truncation and integration of the incoming construct at the new telomeric end.
The main advantages of Cre-mediated integration over other experimental approaches is the ease at which different constructs can be recovered as single-copy integrants at a predetermined genomic site. In the context of the study of genomic imprinting, this could be particularly well suited to the analysis of cis-acting sequences implicated in the mechanism of imprinting. A recent application of Cre-mediated insertion was described in the generation of a GFP insertion within the imprinted domain on distal Chr 7 [68]. In this study, integration events were recovered following the reconstitution of a functional neo selectable marker from complementary non-functional components, using a previously targeted loxP docking site insertion (Figure 1, site 3) next to the Ins2 gene [63]. The integrated construct contained a pCAG-EGFP expression cassette and the authors derived a mouse line carrying this allele, called Tel7KI, and showed that the GFP reporter is regulated by genomic imprinting at this insertion site. Because similar pCAG-based constructs are not subject to imprinting at other integration sites in the genome, this study showed that imprinting signals within the distal Chr 7 imprinted domain are able to act at a distance and impose an imprinted expression pattern onto the inserted GFP reporter.
A variation of the integration reaction was recently developed to study the IC2-independent function of IC1 in the context of distal Chr 7 [63]. In this new Cre/loxP strategy, termed recombinase-mediated chromosome truncation (RMCT), the incoming construct is not a circular molecule but rather a linear construct carrying a terminal array of telomere repeats of the sequence TTAGGG (Figure 5B). In this case, Cre recombination does not lead to integration of the incoming construct but rather to a replacement of the chromosomal domain located distal of the loxP site by the vector sequences. By using a loxP site 5′ of a silent neo selectable marker targeted next to the Ins2 gene (Figure 1, site 3), the authors were able to engineer a truncation of Chr 7 with a breakpoint immediately distal to the IC1-regulated subdomain, deleting the entire IC2 sub-domain and an additional 1.6 Mb of distal Chr 7 sequences. The truncated chromosome, called the DelTel7 allele was shown to be stable in ES cells and germline transmission of the allele was obtained. The paternal heterozygotes are fully viable, showing absence of essential paternally expressed genes on distal Chr 7 or of haploinsufficiency effects for the very distal end of Chr 7. As expected given the large number of maternally expressed genes in the IC2 subdomain, the maternal heterozygotes are embryonic lethal and show placentation defects. The authors were able to show that a paternal deletion of IC2 itself, which leads to reactivation of IC2-regulated genes [13], can rescue this embryonic lethality of the maternally inherited DelTel7 allele. By combining Cre-mediated integration with a replacement reaction, the authors were therefore able to create a unique type of mutation, a chromosomal truncation, which led to important findings on the regulation of imprinting on distal Chr 7.
SUMMARY
Genomic imprinting is a complex epigenetic phenomenon in which specific cis-acting sequences and trans-acting factors regulate the monoallelic expression of important developmental genes. Several mechanistic and functional aspects of imprinting can only be studied via controlled modifications of the genome. The study of genomic imprinting has greatly benefitted from two applications of the Cre/loxP system; conditional mutagenesis as well as engineering of precise chromosomal rearrangements. Together these approaches have provided powerful tools to dissect key aspects of imprinted gene regulation. As more conditional alleles and loxP site insertions are generated, we can expect to see a more widespread application of these technologies to the study of imprinting.
KEY POINTS.
Conditional mutagenesis is the ideal approach to study the roles of cis- and trans-acting factors implicated in the developmental regulation of imprinted expression.
Because several of these genes play important roles in development, conditional approaches will also become more important in the study of imprinted gene function.
Intermolecular recombination can be used to generate uniparental disomies by mitotic recombination or new translocations for the study of imprinted regions.
Intramolecular reactions or trans recombination during meiosis provide alternatives for the generation of large deletions within imprinted domains.
Intermolecular reactions between a targeted loxP site and incoming constructs can be adapted to generate several rearrangements such as insertions and truncations at a predetermined position in an imprinted domain.
Acknowledgments
FUNDING
Canadian Institutes of Health Research (grant MOP-82863 to L.L.). L.L. holds a Canada Research Chair and M.J.J. is the recipient of a scholarship from the Michael Smith Foundation for Health Research.
