

Abstract
Post-translational modification (PTM) of histones plays a central role in genome regulation. Engineering histones with defined PTMs on one or multiple residues is crucial for understanding their function within nucleosomes and chromatin. We introduce a sequential native chemical ligation strategy suitable for the preparation of fully synthetic histone proteins, which allows for site-specific incorporation of varied PTMs throughout the sequence. We demonstrate this method with the generation of histone H3 acetylated at lysine 56 [H3(K56ac)]. H3(K56ac) is essential for transcription, replication, and repair. We examined the influence of H3(K56ac) on the targeting of a model DNA binding factor (LexA) to a site ∼30 bp within the nucleosome. We find that H3(K56ac) increases LexA binding to its DNA target site by 3-fold at physiological ionic strength. We then demonstrate that H3(K56ac) facilitates LexA binding by increasing DNA unwrapping and not by nucleosome repositioning. Furthermore, we find that H3(K56Q) quantitatively imitates H3(K56ac) function. Together these studies introduce powerful tools for the analysis of histone PTM functions.
Keywords: nucleosome, histone post-translational modification, sequential native chemical ligation, fully-synthetic histone, histone H3 lysine 56 acetylation
Introduction
Eukaryotic genomes are organized into long repeats of nucleosomes, which contain ∼147 base pairs of DNA wrapped 1.65 times around a H2A, H2B, H3 and H4 histone octamer1. DNA wrapped into nucleosomes is sterically occluded from DNA interacting proteins2, yet this DNA must be accessed for gene expression, replication and repair. The nucleosome undergoes thermal fluctuations that transiently unwrap nucleosomal DNA to expose it for protein interactions2; 3. The equilibrium between fully wrapped and partially unwrapped DNA is termed “site exposure” and is greatest near the DNA entry-exit region of the nucleosome2. This property of the nucleosome appears to provide access to DNA processing proteins in vivo, such as photolyase access to damaged DNA within chromatin in budding yeast4. In addition, adjacent DNA binding sites within a nucleosome display an inherent cooperativity5; 6 that influences unwrapping and is likely to play an important role in genome-wide transcriptional regulation7; 8.
Nucleosomes contain an enormous number of histone post-translational modifications (PTMs) that appear to function singly and in different combinations9 to silence or activate wrapped nucleosomal DNA10. PTMs on unstructured histone tails appear to function as binding modules for chromatin-associated proteins11 and to influence higher order chromatin structure12. In contrast, PTMs that reside within internal regions of the nucleosome are often inaccessible to binding partners13 but can directly alter inherent nucleosome structure and dynamics14; 15. Nucleosomes with well-defined PTMs are required to quantitatively determine the effect of these modifications on chromatin structure and dynamics, yet the preparation of such nucleosomes has posed a synthetic challenge.
Histones containing defined PTMs have recently been generated by expressed protein ligation (EPL), in which a single synthetic peptide that includes the desired modifications is ligated to an unmodified recombinant protein. This method has been generally limited to one or more modification types located near histone termini12; 14; 16-19. Methylated and acetylated lysines have been introduced chemically as thioether-containing lysine analogs20; 21. More recently, methods have been introduced that exploit pyrrolysl-tRNA synthetase/tRNACUA pairs evolved for specific incorporation of acetylated or mono-methylated lysines into proteins. While these methods can be used to introduce this subset of modifications anywhere in a protein sequence, they are limited to the incorporation of a single modification type in each histone15; 22. However, a typical biologically relevant histone protein often contains multiple simultaneous PTMs throughout the histone sequence23. Here, we demonstrate a sequential native chemical ligation (NCL) strategy24-27 to prepare fully synthetic histones. This opens up the possibility of preparing histones with any desired set of modifications. We applied this scheme to generate full-length histone H3 acetylated at lysine 56.
Acetylation of histone H3 at lysine 56 [H3(K56ac)] is required for a number of DNA processes28; 29. H3(K56) appears to be acetylated prior to deposition onto newly replicated DNA30 and is important for DNA repair31; 32, maintenance of genomic stability33 and transcriptional regulation30; 34; 35. It has been suggested that H3(K56ac) alters chromatin structure and dynamics allowing accessibility to DNA metabolic proteins30 and as a signal to DNA damage checkpoints31.
The preparation of H3(K56ac) using evolved pyrrolysine incorporation machinery was reported recently15. This study suggested that H3(K56ac) modestly increased SWI/SNF and RSC catalyzed nucleosome repositioning, and increased DNA unwrapping at the entry-exit region. They also found H3(K56ac) did not influence chromatin compaction. However, a separate study of the acetylation mimic H3(K56Q) found that it inhibited interactions between chromatin fibers36. Taken together with previous telomere studies, these results are consistent with the notion that H3(K56ac) may function in part as a DNA entry-exit gate for access to nucleosome DNA15; 35. Recently, it was also reported that H3(K56ac) reduces H3-H4 tetramer binding by histone chaperon NAP137. The direct nucleosome characterization in these studies provides a standard by which we may validate the utility of our synthetic strategy. In addition, the influence of H3(K56ac) on protein binding within the nucleosome remains a significant unknown.
Following the incorporation of full-length, fully-synthetic H3(K56ac) into nucleosomes, we tested the DNA entry-exit gate hypothesis using a FRET system to quantitatively detect protein binding to partially unwrapped nucleosomal DNA3. We find that both H3(K56ac) and the acetyl-lysine mimic H3(K56Q) alter nucleosome equilibrium toward DNA unwrapping at the DNA entry-exit region. We also demonstrate that H3(K56ac) and H3(K56Q) increase the accessibility of a model DNA binding protein (LexA) to a target site 27 base pairs into the nucleosome. These results suggest that H3(K56ac) and H3(K56Q) shift the equilibrium of DNA site exposure to increase access of DNA metabolic proteins to DNA sites that are at least 27 base pairs within the nucleosome.
Results
Preparation of fully-synthetic H3(R40C,K56ac,S96C,C110A) that contains two non-native cysteines
Histone H3 is a 135 residue protein that can be easily refolded and incorporated into a nucleosome. This property makes it an excellent candidate for total synthesis by sequential NCL. As the basis for our ligation strategies, we selected the modified X. laevis H3(C110A) sequence, which is commonly used in biophysical studies38. The C110A substitution occurs in yeast and has not previously been reported to affect nucleosome structure, positioning and DNA unwrapping 39; 40.
NCL is the chemoselective reaction between a polypeptide containing an N-terminal Cys with a polypeptide containing a C-terminal thioester that ultimately generates a native peptide bond with a Cys at the ligation site. Cys residues were introduced into H3(C110A) at Arg 40 and Ser 96 based on homology alignments that found H3(R40C) in Cairina moschata41 and H3(S96C) in the H3.1 variant in Homo sapiens, Mus musculus42 and Caenorhabditis elegans43. These Cys residues could be crafted into a two-step NCL strategy in which the longest synthetic segment would be a central 56 residue peptide containing an acetylated-lysine that would eventually become Lys 56 (Fig. 1A). In this strategy, the N-terminal and middle-segments (N1 and M1; see Table 1) were synthesized with C-terminal thioesters. A key feature of the synthesis was the introduction of the N-terminal Cys in the middle segment M1 as a thiazolidine (Thz) moiety24; 44. After the first ligation step that linked the M1 and C1 peptides, the ligation mixture was treated with methoxylamine to unmask the N-terminal Cys of the M1C1 product prior to purification (Fig. 2A). This product was then reacted with peptide N1 to generate the full-length H3(R40C,K56ac,S96C,C110A) (Fig. 1B and 2B). The purified histone showed evidence of methionine oxidation (see Supplemental Information), which required a methionine reduction step followed by final purification and analysis (Fig. 2C-D). While each of the ligation steps proceeded to >70%, the three reverse-phase high performance liquid chromatography (RP-HPLC) purification steps resulted in significant product loss. The full ligation pathway resulted in a yield of 48 μg of H3(R40C,K56ac,S96C,C110A), which represents an overall yield of 2% based on the limiting central peptide. While these yields were low, they were sufficient for initial studies.
Fig. 1.
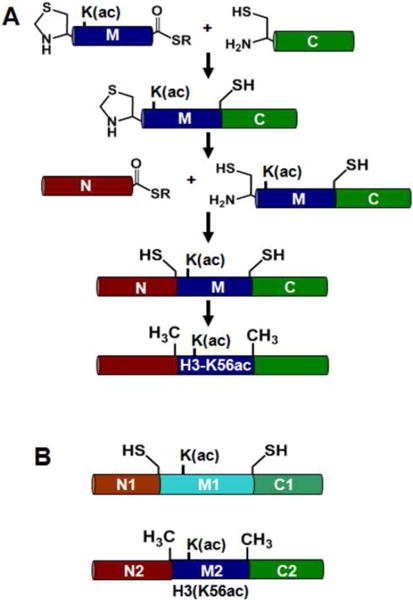
Assembly of modified histone H3 by NCL used in the analysis of reconstituted nucleosomes (A) Schematic representation of total synthesis of H3(K56ac) using sequential NCL in solution phase. Synthesis of (R40C,K56ac,S96C,C110A) proceded through the second ligation step; synthesis of H3(K56ac) used new ligation sites and added the final desulfurization step to generate the native sequence. (B) Proteins generated and characterized: H3(R40C,K56ac,S96C,C110A) and H3(K56ac,C110A).
Table 1.
Sequence of peptide segments utilized in each sequential ligation scheme.
| Peptide | Description | Sequence |
|---|---|---|
| N1 | Unmodified H3(1-39) Thioester | ARTKQTARKSTGGKAPRKQLATKAARKSAPAT GGVKKPH-COSR |
| M1 | H3(40-95) R40Thz,K56ac Thioester | (Thz)YRPGTVALREIRRYQ(Kac)STELLIRKLPQ RLVREIAQDFKTDLRFQSSAVMALQEA-COSR |
| C1 | H3(96-135) S96C,C110A | CEAYLVALFEDTNLAAIHAKRVTIMPKDIQLARR IRGERA-COOH |
| N2 | Unmodified H3(1-46) Thioester | ARTKQTARKSTGGKAPRKQLATKAARKSAPAT GGVKKPHRYRPGTV-COSR |
| M2 | H3(47-90) A47Thz,K56ac Thioester | (Thz)LREIRRYQ(Kac)STELLIRKLPFQRLVREIA QDFKTDLRFQSSAVM-COSR |
| C2 | H3(91-135) A91C,C110A | CLQEASEAYLVALFEDTNLAAIHAKRVTIMPKDI QLARRIRGERA-COOH |
| M3 | H3(47-90) A47Thz Thioester | (Thz)LREIRRYQKSTELLIRKLPFQRLVREIAQD FKTDLRFQSSAVM-COSR |
Fig. 2.
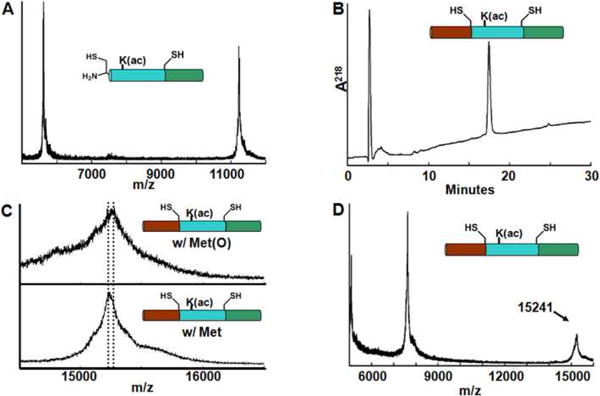
Sequential native chemical ligation to generate H3(R40C,K56ac,S96C,C110A). (A) MALDI-TOF analysis of first ligation product after Thz ring-opening; residues 40-135 of H3(R40C,K56ac,S96C,C110A). A doublet of peaks were observed, corresponding to Met and Met(O) species of the desired product: for Met, m/z expected, 11221; observed 11221. For Met(O) species, m/z expected: 11237, observed 11237. (B) RP-HPLC of final reduced H3(R40C,K56ac,S96C,C110A) with a 27-54% isopropanol / 0.1% TFA gradient at 45°C. (C) Reduction of Met(O) to Met in final ligation product H3(R40C,K56Ac,S96C,C110A). Top: MALDI-TOF MS spectrum of the reduction mixture at 0 min, containing primarily H3(R40C,K56ac,S96C,C110A) Met(O) species (m/z expected 15256, observed 15253). Bottom: MALDI-TOF MS spectrum of the reduction mixture at 60 min, containing primarily H3(R40C,K56ac,S96C,C110A) Met species (m/z expected 15240, observed 15237). (D) MALDI-TOF of reduced H3(R40C,K56ac,S96C,C110A); m/z expected 15240, m/z observed 15241.
The introduction of non-native cysteines in histone H3 alters nucleosome structure
The first generation synthetic H3 retained a Cys at each ligation site. Nucleosomes were reconstituted (Fig. 3) with a 5′-cy3 end-labeled 147 bp 601 nucleosome positioning sequence containing a LexA binding site located between 8-27 bp (Fig. 3A, 601-LexA-end), and histone octamers containing cy5-labeled H2A(K119C) (Fig. 3B) with either unmodified H3, H3(R40C,S96C,C110A), H3(R40C,K56Q,S96C,C110A), or synthetic H3(R40C,K56ac,S96C,C110A) and purified on a sucrose gradient. Following reconstitution, the cy3 and cy5 FRET pairs were juxtaposed near the entry-exit region of the nucleosome (Fig. 3B, green and magenta)3. The placement of one fluorophore on the DNA and the other on the histone octamer allows the detection of DNA movement relative to the histone octamer. Unfortunately, we found that the FRET efficiency decreased from 0.62 ± 0.02 to 0.43 ± 0.01 when wild type H3-containing nucleosomes were compared to the control, H3(R40C,S96C,C110A)-containing nucleosomes (Fig. 4). This observation suggests that the H3(R40C,S96C)-containing nucleosomes display altered structure and/or dynamics. Nucleosomes containing H3(R40C,K56Q,S96C,C110A) or H3(R40C,K56ac,S96C,C110A) resulted in a further decrease in FRET efficiency to 0.38 ± 0.02 and 0.35 ± 0.04, respectively (Fig. 4C). However, this additional reduction in FRET efficiency is significantly less than that induced with the Cys substitutions alone. Our results reveal the potential pitfalls associated with the introduction of non-native histone sequence substitutions on nucleosome structure and/or dynamics.
Fig. 3.
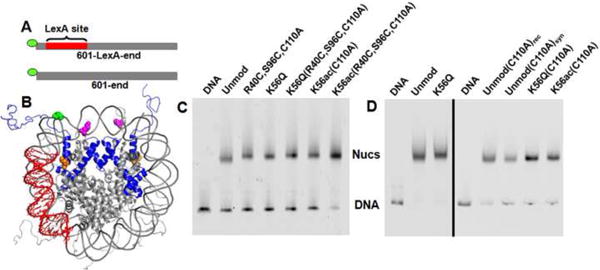
DNA substrates and reconstituted nucleosomes containing cy3 and cy5 for Fluorescence Resonance Energy Transfer (FRET) measurements. (A) The 147 base pair DNA molecules, 601-LexA-end and 601-end, contain the 601 positioning sequence with and without a LexA binding site at base pairs 8-27, respectively, and a cy3 fluorophore attached to the 5′ end. (B) Structure of the nucleosome reconstituted for FRET analysis49 Histone H3 is depicted in blue ribbons with K56 highlighted in orange. Histone H2A(K119C) (magenta) has been modified with Cy5. DNA construct specifically labeled at the 1st base with Cy3 (green) and containing a LexA binding site (red). (C) and (D) are cy3 fluorescence images of PAGE analysis of nucleosome reconstitutions prior and after purification by sucrose gradient centrifugation, respectively.
Fig. 4.
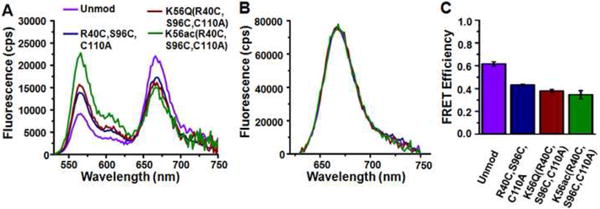
Fluorescence Resonance Energy Transfer (FRET) efficiency and LexA binding are impacted by introduced cysteines in H3 in addition to H3(K56Q) and H3(K56ac). (A) Fluorescence emission spectrum from nucleosomes containing unmodified H3 (purple), H3(R40C, S96C,C110A) (blue), H3(R40C,K56Q,S96C,C110A) (red) or H3(R40C,K56ac,S96C,C110A) (green) when excited at 510 nm (donor excitation). (B) Fluorescence emission spectrum from nucleosomes in A when exited at 610 nm (acceptor only excitation). (C) The energy transfer efficiency for nucleosomes containing unmodified H3, H3(R40C, S96C,C110A), H3(R40C,K56Q,S96C,C110A) or H3(R40C,K56ac,S96C,C110A) determined from the (ratio)A method63. The error bars were determined from the standard deviation of at least three separate measurements.
Preparation of fully synthetic wild type histones H3(K56ac,C110A) and H3(C110A)
Because the introduction of non-native Cys residues significantly influenced nucleosome structure and/or dynamics, we improved our method for preparing fully-synthetic native histones containing defined PTMs. In this second-generation approach we combined sequential NCL with a desulfurization step45 (Fig. 1A). This scheme allows the more common Ala residue to be used as a ligation site. We selected the native Ala residues H3(A47) and H3(A91) and synthesized three peptides (N2, M2, C2; see Table 1). The H3(A47) residue was incorporated as Thz (peptide M2) and H3(A91) as Cys (peptide C2) to allow sequential ligation (Fig. 1). Peptide M2 was mixed with an excess of peptide C2 to form ligation product M2C2 that was purified by RP-HPLC (Fig. 5A). Ring-opening of the Thz was subsequently carried out on the product using methoxylamine to reveal the N-terminal Cys (Fig. 5B). The order of ring-opening and purification minimized reformation of Thz by trace amounts of aldehyde that copurify with the N2 peptide, which renders M2C2 inactive (see Materials and Methods). Peptide N2 was added directly to the mixture, and buffer conditions were adjusted to initiate ligation. Ligation was allowed to proceed for at least four days until no further product formation was observed (Fig. 5C). Free radical desulfurization45 was carried out directly on the crude ligation mixture to convert the ligation site Cys to the Ala found in the native H3 sequence. Desulfurization was monitored by MALDI-TOF MS (Fig. 5D). H3(K56ac,C110A) was purified by RP-HPLC (Fig. 5E) and the product lyophilized for subsequent refolding into nucleosomes. A typical ligation cycle produced 93 μg of H3(K56ac,C110A) at >95% purity from 500 μg of peptide M2 as the limiting reagent. This corresponds to an overall yield of 7% through all synthetic steps. This new scheme provides a reproducible 3-fold increased yield over the original synthetic pathway, allowing us to generate over 0.5 mg of the native histone suitable for detailed biophysical characterization. Moreover, the only limitation on preparing virtually any PTM or combination of PTMs on any histone would appear to be the ability to synthesize appropriately modified ligation peptides.
Fig. 5.
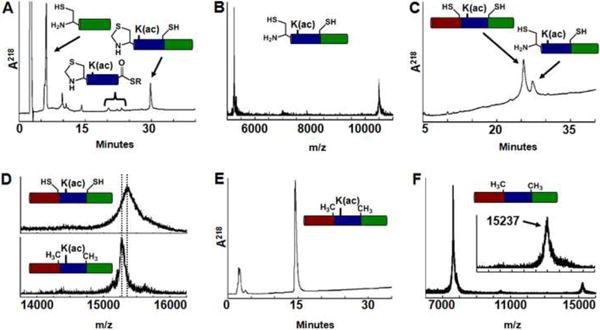
Sequential NCL to generate synthetic H3(K56ac,C110A). (A) RP-HPLC chromatogram of reaction mixture to generate first ligation product, residues 47-135 of H3(Thz47A,K56ac,A91C,C110A); gradient of 27-66% acetonitrile / 0.1% TFA. (B) MALDI-TOF analysis of first ligation product: m/z expected 10491, observed 10492. (C) RP-HPLC of the second ligation step to generate H3(A47C,K56ac,A91C,C110A); gradient of 41-59% acetonitrile / 0.1% TFA. (D) Top: MALDI-TOF analysis of synthetic H3(A47C,K56ac,A91C,C110A) prior to desulfurization, m/z expected 15342; observed 15338. Bottom: Final product H3(K56ac,C110A), m/z expected 15278; observed 15280. (E) RP-HPLC of synthetically generated H3(K56ac,C110A) with a 43-61% acetonitrile, 0.1% TFA gradient. (F) MALDI-TOF analysis of synthetic H3(C110A): m/z expected 15236 observed 15237. (Inset) Magnified view with scale set as in (D).