References
- 1.Sauer B, Henderson N. Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proc Natl Acad Sci USA. 1988;85:5166–70. doi: 10.1073/pnas.85.14.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O’Gorman S, Fox DT, Wahl GM. Recombinase-mediated gene activation and site-specific integration in mammalian cells. Science. 1991;251:1351–5. doi: 10.1126/science.1900642. [DOI] [PubMed] [Google Scholar]
- 3.Belteki G, Gertsenstein M, Ow D, et al. Site-specific cassette exchange and germline transmission with mouse ES cells expressing phiC31 integrase. Nat Biotechnol. 2003;21:321–4. doi: 10.1038/nbt787. [DOI] [PubMed] [Google Scholar]
- 4.Groth AC, Olivares EC, Thyagarajan B, et al. A phage integrase directs efficient site-specific integration in human cells. Proc Natl Acad Sci USA. 2000;97:5995–6000. doi: 10.1073/pnas.090527097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Olivares EC, Hollis RP, Calos MP. Phage R4 integrase mediates site-specific integration in human cells. Gene. 2001;278:167–76. doi: 10.1016/s0378-1119(01)00711-9. [DOI] [PubMed] [Google Scholar]
- 6.Morison I, Ramsay JP, Spencer HG. A census of mammalian imprinting. Trends Genet. 2005;21:457–65. doi: 10.1016/j.tig.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 7.Reik W, Walter J. Genomic imprinting: parental influence on the genome. Nat Rev Genet. 2001;2:21–32. doi: 10.1038/35047554. [DOI] [PubMed] [Google Scholar]
- 8.Bartolomei M. Genomic imprinting: employing and avoiding epigenetic processes. Genes Dev. 2009;23:2124–33. doi: 10.1101/gad.1841409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fowden AL, Sibley C, Reik W, et al. Imprinted genes, placental development and fetal growth. Horm Res. 2006;65 (Suppl 3):50–8. doi: 10.1159/000091506. [DOI] [PubMed] [Google Scholar]
- 10.Wagschal A, Feil R. Genomic imprinting in the placenta. Cytogenet Genome Res. 2006;113:90–8. doi: 10.1159/000090819. [DOI] [PubMed] [Google Scholar]
- 11.Murrell A, Heeson S, Reik W. Interaction between differentially methylated regions partitions the imprinted genes Igf2 and H19 into parent-specific chromatin loops. Nat Genet. 2004;36:889–93. doi: 10.1038/ng1402. [DOI] [PubMed] [Google Scholar]
- 12.Reese KJ, Lin S, Verona R, et al. Maintenance of paternal methylation and repression of the imprinted H19 gene requires MBD3. PLoS Genet. 2007;3:e137. doi: 10.1371/journal.pgen.0030137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fitzpatrick G, Soloway P, Higgins M. Regional loss of imprinting and growth deficiency in mice with a targeted deletion of KvDMR1. Nat Genet. 2002;32:426–31. doi: 10.1038/ng988. [DOI] [PubMed] [Google Scholar]
- 14.Mancini-Dinardo D, Steele SJ, Levorse J, et al. Elongation of the Kcnq1ot1 transcript is required for genomic imprinting of neighboring genes. Genes Dev. 2006;20:1268–82. doi: 10.1101/gad.1416906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gu H, Marth JD, Orban PC, et al. Deletion of a DNA polymerase beta gene segment in T cells using cell type-specific gene targeting. Science. 1994;265:103–6. doi: 10.1126/science.8016642. [DOI] [PubMed] [Google Scholar]
- 16.Copp AJ. Death before birth: clues from gene knockouts and mutations. Trends Genet. 1995;11:87–93. doi: 10.1016/S0168-9525(00)89008-3. [DOI] [PubMed] [Google Scholar]
- 17.Okano M, Bell DW, Haber DA, et al. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–57. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 18.Dodge JE, Okano M, Dick F, et al. Inactivation of Dnmt3b in mouse embryonic fibroblasts results in DNA hypomethylation, chromosomal instability, and spontaneous immortalization. J Biol Chem. 2005;280:17986–91. doi: 10.1074/jbc.M413246200. [DOI] [PubMed] [Google Scholar]