We repeated the sequential ligation using synthetic segment M3, bearing the unmodified K56 residue, to generate 120 μg of the chemically synthesized histone H3(C110A)syn (Fig. 5F). Nucleosomes containing this synthetic unmodified protein were directly compared to nucleosomes reconstituted with recombinantly expressed H3(C110A)rec to demonstrate that the synthetic process did not introduce any undesired modifications.
Acetylation of H3(K56) reduces DNA wrapping at the entry-exit region of the nucleosome
We examined the biophysical properties of H3(K56ac) and H3(K56Q) using the FRET system described above (Fig. 3A, 601-LexA-end). We determined the FRET efficiency at low ionic strength (0.5× TE with 1 mM Na+ from disodium-EDTA) for nucleosomes containing unmodified H3, H3(K56Q), H3(C110A)rec, H3(C110A)syn, H3(K56Q,C110A) and H3(K56ac,C110A) and at physiological ionic strength (0.5× TE with 75 mM or 130 mM NaCl) for nucleosomes containing H3(C110A)rec, H3(C110A)syn, H3(K56Q,C110A) and H3(K56ac,C110A) (Fig. 6, Table 2). We find that the FRET efficiency is reduced by about 15 to 20 percent for nucleosomes containing H3(K56ac,C110A), H3(K56Q) and H3(K56Q,C110A). In contrast, FRET efficiency is increased by 6% with H3(C110A)rec and unaltered for H3(C110A)syn with respect to unmodified H3 (Fig. 6C). These results indicate that H3(K56ac) increases the average distance between the DNA and the histone surface at the entry-exit region under low and physiological ionic strengths. In addition, H3(K56Q) appears to quantitatively mimic the effects of H3(K56ac) on the steady state structure and that this difference does not depend on ionic strength.
Fig. 6.
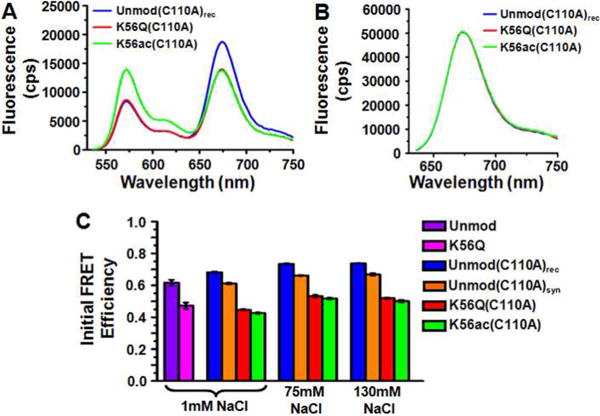
Fluorescence Resonance Energy Transfer (FRET) efficiency is reduced by H3(K56Q) and H3(K56ac). (A) Fluorescence emission spectra from nucleosomes containing unmodified H3(C110A) (blue), H3(K56Q,C110A) (red) and H3(K56ac,C110A) (blue) when excited at 510 nm (donor excitation) with 130 mM NaCl. (B) Fluorescence emission spectrum from nucleosomes in A when exited at 610 nm (acceptor excitation). (C) The FRET efficiency as determined by the (ratio)A method63 of nucleosomes containing unmodified H3 (purple) and H3(K56Q) (magenta) at 1 mM NaCl, and unmodified H3(C110A)rec(blue), unmodified H3(C110A)syn(orange), H3(K56Q,C110A) (red) and H3(K56ac,C110A) (green) at 1 mM, 75 mM and 130 mM NaCl. The error bars were determined from the standard deviation of three separate measurements.
Table 2.
The FRET efficiency without LexA (E0), the concentration of half saturation by LexA to the nucleosome (S0.5-nuc), the nucleosome site exposure equilibrium constant of the LexA target site (Keq), and the relative nucleosome site exposure equilibrium constant with respect to recombinant H3 or H3(C110A)rec (Keq-rel).
| 1mM Na+ | 75mM NaCl | 130mM NaCl | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| H3 histone | E0 | S0.5-nuc (nM) | Keq | Keq-rel | E0 | S0.5-nuc (nM) | Keq-rel | E0 | S0.5-nuc (nM) | Keq-rel |
| H3 | 0.62 ± 0.02 | 43 ± 3 | 0.0033 ± 0.0005 | 1 | - | - | - | - | - | - |
| H3(K56Q) | 0.47 ± 0.02 | 22 ± 2 | 0.0063 ± 0.0010 | 1.9 ± 0.4 | - | - | - | - | - | - |
| H3(C110A)rec | 0.68 ± 0.01 | 58 ± 6 | 0.0056 ± 0.0009 | 1 | 0.73 ± 0.01 | 3240± 181 | 1 | 0.74 ± 0.01 | 13122 ± 536 | 1 |
| H3(C110A)syn | 0.61 ± 0.01 | 62 ± 7 | 0.0052 ± 0.0008 | 0.9 ± 0.2 | 0.66 ± 0.01 | - | - | 0.67 ± 0.01 | 11849 ± 876 | 1.1 ± 0.2 |
| H3(K56Q, C110A) | 0.45 ± 0.01 | 32 ± 3 | 0.0102 ± 0.0014 | 1.8 ± 0.4 | 0.53 ± 0.01 | 1276 ± 150 | 2.5 ± 0.6 | 0.52 ± 0.01 | 5176 ± 524 | 2.5 ± 0.5 |
| H3(K56ac, C110A) | 0.43 ± 0.01 | 32 ± 3 | 0.0102 ± 0.0015 | 1.8 ± 0.4 | 0.52 ± 0.01 | 991 ±115 | 3.3 ± 0.7 | 0.50 ± 0.01 | 3975 ± 488 | 3.3 ± 0.7 |
Nucleosomes containing H3(K56ac) or H3(K56Q) showed a slight shift in electrophoretic mobility. Altered mobility could be explained by the increase in DNA unwrapping consistent with our FRET measurements; alternatively, it could be attributed to a shift in nucleosome position. We therefore determined the positions of nucleosomes containing H3(C110A)rec and H3(K56Q,C110A) by hydroxyl radical cleavage39 using FeBABE; this label did not alter the gel mobility of nucleosomes containing H3(C110A)rec or H3(K56Q,C110A) (Fig. 7B). We found that the cleavage pattern was indistinguishable between these nucleosomes (Fig. 7C, D). This indicates that the observed altered mobility and reduced FRET of nucleosomes containing H3(K56Q) and, by extension, H3(K56ac), is not due to nucleosome repositioning but rather due to increased DNA unwrapping.
Figure 7.
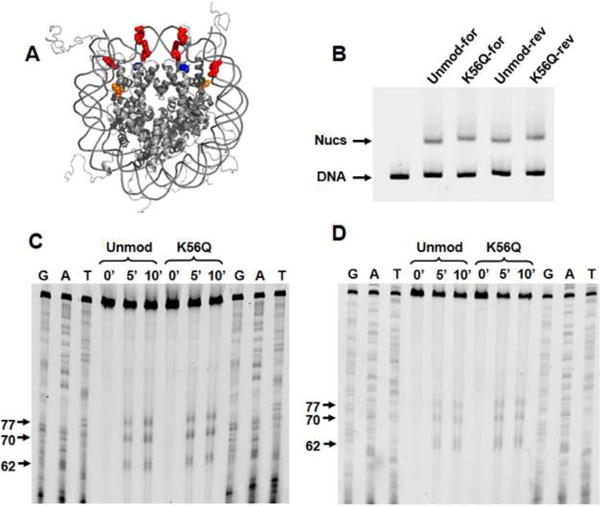
Nucleosome positioning is not influenced by K56Q. (A) The crystal structure of the nucleosome with H3(K56) shown in orange, H4(S47), which is replaced with a cysteine and labeled with FeBABE, shown in blue, and the bases that are cleaved by FeBABE shown in red. (B) EMSA of nucleosomes labeled with FeBABE at H4(S47C). Lane 1 contains the DNA substraite. Lanes 2 and 4 contain nucleosomes with H3(C110A)rec with the top and bottom DNA strand 5 prime labeled with cy3, respectively. Lanes 3 and 5 contain nucleosomes with H3(K56Q,C110A) with the top and bottom DNA strand 5 prime labeled with cy3, respectively. (C) and (D) Denaturing polyacrylimide gel electorphoresis of the nucleosomal DNA cleaved by FeBABE for 0, 5 and 10 minutes. Within each gel, lanes 1-3 and 10-12 contain sequencing tracks terminated with ddGTP,ddATP and ddTTP respectively, lanes 4-6 contain nucleosomes with H3(C110A)rec and lanes 7-9 contain nucleosomes with H3(K56Q,C110A). (C) and (D) are images of denaturing gels where the top and bottom DNA strands, respectively, are visualized by cy3 fluorescence.
Acetylation of H3(K56) facilitates protein binding within the nucleosome at low ionic strength
We initially determined the influence of H3(K56ac) and H3(K56Q) on DNA unwrapping and protein binding to a DNA target site buried within the nucleosome at low ionic strength since previous FRET studies of DNA unwrapping have been carried out under these conditions3; 15; 46-48. We performed LexA binding studies3 by detecting the reduction in FRET efficiency that is due to LexA binding to its target sequence, which is located within the nucleosome between base pairs 8-27 of the 147 base pair 601 nucleosome positioning sequence (Fig. 3A, 8A-C). We initially titrated LexA from 0 to 3 μM in the presence of 1.0 mM Na+ and find that at high concentrations of LexA the FRET efficiency reduces to approximately 0.2 (Fig. 8D). Such a non-zero FRET efficiency at high LexA concentrations is concordant with previous site accessibility measurements3. These results are consistent with the conclusion that unmodified nucleosomes and nucleosomes containing H3(K56ac) or H3(K56Q) are not disassembled by LexA binding.
Fig. 8.
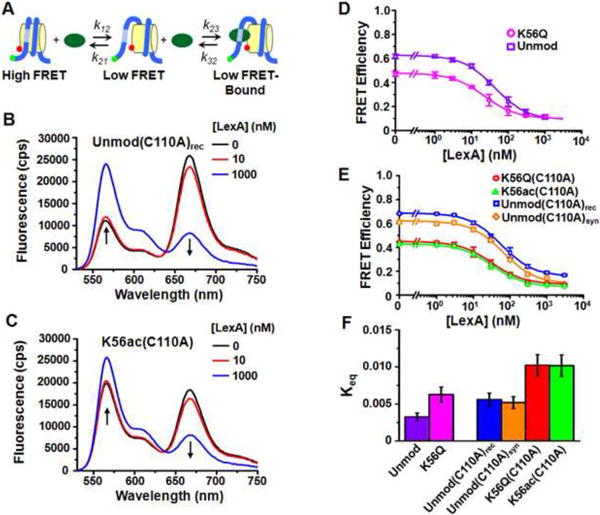
H3(K56Q) and H3(K56ac) increase the site exposure equilibrium constant which facilitates LexA binding within nucleosomes at 1 mM Na+. (A) A three state model for LexA binding to its target site within a nucleosome. (B) and (C) are fluorescence emission spectra at 0 nM (black), 10 nM (blue) and 1000 nM (red) of LexA with nucleosomes containing unmodified H3(C110A)rec or H3(K56ac,C110A), respectively. The samples were excited at 510 nm (donor excitation). (D) Energy transfer efficiency, as determined by the (ratio)A method, versus LexA concentration for nucleosomes containing unmodified H3 (purple) and H3(K56Q) (magenta). (E) Energy transfer efficiency, as determined by the (ratio)A method, versus LexA concentration for nucleosomes containing unmodified, recombinant H3(C110A)rec (blue), unmodified, synthetic H3(C110A)syn (orange), H3(K56Q,C110A) (red) and H3(K56ac,C110A) (green). Plots in (D) and (E) are the average of three LexA titrations and the error bars were determine from the standard deviation of the three measurements. The data were fit to a non-cooperative binding curve, which determines S0.5-nuc, the LexA concentration at which 50% of the nucleosomes are bound by LexA. (F) Equilibrium constants for site exposure for nuclesomes containing unmodified H3 (0.0033 ± 0.0005), H3(K56Q) (0.006 ± 0.001), unmodified H3(C110A)rec (0.0055 ± 0.0009), unmodified H3(C110A)syn (0.0052 ± 0.0008), H3(K56Q,C110A) (0.010 ± 0.002) and H3(K56ac) (0.010 ± 0.002). The equilibrium constants were determined from the ratio: Keq = S0.5-DNA / S0.5-nuc (see Methods for details).
The FRET efficiencies in the presence of LexA were fit to a non-cooperative binding curve and the concentration of half saturation by LexA (S0.5-nuc) was determined for nucleosomes containing unmodified H3, H3(K56Q), H3(C110A)rec, H3(C110A)syn, H3(K56Q,C110A) and H3(K56ac,C110A) (Fig. 8D-E, Table 2). The concentration of half saturation by LexA binding to its site within naked DNA was determined by gel shift analysis3. We used two separate preparations of LexA with S0.5-DNA of 0.14 ± 0.02 nM and 0.32 ± 0.04 (see Supplemental Information).
We determined the site exposure equilibrium constant, Keq, from the half saturation value of LexA binding to its target sequence within the nucleosome and to naked DNA, since S0.5-nuc = S0.5-DNA / Keq, in the limit that Keq is much less than 1 (See Materials and Methods for details). From this equation, we determined the equilibrium constant for site exposure for nucleosomes containing unmodified H3, H3(K56Q), H3(C110A)rec, H3(C110A)syn, H3(K56Q,C110A) and H3(K56ac,C110A) (Fig. 8F, Table 2) at low ionic strength (1 mM Na+).
We confirmed that the reduction in FRET efficiency is due to LexA binding by Electrophoretic Mobility Shift Assays (EMSA) (see Supplemental Information). We find, as previously reported3, that the LexA-nucleosome complex is not stable under electrophoresis conditions. Therefore, we used glutaraldehyde to crosslink the nucleosome/LexA complex to prevent dissociation. We find that S0.5-nuc as determined by EMSA is consistent with the measured reduction of FRET efficiencies for nucleosomes containing H3(C110A)rec, H3(K56Q,C110A) and H3(K56ac,C110A). Furthermore, the increase in Keq by H3(K56Q,C110A) and H3(K56ac,C110A) measured by EMSA is consistent with the increased Keq determined by FRET efficiency measurements.
We controlled for the effects of nonspecific LexA binding on FRET efficiency by determining the FRET efficiency of nucleosomes that did not contain the LexA target sequence (Fig 3A, 601-end). We find no decrease in FRET efficiency in the presence of up to 1 μM LexA (see Supplemental Information). This concentration of LexA fully reduces the FRET efficiency of nucleosomes that contain the LexA target sequence (Fig. 8D-E). These results confirm that the reduction in FRET efficiency is due to LexA binding to its target sequence within the nucleosome.
The change in the site exposure equilibrium of modified nucleosomes relative to unmodified nucleosomes is equal to the change in probability that LexA can bind to its site that extends 27 base pairs into the nucleosome. H3(K56ac,C110A) increases this value by 1.8 ± 0.4 times. The H3(K56Q) substitution, which has been used in numerous genetic studies as a mimic of H3(K56ac), increases the probability of LexA binding by 1.8 ± 0.4 and 1.9 ± 0.5 with and without H3(C110A), respectively. This demonstrates that the H3(C110A) mutation does not alter the influence of H3(K56Q) on nucleosomal DNA unwrapping, which is consistent with FRET efficiency measurements in the absence of LexA. H3(C110A) does appear to modestly increase the absolute value of DNA site accessibility. However, by comparing H3(K56ac,C110A) and H3(K56Q,C110A) to H3(C110A)rec and H3(C110A)syn, we control for this effect.
Acetylation of H3(K56) facilitates protein binding by increasing the probability that the nucleosome is partially unwrapped
The increased protein accessibility induced by H3(K56ac) could result from changes in unwrapping, nucleosome DNA repositioning, or both. To resolve these possibilities, we determined the position of nucleosomes containing H3(C110A)rec and H3(K56Q,C110A) in the presence of 1 μM LexA by hydroxyl radical mapping. We found that nucleosomes in the presence of 1 μM LexA retained an identical cleavage pattern to nucleosomes without LexA as measured by denaturing PAGE (see Supplemental Information). LexA at this concentration of 1 μM is bound to its target sequence within nucleosomes as measured by FRET efficiency (Fig. 8) and EMSA (Supplemental Information).
In addition, we carried out FRET efficiency studies with nucleosomes that were labeled at the 80th base pair with Cy3 (Fig. 9A). Based on the nucleosome crystal structure 49, the distance between the Cy3 molecule and the nearest Cy5 molecule is about 2.3 nm, which converts to a FRET efficiency of 0.99; we anticipate that this efficiency would be slightly reduced due to the 6-carbon linker used to attach cy3 to the thymine base. If the LexA site were exposed only by repositioning, the distance between Cy3 and the nearest Cy5 would increase to 6.2 nm, which converts to a FRET efficiency of 0.45. We find that the FRET efficiency remains constant at 0.8 for nucleosomes containing unmodified H3, H3(K56Q), H3(C110A)rec, H3(K56Q,C110A) and H3(K56ac,C110A) under conditions consistent with full occupancy of the LexA binding site (Fig. 9).
Fig. 9.
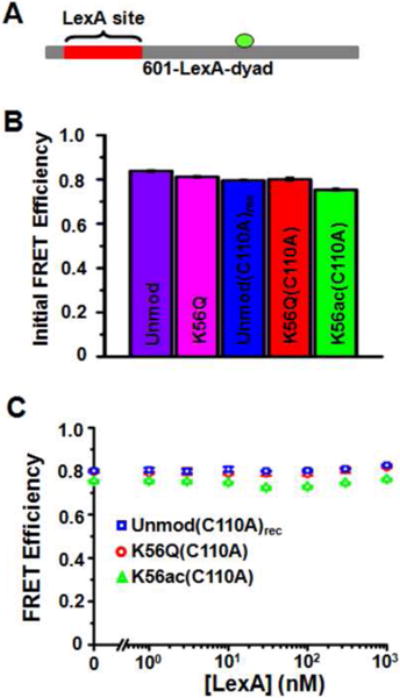
(A) The 147 base pair DNA molecule, 601-LexA-dyad, contains the 601 positioning sequence with LexA binding site at base pairs 8-27 and a cy3 fluorophore attached to 80th base pair of the DNA molecule. (B) The FRET efficiency as determined by the (ratio)A method of nucleosomes containing unmodified H3, H3(K56Q), H3(C110A)rec, H3(K56Q,C110A) and H3(K56ac,C110A). Each nucleosome contained the 601-LexA-dyad DNA molecule. The error bars were determined from the standard deviation of three separate measurements. (C) The energy transfer efficiency determined by the (ratio)A method versus LexA concentration for H3(C110A)rec, H3(K56Q,C110A) and H3(K56ac,C110A) nucleosomes containing the 601-dyad DNA construct (Fig. 3A). Each plot is the average of at least three LexA titrations and the error bars were determine from the standard deviation of the three measurements.
The combination of the FRET studies and hydroxyl radical mapping suggests that H3(K56Q) and H3(K56ac) do not increase DNA site accessibility via a nucleosome repositioning model. Instead, K56 acetylation and its mimic appear to increase LexA accessibility by increasing the probability that the nucleosome is partially unwrapped.
Acetylation of H3(K56) facilitates accessibility to DNA within nucleosomes at physiological ionic strength
Our initial studies were carried out at low ionic strength, but the physiologically relevant concentration of monovalent ions is 130 mM to 150 mM. Therefore, we carried out LexA binding studies with fluorophore labeled nucleosomes at both 75 mM and 130 mM NaCl as described above for 1 mM Na+. We determined at 75 mM and 130 mM NaCl the S0.5-nuc for H3(C110A)rec, H3(C110A)syn, H3(K56Q,C110A) and H3(K56ac,C110A) (Fig. 10A-B, Table 2). We were unable to determine the S0.5-DNA by EMSA because of the increase in NaCl. Therefore, we determined the Keq of H3(K56Q,C110A) and H3(K56ac,C110A) nucleosomes relative to unmodified nucleosomes. We find at 75 mM, Keq-H3(K56ac) / Keq-Unmod = 3.3 ± 0.4 and Keq-H3(K56Q) / Keq-Unmod = 2.5 ± 0.3, while at 130 mM, Keq-H3(K56ac) / Keq-Unmod = 3.3 ± 0.4 and Keq-H3(K56Q) / Keq-Unmod = 2.5 ± 0.3 (Figure 10C, Table 2).
Fig. 10.