- 19.Kaneda M, Okano M, Hata K, et al. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature. 2004;429:900–3. doi: 10.1038/nature02633. [DOI] [PubMed] [Google Scholar]
- 20.Lomelí H, Ramos-Mejía V, Gertsenstein M, et al. Targeted insertion of Cre recombinase into the TNAP gene: excision in primordial germ cells. Genesis. 2000;26:116–7. [PubMed] [Google Scholar]
- 21.Bourc’his D, Xu GL, Lin CS, et al. Dnmt3L and the establishment of maternal genomic imprints. Science. 2001;294:2536–9. doi: 10.1126/science.1065848. [DOI] [PubMed] [Google Scholar]
- 22.Hata K, Okano M, Lei H, et al. Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development. 2002;129:1983–93. doi: 10.1242/dev.129.8.1983. [DOI] [PubMed] [Google Scholar]
- 23.de Vries WN, Binns LT, Fancher KS, et al. Expression of Cre recombinase in mouse oocytes: a means to study maternal effect genes. Genesis. 2000;26:110–2. [PubMed] [Google Scholar]
- 24.Hirasawa R, Chiba H, Kaneda M, et al. Maternal and zygotic Dnmt1 are necessary and sufficient for the maintenance of DNA methylation imprints during preimplantation development. Genes Dev. 2008;22:1607–16. doi: 10.1101/gad.1667008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jackson-Grusby L, Beard C, Possemato R, et al. Loss of genomic methylation causes p53-dependent apoptosis and epigenetic deregulation. Nat Genet. 2001;27:31–9. doi: 10.1038/83730. [DOI] [PubMed] [Google Scholar]
- 26.Kaneda M, Hirasawa R, Chiba H, et al. Genetic evidence for Dnmt3a-dependent imprinting during oocyte growth obtained by conditional knockout with Zp3-Cre and complete exclusion of Dnmt3b by chimera formation. Genes Cells. 2010 doi: 10.1111/j.1365-2443.2009.01374.x. [DOI] [PubMed] [Google Scholar]
- 27.Thorvaldsen JL, Duran KL, Bartolomei M. Deletion of the H19 differentially methylated domain results in loss of imprinted expression of H19 and Igf2. Genes Dev. 1998;12:3693–702. doi: 10.1101/gad.12.23.3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Srivastava M, Hsieh S, Grinberg A, et al. H19 and Igf2 monoallelic expression is regulated in two distinct ways by a shared cis acting regulatory region upstream of H19. Genes Dev. 2000;14:1186–95. [PMC free article] [PubMed] [Google Scholar]
- 29.Lakso M, Pichel JG, Gorman JR, et al. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci USA. 1996;93:5860–5. doi: 10.1073/pnas.93.12.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brüning JC, Michael MD, Winnay JN, et al. A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Molecular Cell. 1998;2:559–69. doi: 10.1016/s1097-2765(00)80155-0. [DOI] [PubMed] [Google Scholar]
- 31.Srivastava M, Frolova E, Rottinghaus B, et al. Imprint control element-mediated secondary methylation imprints at the Igf2/H19 locus. J Biol Chem. 2003;278:5977–83. doi: 10.1074/jbc.M208437200. [DOI] [PubMed] [Google Scholar]
- 32.Yoon B, Herman H, Sikora A, et al. Regulation of DNA methylation of Rasgrf1. Nat Genet. 2002;30:92–6. doi: 10.1038/ng795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoon B, Herman H, Hu B, et al. Rasgrf1 imprinting is regulated by a CTCF-dependent methylation-sensitive enhancer blocker. Mol Cell Biol. 2005;25:11184–90. doi: 10.1128/MCB.25.24.11184-11190.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holmes R, Chang Y, Soloway P. Timing and sequence requirements defined for embryonic maintenance of imprinted DNA methylation at Rasgrf1. Mol Cell Biol. 2006;26:9564–70. doi: 10.1128/MCB.00058-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wylie AA, Pulford DJ, McVie-Wylie AJ, et al. Tissue-specific inactivation of murine M6P/IGF2R. Am J Pathol. 2003;162:321–8. doi: 10.1016/S0002-9440(10)63823-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lau M, Stewart C, Liu Z, et al. Loss of the imprinted IGF2/cation-independent mannose 6-phosphate receptor results in fetal overgrowth and perinatal lethality. Genes Dev. 1994;8:2953–63. doi: 10.1101/gad.8.24.2953. [DOI] [PubMed] [Google Scholar]