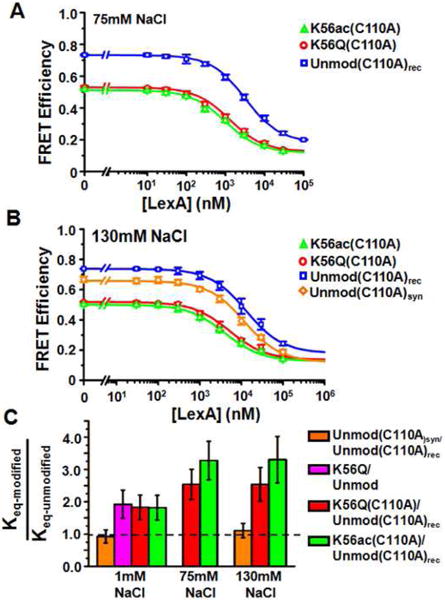
H3(K56ac) and H3(K56Q) enhances LexA binding to its DNA target sequence within nucleosomes at physiological ionic strength. (A) and (B) Energy transfer efficiency in the presence of 75 mM and 130 mM NaCl, respectively, as determined by the (ratio)A method, versus LexA concentration for nucleosomes containing unmodified and recombinant H3(C110A)rec (blue), unmodified and synthetic H3(C110A)syn (orange), H3(K56Q,C110A) (red), and H3(K56ac,C110A) (green). Plots in (A) and (B) are the average of three LexA titrations and the error bars were determine from the standard deviation of the three measurements. The data were fit to a non-cooperative binding curve, which determines S0.5-nuc, the LexA concentration at which 50% of the nucleosomes are bound by LexA.. (F) Equilibrium constants of H3(C110A)syn relative to H3(C110A)rec (0.93 ± 0.20), H3(K56Q) relative to H3 (1.9 ± 0.4), H3(K56Q,C110A) relative to H3(C110A)rec (1.8 ± 0.4) and H3(K56ac,C110A) relative to H3(C110A)rec (1.8 ± 0.4) at 1 mM Na+; H3(K56Q,C110A) relative to H3(C110A)rec (2.5 ± 0.3), and H3(K56ac,C110A) relative to H3(C110A)rec (3.3 ± 0.4) at 75 mM NaCl; synthetic H3(C110A) relative to H3(C110A)rec (1.11 ± 0.22), H3(K56Q,C110A) relative to H3(C110A)rec (2.5 ± 0.3), and H3(K56ac,C110A) relative to H3(C110A)rec (3.3 ± 0.4) at 130 mM NaCl.
These results imply that at the physiological ionic strength of 130 mM, H3(K56ac) increases DNA unwrapping fluctuations that expose the LexA target site 3-fold, which in turn results in a 3-fold increase in LexA binding to its target site. Furthermore, we find that H3(K56Q) increases DNA site exposure similarly to H3(K56ac) at physiological ionic strength, which suggests that H3(K56Q) is a good acetylation mimic of H3(K56ac) for in vivo studies.
Discussion
PTMs of histones occur throughout the protein sequence with multiple disparate types of modification often detected on a single histone 9; 50. Current methods to prepare modified histones are limited to the site-specific introduction of a single type of modification within a histone protein 15; 20; 21, or a variety of modifications within a localized region of the histone protein 12; 14; 16-18; 51. We have established a method for incorporating one or several PTMs into a histone protein by sequential NCL, targeting the common Ala residue as a ligation junction. The procedure thus generates a nativelike histone containing only the PTM(s) of interest with no non-native residues other than the well-studied C110A. In other studies, the introduction of select non-native Cys residues through ligation in the unstructured nucleosome tail has had a negligible effect on nucleosome dynamics 12; 16; 51. Similarly, desulfurization has been coupled with ligation in the context of introducing modifications into N-terminal and C-terminal tails of semi-synthetic histones17-19. Our work demonstrates that the semi-conservative introduction of Cys residues into the nucleosome core can perturb DNA wrapping. In the context of synthetic histones, this effect can be mitigated by conversion to a native Ala residue. However, the impact of these substitutions must be considered when interpreting biochemical or biophysical measurements which require the introduction of non-native Cys sites throughout the nucleosome.
Although our initial study was restricted to the synthesis and characterization of unmodified H3 and H3(K56ac), this method is limited only by the synthesis of individual peptide segments and would allow for the introduction of PTMs throughout histone H3. As H3 is the largest of the core histone proteins, our success suggests that the total synthesis strategy may be applied to all of the histone proteins, including rare variants. These methods should therefore be useful in determining the function and biophysical properties associated with the voluminous numbers of cellular PTMs.
Using sequential NCL methodology we have engineered and characterized nucleosomes containing K56ac within histone H3. Our measurements of nucleosomes containing K56ac are in agreement with the results of Neumann et al., which show that K56ac increases the population of nucleosomes that are partially unwrapped at the DNA entry-exit region by up to 7 times15 at low ionic strength We extend these studies to demonstrate that DNA unwrapping in the DNA entry-exit region facilitates protein binding 27 base pairs into the nucleosome by a factor of 1.8 at low ionic conditions (0.5× TE). In addition, we determined the influence of K56ac on DNA unwrapping and protein binding within the nucleosome at physiological ionic strength (130 mM NaCl). We find that this enhances the influence of K56ac on DNA unwrapping such that protein binding is increased by 3.3 times to 27 base pairs into the nucleosome. These studies are consistent with enhanced accessibility to transcription factors and DNA repair components in chromatin regions containing K56ac within histone H3.
Interestingly, Neumann et al. found that the FRET distribution was not altered 27 base pairs into the nucleosomes with K56ac. This appears to be in contrast to our result that K56ac facilitates protein binding to a site that extends 27 base pairs into the nucleosome. We can understand this apparent discrepancy by considering the previously reported cooperativity of adjacent DNA target sites within a nucleosome5; 6. Protein binding to the outer DNA target site within the nucleosome facilitates binding to the inner target site. In our case, K56ac appears to act as the outer adjacent site that facilitates LexA binding to its target site within the nucleosome.
The acetylation of H3(K56) has also been shown to be important for transcriptional regulation30; 34; 35; 52. For example, K56ac within H3 has been implicated in the transcriptional regulation of the HTA1 and SUC2 genes30. Interestingly, studies carried out using ChIP have demonstrated that the occupancy of SWI/SNF chromatin remodeler Snf5 is reduced 2-3 fold by the H3(K56R) substitution that mimics unacetylated lysine in the promoter region and the coding region of HTA1, as well as in the SUC2 gene30. Such a reduction in Snf5 occupancy could be explained by the 3-fold reduction we observe in site accessibility when nucleosomes are not acetylated at H3(K56). However, it is also possible that these changes in occupancy result from indirect effects of acetylation, since SWI/SNF is known to contact the nucleosome core particle over a large surface area rather than through a specific interaction with H3(K56)53. For example, we find that acetylation loosens nucleosomal DNA in the entry-exit region, which could influence the formaldehyde crosslinking required to detect SWI/SNF binding in this study.
Our quantitative measurements of LexA protein accessibility by K56ac and K56Q are in agreement with multiple studies. During DNA replication, nucleosomes are assembled with H3(K56ac)30. Polymerase misincorporation errors and DNA lesions result in mismatched nucleotides, replication fork collapse and DNA double strand breaks that must be repaired to ensure genomic stability54. Deletion of rtt109, which acetylates H3(K56), or mutation of H3(K56) to Arg [H3(K56R)] that mimics unacetylated H3, both cause large defects in post-replication DNA repair32 and lead to genomic instability33. Recently we have shown that the DNA mismatch recognition complex hMSH2-hMSH6 can remodel nucleosomes near a mismatch and that this activity is enhanced 2-fold for nucleosomes containing H3(K56Q)55. This result is consistent with our observation that H3(K56Q)-containing nucleosomes increase DNA accessibility by a factor of 3 at physiological ionic strength. Together, these observations suggest that increased DNA site accessibility near the DNA entry-exit region associated with H3(K56ac) facilitates nucleosome remodeling by hMSH2-hMSH6.
Numerous studies use the H3(K56Q) substitution to mimic lysine acetylation, including a number of genetic studies that found phenotypes in both gene expression and DNA repair31; 35. Our study suggests that similar phenotypes will result in cells that are constitutively acetylated at H3(K56). Recently, the crystal structure of nucleosomes containing H3(K56Q) was reported by Watanabe et al.36 They found that K56Q did not impact the structure of the full wrapped state of the nucleosome, which is consistent with a role for K56 acetylation in nucleosome dynamics. They also reported that H3(K56Q) did not influence compaction of nucleosome arrays regardless of nucleosome density. However, H3(K56Q) did dramatically reduce interactions between multiple arrays of nucleosomes and thus H3(K56ac) may function to reduce chromatin-chromatin interactions to help keep nucleosome-free regions accessible for DNA replication and repair. Their studies relied on the assumption that H3(K56Q) accurately mimics H3(K56ac). Our studies, which demonstrate the H3(K56Q) mimics H3(K56ac), indicates that this assumption is correct.
In conclusion, our studies demonstrate the power of sequential NCL to engineer fully-synthetic histones with precise PTMs that can be combined with quantitative biophysical methodologies to determine the function of these PTMs in the context of nucleosomes. Ongoing studies will test the function of multiple PTMs found in biologically relevant processes.
Materials and Methods
Peptide synthesis
Peptides (Table 1) were synthesized manually using standard Boc-N-α protection strategies and in situ neutralization protocols56 utilizing HBTU activation. C-terminal peptides C1 and C2 were synthesized on pre-loaded Boc-Ala-PAM resin (Novabiochem). Thioester peptides N1, N2, M1 and M2 were synthesized on MBHA resin with a mercaptopropionamide linker to generate the C-terminal thioester moiety necessary for subsequent ligation57. Acetylated lysine was incorporated as the commercially available Boc-protected derivative (Novabiochem, N-α-t.-Boc-acetyllysine), and the protected N-terminal Cys was incorporated as thiaproline (Boc-L-thiazolidine-4-carboxylic acid, Bachem). Peptides were cleaved from the solid support with standard anhydrous hydrogen fluoride (HF) cleavage conditions utilizing p-cresol as scavenger. Following synthesis and purification, all peptide purities were assessed by RP-HPLC as >95% with the exception of peptide M1, which contained a mixture of Met and Met(O) species.
Synthesis of H3(R40C,K56ac,S96C,C110A)
Synthetic H3(R40C,K56ac,S96C,C110A) proteins were generated by sequential native chemical ligation (Fig. 1 A). In the first step of the ligation, peptide M1 propionamide thioester was resuspended with 5-10 fold molar excess of peptide C1 in 100 mM HEPES pH 7.5, 1M NaCl, 50 mM MESNA and 6 M Guanidine (Gdn) HCl and reacted for 2 days at 25°C. Upon completion, direct addition of 500 mM methoxylamine HCl to the ligation mixture generated the free N-terminal Cys by unmasking the Thz24. Complete conversion to the desired terminal Cys was observed within 6 hours. The M1C1 product was purified to >95% by RP-HPLC over a gradient of 22.5-50% isopropanol / 0.1%TFA with a Vydac C4 column at 45°C (Fig. 2A) and the product identity was confirmed by MALDI-TOF MS.
Purified ligation product M1C1 was resuspended with a 20-fold molar excess of peptide N1 in 100 mM HEPES pH 7.5, 1 M NaCl, 50 mM MESNA, 10 mM TCEP and 6 M Gdn-HCl. The ligation mixture was nutated for 24 hours at 25°C to generate the site specifically modified H3(R40C,K56ac,S96C,C110A). The final product was purified by RP-HPLC with a step gradient of acetonitrile / 0.1% TFA on a Supelco Widebore C18 column at 25°C as follows: 11-16% over 5 min, 16-43% over 5 min and 43-66% over 30 min. Fractions identified by MALDI-TOF MS to contain full length H3(R40C,K56ac,S96C) with and without the Met(O) species were pooled and lyophilized.
Reduction of Met(O) to Met in H3(R40C,K56ac,S96C,C110A) was accomplished by suspending the lyophilized protein in 200μL TFA with 25μL dimethylsulfide and 0.045 M sodium iodide (NaI)58. Reduction was allowed to proceed for 1 hr on ice until complete as monitored by MALDI-TOF MS (Fig. 2C). Reduced protein was precipitated and washed with cold anhydrous diethyl ether, then purified by RP-HPLC on a Supelco Widebore C18 column with a gradient of 43-66% acetonitrile / 0.1% TFA (Fig. 2D). MALDI-TOF MS confirmed that the Met(O) species was absent in the collected fraction. The total synthesis of H3(R40C,K56ac,S96C,C110A) as described provided an overall ligation yield of 2% (measured by UV quantification).
Synthesis of H3(A47C,K56ac,A91C,C110A)
Sequential NCL was employed to generate synthetic H3(A47C,K56ac,A91C,C110A) (Fig. 1). Thioester peptide M2 (typically 0.5 mg) was resuspended with 2.5 molar excess of C2 in 100 mM phosphate pH 7.5, 1 M NaCl, 60 mM 4-mercaptophenylacetic acid (MPAA), 20 mM TCEP and 50% trifloroethanol (TFE) and reacted for 48 hours at 25°C (Fig. 5A). We determined that if ring opening was carried out prior to the first purification step, we observed back-formation of Thz-M2C2 over the course of the second ligation reaction. We attribute this to copurification of trace amounts of formaldehyde generated in cleavage of a His(Bom) side chain protecting group in peptide N2. This back ring closure was minimized by the presence of methoxylamine in the ligation reaction. We therefore purified Thz-M2C2 prior to the ring-opening step. While the ligation product partially precipitated under these buffer conditions, reaction in a phosphate/Gdn buffer prevented this problem and was used in future ligation iterations. Product was resusupended in 100 mM phosphate pH 7.5, 1 M NaCl, 6 M Gdn HCl with 60 mM TCEP, and purified by RP-HPLC on a Supelco Widebore C18 column with a 32-59% acetonitrile / 0.1% TFA gradient.
Lyophilized ligation product Thz-M2C2 was resuspended with 100 mM phosphate pH 7.5, 1 M NaCl, 6 M Gdn-HCl, 20 mM TCEP. 350 mM methoxylamine hydrochloride was added to convert the N-terminal Thz to Cys. Deprotection was allowed to proceed for 6 hrs at 25°C (Fig. 5B). The mixture was adjusted to pH 7.5 with NaOH and then MESNA was added to the mixture to a final concentration of 100 mM. The ligation was initiated with the addition of a 5-fold molar excess of peptide N2. Ligations were monitored by RP-HPLC and SDS-PAGE for 4 to 6 days at 25°C until no additional product formation was observed (Fig. 5C). We attribute the slow product formation to the C-terminal residue of peptide N2 (Val), which is known to result in slow ligation59. The crude ligation mixture was carried forward for desulfurization to reveal the final H3(K56ac,C110A) protein.
Desulfurization of H3(A47C,K56ac,A91C,C110A) (N2M2C2) to yield H3(K56ac,C110A)
The H3(A47C,K56ac,A91C,C110A) ligation mixture was directly desulfurized prior to purification using free radical desulfurization conditions45. The mixture was adjusted to final concentrations of 50 mM Phosphate pH 7.5, 500 mM NaCl, 0.3 M TCEP, 100 mM MESNA, 5 M Gdn-HCl and the sample was sparged with argon for 30 min. Desulfurization was initiated with the addition of VA-044US (Wako Chemical) to a final concentration of 10 mM at 42°C, and the reaction was allowed to proceed until complete as monitored by MALDI-TOF analysis (minimum of 3 hrs; Fig. 5D). The final desulfurized product H3(K56ac,C110A) was purified by RP-HPLC with a gradient of 41-59% acetonitrile / 0.1% TFA on a Supelco Widebore C18 column (Fig. 5E). A typical ligation began with 0.5 mg of limiting peptide M2. The ligation and desulfurization procedures described yielded 93 μg of final product H3(K56ac,C110A) as determined by UV quantification on a NanoDrop 1000 (Thermo Scientific): for an overall synthetic yield of 7%. We attribute this increase in synthetic yield to the use of only two chromatography steps through our ligation pathway: purification of Thz-M2C2, and of the final desulfurized product H3(K56ac,C110A). If Met(O) species was observed in the full-length native H3-K56ac, reduction was carried out on the purified product without the need for further purification.
Total Synthesis of H3(C110A)
Peptide M3, the unmodified variant of peptide M2, was synthesized and used for the total synthesis of H3(C110A)syn using the preceding conditions with the following changes. Following Thz deprotection of purified ligation product Thz-M3C2, the pH was adjusted to 7.5 and the ligation was initiated by addition of 75 mM MPAA and a 20-fold molar excess of peptide N2. Ligation progress was monitored for three days and the resulting reaction mixture was dialyzed to remove MPAA prior to desulfurization as described. Fully synthetic H3(C110A)syn was purified by RP-HPLC to yield 120 μg as determined by UV quantitation, and the protein identity was confirmed by MALDI-TOF. (Fig. 5F)
Preparation of DNA constructs
The DNA molecules 601-end, 601-LexA-end (Fig. 3A), and 601-LexA-dyad (Fig. 10A) were prepared by PCR with Cy3 labeled oligonucleotides from plasmid containing the 601 positioning sequence with or without a LexA binding site at bases 8-273. Oligonucleotides were labeled with a Cy3 NHS ester (GE healthcare) at an amino group attached at the 5‘-end or to a modified internal thymine and then purified by RPLC on a C18 Vydac column. The oligos used to amplify 601-LexA-end were Cy3-CTGGAGATACTGTATGAGCATAC AGTACAATTGGTC and ACAGGATGTATATATCTGACACGTGCCTGGAGACTA; 601-LexA-dyad were CTGGAGATACTGTATGAGCATACAGTACAATTGGTCGTAGCA and ACAGGATGTATATATCTGACACGTGCCTGGAGACTAGGGAGTAATCCCCTTGGCG GTTAAAACGCGG(T-Cy3)GGACA; 601-end were Cy3-CTGGAGAATCCCGGTGCCG and TCAGGATGTATATATCTGACACGTGCCTGGAGACTA. Following PCR amplification, each DNA molecule was purified by HPLC with a Gen-Pak Fax column (Waters).
Preparation of histone octamers and LexA protein
Recombinant histones were expressed and purified as previously described60. Plasmids encoding histones H2A(K119C), H2B, H3, and H4 were generous gifts from Dr. Karolin Luger (Colorado State University) and Dr. Jonathan Widom (Northwestern University). Mutations H3(R40C), H3(S96C), H3(C110A), H3(K56Q), and H4(S47C) were introduced by site-directed mutagenesis (Stratagene). H2A(K119C) was labeled before or after histone octamer refolding with Cy5-maleamide (GE Healthcare). We achieved labeling efficiency of 75%-90% as determined by mass spectrometry and UV absorption. Histones: H2A(K119C), H2B, H4 and either H3, H3(C110A)rec, H3(C110A)syn, H3(K56Q), H3(K56Q,C110A) or H3(K56ac,C110A) were combined at equal molar ratios and refolded as previously described3. The purity of each octamer was confirmed by SDS-PAGE and mass spectrometry. LexA protein was expressed and purified from the pJWL288 plasmid (gift from Dr. Jonathan Widom) as previously described61.
H2A histone labeling
H2A was labeled with Cy5 before octamer refolding for octamers that contained H3(C110), while H2A was labeled either before or after refolding for octamers that contained H3(C110A). We found the labeling method did not influence our FRET measurements. H2A was labeled before octamer refolding by first resuspending H2A(K119C) to 1.2 mg/ml in 1.5 M Gdn-HCl, 800 mM HEPES, pH 7.1 and purging under Argon atmosphere with stirring for 1 hr at 25°C. TCEP pH 7.1 was added to 0.7 mM final concentration and incubated 20 min at 25°C with stirring under Argon. Cy5-maleamide (GE Healthcare) was resuspended to 7 mg/ml in anhydrous DMF and added dropwise with stirring to 1.1 mg/ml final concentration. The reaction was allowed to proceed for 5 hrs at 25°C with stirring under Argon before quenching with 10 mM dithiothreitol (DTT). Unreacted dye was removed from conjugated protein on a Sephadex G-25 column (Amersham) at 1 ml/min equilibrated with TU1000 buffer (6M Urea, 1M NaCl, 10mM Tris pH 9, 1mM BME). Purified fractions were dialyzed extensively against 2mM BME before lyophilization.
H2A was Cy5 labeled following octamer refolding by resuspending purified histone octamer containing H2A(K119C) to 1mg/ml in 2M NaCl, 200mM HEPES pH 7.1, 1mM EDTA and then purging under argon atmosphere without stirring for 1 hr at 4°C. TCEP pH 7.1 was added to 0.7mM final concentration and incubated under argon atmosphere for 20 min at 4°C. Cy5-maleamide (GE Healthcare) was resuspended to 2.5mg/ml in anhydrous DMF and added dropwise with thorough mixing to 0.35mg/ml final concentration. The reaction was allowed to proceed for 2 hrs at 25°C on a shaker rotisserie and then transferred to 4°C overnight before quenching with 10mM DTT. Unreacted dye was removed by sucrose gradient purification of reconstituted nucleosome (see below).