- 37.Cattanach BM, Kirk M. Differential activity of maternally and paternally derived chromosome regions in mice. Nature. 1985;315:496–8. doi: 10.1038/315496a0. [DOI] [PubMed] [Google Scholar]
- 38.Cattanach BM. Parental origin effects in mice. J Embryol Exp Morphol. 1986;97 (Suppl):137–50. [PubMed] [Google Scholar]
- 39.Koike H, Horie K, Fukuyama H, et al. Efficient biallelic mutagenesis with Cre/loxP-mediated inter-chromosomal recombination. EMBO Reports. 2002;3:433–7. doi: 10.1093/embo-reports/kvf097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu P, Jenkins N, Copeland N. Efficient Cre-loxP-induced mitotic recombination in mouse embryonic stem cells. Nat Genet. 2002;30:66–72. doi: 10.1038/ng788. [DOI] [PubMed] [Google Scholar]
- 41.Ramírez-Solis R, Liu P, Bradley A. Chromosome engineering in mice. Nature. 1995;378:720–4. doi: 10.1038/378720a0. [DOI] [PubMed] [Google Scholar]
- 42.Lefebvre L, Dionne N, Karaskova J, et al. Selection for transgene homozygosity in embryonic stem cells results in extensive loss of heterozygosity. Nat Genet. 2001;27:257–8. doi: 10.1038/85808. [DOI] [PubMed] [Google Scholar]
- 43.Fukushige S, Sauer B. Genomic targeting with a positive-selection lox integration vector allows highly reproducible gene expression in mammalian cells. Proc Natl Acad Sci USA. 1992;89:7905–9. doi: 10.1073/pnas.89.17.7905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lefebvre L, Mar L, Bogutz A, et al. The interval between Ins2 and Ascl2 is dispensable for imprinting centre function in the murine Beckwith-Wiedemann region. Hum Mol Genet. 2009;18:4255–67. doi: 10.1093/hmg/ddp379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adams D, Bradley A. Induced mitotic recombination: a switch in time. Nat Genet. 2002;30:6–7. doi: 10.1038/ng0102-6. [DOI] [PubMed] [Google Scholar]
- 46.Armakolas A, Klar AJ. Cell type regulates selective segregation of mouse chromosome 7 DNA strands in mitosis. Science. 2006;311:1146–9. doi: 10.1126/science.1120519. [DOI] [PubMed] [Google Scholar]
- 47.LaSalle JM, Lalande M. Homologous association of oppositely imprinted chromosomal domains. Science. 1996;272:725–8. doi: 10.1126/science.272.5262.725. [DOI] [PubMed] [Google Scholar]
- 48.Riesselmann L, Haaf T. Preferential S-phase pairing of the imprinted region on distal mouse chromosome 7. Cytogenet Cell Genet. 1999;86:39–42. doi: 10.1159/000015426. [DOI] [PubMed] [Google Scholar]
- 49.Muzumdar MD, Luo L, Zong H. Modeling sporadic loss of heterozygosity in mice by using mosaic analysis with double markers (MADM) Proc Natl Acad Sci USA. 2007;104:4495–500. doi: 10.1073/pnas.0606491104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun L, Wu X, Han M, et al. A mitotic recombination system for mouse chromosome 17. Proc Natl Acad Sci USA. 2008;105:4237–41. doi: 10.1073/pnas.0800798105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang W, Warren M, Bradley A. Induced mitotic recombination of p53 in vivo. Proc Natl Acad Sci USA. 2007;104:4501–5. doi: 10.1073/pnas.0607953104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zong H, Espinosa JS, Su HH, et al. Mosaic analysis with double markers in mice. Cell. 2005;121:479–92. doi: 10.1016/j.cell.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 53.Smith AJ, De Sousa MA, Kwabi-Addo B, et al. A site-directed chromosomal translocation induced in embryonic stem cells by Cre-loxP recombination. Nat Genet. 1995;9:376–85. doi: 10.1038/ng0495-376. [DOI] [PubMed] [Google Scholar]
- 54.Van Deursen J, Fornerod M, Van Rees B, et al. Cre-mediated site-specific translocation between nonhomologous mouse chromosomes. Proc Natl Acad Sci USA. 1995;92:7376–80. doi: 10.1073/pnas.92.16.7376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Buchholz F, Refaeli Y, Trumpp A, et al. Inducible chromosomal translocation of AML1 and ETO genes through Cre/loxP-mediated recombination in the mouse. EMBO Reports. 2000;1:133–9. doi: 10.1093/embo-reports/kvd027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Collins EC, Pannell R, Simpson EM, et al. Inter-chromosomal recombination of Mll and Af9 genes mediated by cre-loxP in mouse development. EMBO Reports. 2000;1:127–32. doi: 10.1093/embo-reports/kvd021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cleary MA, van Raamsdonk CD, Levorse J, et al. Disruption of an imprinted gene cluster by a targeted chromosomal translocation in mice. Nat Genet. 2001;29:78–82. doi: 10.1038/ng715. [DOI] [PubMed] [Google Scholar]
- 58.Zheng B, Sage M, Sheppeard EA, et al. Engineering mouse chromosomes with Cre-loxP: range, efficiency, and somatic applications. Mol Cell Biol. 2000;20:648–55. doi: 10.1128/mcb.20.2.648-655.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hérault Y, Rassoulzadegan M, Cuzin F, et al. Engineering chromosomes in mice through targeted meiotic recombination (TAMERE) Nat Genet. 1998;20:381–4. doi: 10.1038/3861. [DOI] [PubMed] [Google Scholar]
- 60.Vidal F, Sage J, Cuzin F, et al. Cre expression in primary spermatocytes: a tool for genetic engineering of the germ line. Mol Reprod Dev. 1998;51:274–80. doi: 10.1002/(SICI)1098-2795(199811)51:3<274::AID-MRD6>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 61.Kmita M, Fraudeau N, Hérault Y, et al. Serial deletions and duplications suggest a mechanism for the collinearity of Hoxd genes in limbs. Nature. 2002;420:145–50. doi: 10.1038/nature01189. [DOI] [PubMed] [Google Scholar]
- 62.Tarchini B, Huynh TH, Cox GA, et al. HoxD cluster scanning deletions identify multiple defects leading to paralysis in the mouse mutant Ironside. Genes Dev. 2005;19:2862–76. doi: 10.1101/gad.351105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oh R, Ho R, Mar L, et al. Epigenetic and phenotypic consequences of a truncation disrupting the imprinted domain on distal mouse chromosome 7. Mol Cell Biol. 2008;28:1092–103. doi: 10.1128/MCB.01019-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sauer B, Henderson N. Targeted insertion of exogenous DNA into the eukaryotic genome by the Cre recombinase. New Biol. 1990;2:441–9. [PubMed] [Google Scholar]
- 65.Hardouin N, Nagy A. Gene-trap-based target site for cre-mediated transgenic insertion. genesis. 2000;26:245–52. doi: 10.1002/(sici)1526-968x(200004)26:4<245::aid-gene50>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 66.Araki K, Araki M, Yamamura K. Targeted integration of DNA using mutant lox sites in embryonic stem cells. Nucleic Acids Res. 1997;25:868–72. doi: 10.1093/nar/25.4.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Araki K, Araki M, Yamamura K. Site-directed integration of the cre gene mediated by Cre recombinase using a combination of mutant lox sites. Nucleic Acids Res. 2002;30:e103. doi: 10.1093/nar/gnf102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jones MJ, Lefebvre L. An imprinted GFP insertion reveals long-range epigenetic regulation in embryonic lineages. Dev Biol. 2009;336:42–52. doi: 10.1016/j.ydbio.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]