Nucleosome preparation
Nucleosomes were reconstituted by salt double dialysis62 with 7μg of DNA and 5μg of histone octamer (HO). The DNA and HO were mixed in 50 μl of 0.5× TE pH 8.0, 2 M NaCl and 1 mM BZA (benzamidine). The sample was loaded into an engineered 50 μl dialysis chamber which was placed in a large dialysis tube with 80 ml of 0.5× TE pH 8.0, 2 M NaCl and 1 mM BZA. The large tube was extensively dialyzed against 0.5× TE pH 8.0 with 1 mM BZA. The 50 μl sample was extracted from the dialysis button and purified by sucrose gradient centrifugation (Fig. 3D).
Mapping nucleosome positions with hydroxyl radical cleavage
Nucleosome positions were mapped using Fe(III) (s)-1-(p-bromoacetamidobenzyl) ethylenediamine tetraacetic acid (FeBABE protein cutting reagent, Thermo Scientific) conjugated to H4(S47C). Histones: H2A, H2B, H4(S47C) and either H3(C110A)rec or H3(K56Q,C110A) were combined at equal molar ratios and refolded as previously described3. Histone octamer (HO) was extensively dialyzed against “labeling buffer” (2M NaCl, 30mM HEPES pH 8.2, 5% glycerol, 4mM EDTA). For FeBABE conjugation, HO was resuspended to 1mg/ml in labeling buffer and purged under argon atmosphere without stirring for 1hr at 4°C. FeBABE was resuspended to 5mg/ml in degassed labeling buffer and added dropwise to the HO with thorough mixing to 0.8mg/ml final concentration. The reaction was allowed to proceed for 2 hrs at 25°C on a shaker rotisserie and then extensively dialyzed against 2M NaCl, 50mM Tris pH 8.2, 50% glycerol, 0.1mM EDTA at 4°C to removed unconjugated FeBABE.
Nucleosomes containing 601-LexA with Cy3 on the 5′ end of the forward or reverse strand were reconstituted by salt double dialysis62 as previously described (Fig. 7B). To perform hydroxyl radical mapping, nucleosomes were resuspended to 25nM on ice in degassed 20mM Tris pH 7.5, 0.1mM EDTA, 10% glycerol. Then 40mM L-ascorbic acid in degassed 20mM Tris pH 7.5, 10mM EDTA and 80mM hydrogen peroxide in degassed 20mM Tris pH 7.5, 10mM EDTA were added in quick succession to the nucleosomes with thorough mixing to 4mM and 8mM final concentration, respectively. The reaction was allowed to proceed for 10 and 20 minutes before transferring 7ul of reaction mixture to 3ul of 1.3M Tris pH 7.5. Samples were mixed with and equal volume of formamide and resolved by 12% denaturing PAGE in 7 m urea and 1 × TBE. The sequence markers were prepared with a SequiTherm Excel II DNA sequencing kit (Epicenter) using Cy3-labeled primers, a 601-LexA DNA template, and either ddGTP, ddATP or ddTTP. Results were imaged by a Typhoon 8600 variable mode imager (GE Healthcare) (Fig. 7).
Fluorescence Resonance Energy Transfer (FRET) efficiency measurements
FRET efficiency measurements were determined by the (ratio)A method as previously described63. The fluorescence emission spectra were measured at 25°C with a Fluoromax-3 (Horiba) photon counting steady-state fluorometer. Cy3/Cy5 labeled nucleosomes at 5 nM were excited at 510nm and at 610 nm, while the emission spectra were collected from 530-750nm and 630-750nm, respectively. The FRET efficiencies (E) were measured from acceptor emission using the (ratio)A equation: E = 2[(εA(u′′)FA(u′)/FA(u′′) − εA(u′)] / [(εD(u′) d+], where u′ = 510nm for donor (D) excitation and u′′ = 610nm for director acceptor (A) excitation. The prefactor of 2 reflects the presence of 2 acceptor molecules per donor molecule. FA(u′) is the is the fluorescence emission of A after subtraction of overlapping D emission when excited at 510nm. FA (u′′) is the fluorescence emission of A when excited at 610nm. εD(u′), εA(u′), and εA(u′′) are the molar extinction coeffieients of D and A at u′ and u′′. d+ is the fractional labeling of D, which is 1.
Site accessibility equilibrium measurements
The equilibrium constants for site accessibility were determined from the reduction in FRET efficiency as LexA binds to its target site buried within the nucleosome (Fig. 4 and 8)3. LexA was titrated from 0 to 3 μM with 5 nM Cy3/Cy5 labeled nucleosomes in 0.5× TE. The FRET efficiency was determined by the (ratio)A method in triplicate for each LexA concentration. The average FRET efficiency vs. LexA concentration was fit to a non-cooperative binding isotherm: E = EF + (E0 − EF)/(1 + [LexA]/S0.5), where E is the FRET efficiency, E0 is the FRET efficiency without LexA, EF is the FRET efficiency at high LexA concentration and S0.5-nuc is the LexA concentration at which the FRET efficiency has been reduced by half, i.e. E = (E0 + EF)/2. The equilibrium constant, Keq, was determined from Keq = S0.5-DNA / S0.5-nuc, which is true for the 3 state model (Fig. 8A) when S0.5-DNA ≪ S0.5-nuc, as is the case here. S0.5-DNA is the LexA concentration at which its target site within naked DNA is 50% bound by LexA and was determined by gel shift on a polyacrylimide gel (See Supplemental Information).
Supplementary Material
Acknowledgments
We thank Richard Fishel for extensive discussions and careful editing of the manuscript. We thank Jonathan Widom and Karolin Luger for the X. laevis histone and LexA expression vectors. We are grateful to Dongping Zhong for access to a Fluoromax-3, to Karin Musier-Forsyth for access to a Typhoon Trio fluorescence scanner and a fluorescence plate reader, and to Ross Dalbey for access to a table top ultracentrifuge. We gratefully acknowledge support from an American Heart Association Predoctoral Fellowship 0815460D (JAN); NIH GM083055 (MGP and JJO); NSF CAREER MCB0845696 (JJO), and a Career Award in the Basic Biomedical Sciences from the Burroughs Wellcome Fund (MGP).
Abbreviations
- BZA
benzamidine
- EDTA
ethylenediaminetetraacetic acid
- EMSA
electromobility shift assay
- EPL
expressed protein ligation
- FRET
fluorescence resonance energy transfer
- Gdn-HCl
guanidinium hydrochloride
- H3(C110A)rec
recombinant H3(C110A)
- H3(C110A)syn
synthetic H3(C110A)
- HO
Histone Octamer
- MESNA
sodium mercaptoethanesulfonate
- MPAA
4-mercaptophenylacetic acid
- NCL
native chemical ligation
- PTM
post-translational modification
- RP-HPLC
reverse phase high performance liquid chromatography
- TCEP
20 mM tris(2-carboxyethyl)phosphine
- Thz
thiazolidine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Van Holde KE. Chromatin. Springer-Verlag; New York: 1989. [Google Scholar]
- 2.Polach KJ, Widom J. Mechanism of protein access to specific DNA sequences in chromatin: a dynamic equilibrium model for gene regulation. J Mol Biol. 1995;254:130–49. doi: 10.1006/jmbi.1995.0606. [DOI] [PubMed] [Google Scholar]
- 3.Li G, Widom J. Nucleosomes facilitate their own invasion. Nat Struct Mol Biol. 2004;11:763–9. doi: 10.1038/nsmb801. [DOI] [PubMed] [Google Scholar]
- 4.Bucceri A, Kapitza K, Thoma F. Rapid accessibility of nucleosomal DNA in yeast on a second time scale. Embo J. 2006;25:3123–32. doi: 10.1038/sj.emboj.7601196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adams CC, Workman JL. Binding of disparate transcriptional activators to nucleosomal DNA is inherently cooperative. Mol Cell Biol. 1995;15:1405–21. doi: 10.1128/mcb.15.3.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Polach KJ, Widom J. A model for the cooperative binding of eukaryotic regulatory proteins to nucleosomal target sites. J Mol Biol. 1996;258:800–12. doi: 10.1006/jmbi.1996.0288. [DOI] [PubMed] [Google Scholar]
- 7.Bernstein BE, Liu CL, Humphrey EL, Perlstein EO, Schreiber SL. Global nucleosome occupancy in yeast. Genome Biol. 2004;5:R62. doi: 10.1186/gb-2004-5-9-r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tirosh I, Barkai N. Two strategies for gene regulation by promoter nucleosomes. Genome Res. 2008;18:1084–91. doi: 10.1101/gr.076059.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Su X, Zhang L, Lucas DM, Davis ME, Knapp AR, Green-Church KB, Marcucci G, Parthun MR, Byrd JC, Freitas MA. Histone H4 acetylation dynamics determined by stable isotope labeling with amino acids in cell culture and mass spectrometry. Anal Biochem. 2007;363:22–34. doi: 10.1016/j.ab.2006.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fischle W, Wang Y, Allis CD. Histone and chromatin cross-talk. Curr Opin Cell Biol. 2003;15:172–83. doi: 10.1016/s0955-0674(03)00013-9. [DOI] [PubMed] [Google Scholar]
- 11.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14:1025–40. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844–7. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 13.Cosgrove MS, Boeke JD, Wolberger C. Regulated nucleosome mobility and the histone code. Nat Struct Mol Biol. 2004;11:1037–43. doi: 10.1038/nsmb851. [DOI] [PubMed] [Google Scholar]
- 14.Manohar M, Mooney AM, North JA, Nakkula RJ, Picking JW, Edon A, Fishel R, Poirier MG, Ottesen JJ. Acetylation of histone H3 at the nucleosome dyad alters DNA-histone binding. J Biol Chem. 2009;284:23312–21. doi: 10.1074/jbc.M109.003202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neumann H, Hancock SM, Buning R, Routh A, Chapman L, Somers J, Owen-Hughes T, van Noort J, Rhodes D, Chin JW. A method for genetically installing site-specific acetylation in recombinant histones defines the effects of H3 K56 acetylation. Mol Cell. 2009;36:153–63. doi: 10.1016/j.molcel.2009.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shogren-Knaak MA, Fry CJ, Peterson CL. A native peptide ligation strategy for deciphering nucleosomal histone modifications. J Biol Chem. 2003;278:15744–8. doi: 10.1074/jbc.M301445200. [DOI] [PubMed] [Google Scholar]
- 17.He S, Bauman D, Davis JS, Loyola A, Nishioka K, Gronlund JL, Reinberg D, Meng F, Kelleher N, McCafferty DG. Facile synthesis of site-specifically acetylated and methylated histone proteins: reagents for evaluation of the histone code hypothesis. Proc Natl Acad Sci U S A. 2003;100:12033–8. doi: 10.1073/pnas.2035256100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McGinty RK, Kim J, Chatterjee C, Roeder RG, Muir TW. Chemically ubiquitylated histone H2B stimulates hDot1L-mediated intranucleosomal methylation. Nature. 2008;453:812–6. doi: 10.1038/nature06906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiang KP, Jensen MS, McGinty RK, Muir TW. A semisynthetic strategy to generate phosphorylated and acetylated histone H2B. Chembiochem. 2009;10:2182–7. doi: 10.1002/cbic.200900238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simon MD, Chu F, Racki LR, de la Cruz CC, Burlingame AL, Panning B, Narlikar GJ, Shokat KM. The site-specific installation of methyl-lysine analogs into recombinant histones. Cell. 2007;128:1003–12. doi: 10.1016/j.cell.2006.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo J, Wang J, Lee JS, Schultz PG. Site-specific incorporation of methyl- and acetyl-lysine analogues into recombinant proteins. Angew Chem Int Ed Engl. 2008;47:6399–401. doi: 10.1002/anie.200802336. [DOI] [PubMed] [Google Scholar]
- 22.Nguyen DP, Alai MM, Kapadnis PB, Neumann H, Chin JW. Genetically encoding N(epsilon)-methyl-L-lysine in recombinant histones. J Am Chem Soc. 2009;131:14194–5. doi: 10.1021/ja906603s. [DOI] [PubMed] [Google Scholar]
- 23.Lennartsson A, Ekwall K. Histone modification patterns and epigenetic codes. Biochim Biophys Acta. 2009;1790:863–8. doi: 10.1016/j.bbagen.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 24.Bang D, Kent SB. A one-pot total synthesis of crambin. Angew Chem Int Ed Engl. 2004;43:2534–8. doi: 10.1002/anie.200353540. [DOI] [PubMed] [Google Scholar]
- 25.Kent SB. Total chemical synthesis of proteins. Chem Soc Rev. 2009;38:338–51. doi: 10.1039/b700141j. [DOI] [PubMed] [Google Scholar]
- 26.Ottesen JJ, Bar-Dagan M, Giovani B, Muir TW. An amalgamation of solid phase peptide synthesis and ribosomal peptide synthesis. Biopolymers. 2008;90:406–14. doi: 10.1002/bip.20810. [DOI] [PubMed] [Google Scholar]
- 27.Yamamoto N, Tanabe Y, Okamoto R, Dawson PE, Kajihara Y. Chemical synthesis of a glycoprotein having an intact human complex-type sialyloligosaccharide under the Boc and Fmoc synthetic strategies. J Am Chem Soc. 2008;130:501–10. doi: 10.1021/ja072543f. [DOI] [PubMed] [Google Scholar]
- 28.Downs JA. Histone H3 K56 acetylation, chromatin assembly, and the DNA damage checkpoint. DNA Repair (Amst) 2008;7:2020–4. doi: 10.1016/j.dnarep.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 29.Ozdemir A, Masumoto H, Fitzjohn P, Verreault A, Logie C. Histone H3 lysine 56 acetylation: a new twist in the chromosome cycle. Cell Cycle. 2006;5:2602–8. doi: 10.4161/cc.5.22.3473. [DOI] [PubMed] [Google Scholar]
- 30.Xu F, Zhang K, Grunstein M. Acetylation in histone H3 globular domain regulates gene expression in yeast. Cell. 2005;121:375–85. doi: 10.1016/j.cell.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 31.Chen CC, Carson JJ, Feser J, Tamburini B, Zabaronick S, Linger J, Tyler JK. Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell. 2008;134:231–43. doi: 10.1016/j.cell.2008.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Q, Zhou H, Wurtele H, Davies B, Horazdovsky B, Verreault A, Zhang Z. Acetylation of histone H3 lysine 56 regulates replication-coupled nucleosome assembly. Cell. 2008;134:244–55. doi: 10.1016/j.cell.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Celic I, Verreault A, Boeke JD. Histone H3 K56 hyperacetylation perturbs replisomes and causes DNA damage. Genetics. 2008;179:1769–84. doi: 10.1534/genetics.108.088914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Williams SK, Truong D, Tyler JK. Acetylation in the globular core of histone H3 on lysine-56 promotes chromatin disassembly during transcriptional activation. Proc Natl Acad Sci U S A. 2008;105:9000–5. doi: 10.1073/pnas.0800057105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu F, Zhang Q, Zhang K, Xie W, Grunstein M. Sir2 deacetylates histone H3 lysine 56 to regulate telomeric heterochromatin structure in yeast. Mol Cell. 2007;27:890–900. doi: 10.1016/j.molcel.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watanabe S, Resch M, Lilyestrom W, Clark N, Hansen JC, Peterson C, Luger K. Structural characterization of H3K56Q nucleosomes and nucleosomal arrays. Biochim Biophys Acta. 1799:480–6. doi: 10.1016/j.bbagrm.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Andrews AJ, Chen X, Zevin A, Stargell LA, Luger K. The histone chaperone Nap1 promotes nucleosome assembly by eliminating nonnucleosomal histone DNA interactions. Mol Cell. 37:834–42. doi: 10.1016/j.molcel.2010.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luger K, Rechsteiner TJ, Flaus AJ, Waye MM, Richmond TJ. Characterization of nucleosome core particles containing histone proteins made in bacteria. J Mol Biol. 1997;272:301–11. doi: 10.1006/jmbi.1997.1235. [DOI] [PubMed] [Google Scholar]
- 39.Flaus A, Luger K, Tan S, Richmond TJ. Mapping nucleosome position at single base-pair resolution by using site-directed hydroxyl radicals. Proc Natl Acad Sci U S A. 1996;93:1370–5. doi: 10.1073/pnas.93.4.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Poirier MG, Oh E, Tims HS, Widom J. Dynamics and function of compact nucleosome arrays. Nat Struct Mol Biol. 2009;16:938–44. doi: 10.1038/nsmb.1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tonjes R, Doenecke D. Structure of a duck H3 variant histone gene: a H3 subtype with four cysteine residues. Gene. 1985;39:275–9. doi: 10.1016/0378-1119(85)90323-3. [DOI] [PubMed] [Google Scholar]
- 42.Marzluff WF, Gongidi P, Woods KR, Jin J, Maltais LJ. The human and mouse replication-dependent histone genes. Genomics. 2002;80:487–98. [PubMed] [Google Scholar]
- 43.Roberts SB, Emmons SW, Childs G. Nucleotide sequences of Caenorhabditis elegans core histone genes. Genes for different histone classes share common flanking sequence elements. J Mol Biol. 1989;206:567–77. doi: 10.1016/0022-2836(89)90566-4. [DOI] [PubMed] [Google Scholar]
- 44.Villain M, Vizzavona J, Rose K. Covalent capture: a new tool for the purification of synthetic and recombinant polypeptides. Chem Biol. 2001;8:673–9. doi: 10.1016/s1074-5521(01)00044-8. [DOI] [PubMed] [Google Scholar]
- 45.Wan Q, Danishefsky SJ. Free-radical-based, specific desulfurization of cysteine: a powerful advance in the synthesis of polypeptides and glycopolypeptides. Angew Chem Int Ed Engl. 2007;46:9248–52. doi: 10.1002/anie.200704195. [DOI] [PubMed] [Google Scholar]
- 46.Koopmans WJ, Buning R, Schmidt T, van Noort J. spFRET using alternating excitation and FCS reveals progressive DNA unwrapping in nucleosomes. Biophys J. 2009;97:195–204. doi: 10.1016/j.bpj.2009.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kruithof M, van Noort J. Hidden Markov analysis of nucleosome unwrapping under force. Biophys J. 2009;96:3708–15. doi: 10.1016/j.bpj.2009.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li G, Levitus M, Bustamante C, Widom J. Rapid spontaneous accessibility of nucleosomal DNA. Nat Struct Mol Biol. 2005;12:46–53. doi: 10.1038/nsmb869. [DOI] [PubMed] [Google Scholar]
- 49.Richmond TJ, Davey CA. The structure of DNA in the nucleosome core. Nature. 2003;423:145–50. doi: 10.1038/nature01595. [DOI] [PubMed] [Google Scholar]
- 50.Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, Zhao K. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ferreira H, Flaus A, Owen-Hughes T. Histone modifications influence the action of Snf2 family remodelling enzymes by different mechanisms. J Mol Biol. 2007;374:563–79. doi: 10.1016/j.jmb.2007.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Adkins MW, Howar SR, Tyler JK. Chromatin disassembly mediated by the histone chaperone Asf1 is essential for transcriptional activation of the yeast PHO5 and PHO8 genes. Mol Cell. 2004;14:657–66. doi: 10.1016/j.molcel.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 53.Dechassa ML, Zhang B, Horowitz-Scherer R, Persinger J, Woodcock CL, Peterson CL, Bartholomew B. Architecture of the SWI/SNF-nucleosome complex. Mol Cell Biol. 2008;28:6010–21. doi: 10.1128/MCB.00693-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Friedberg EC. DNA repair and mutagenesis. 2nd. ASM Press; Washington, DC: 2006. [Google Scholar]
- 55.Javaid S, Manohar M, Punja N, Mooney AM, Ottesen JJ, Poirier MG, Fishel R. Nucleosome Remodeling by hMSH2-hMSH6. Mol Cell. 2009;36:1086–1094. doi: 10.1016/j.molcel.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schnolzer M, Alewood P, Jones A, Alewood D, Kent SB. In situ neutralization in Boc-chemistry solid phase peptide synthesis. Rapid, high yield assembly of difficult sequences. Int J Pept Protein Res. 1992;40:180–93. doi: 10.1111/j.1399-3011.1992.tb00291.x. [DOI] [PubMed] [Google Scholar]
- 57.Blake J. Peptide segment coupling in aqueous medium: silver ion activation of the thiolcarboxyl group. Int J Pept Protein Res. 1981;17:273–4. doi: 10.1111/j.1399-3011.1981.tb01992.x. [DOI] [PubMed] [Google Scholar]
- 58.Hackenberger CP. The reduction of oxidized methionine residues in peptide thioesters with NH4I-Me2S. Org Biomol Chem. 2006;4:2291–5. doi: 10.1039/b603543d. [DOI] [PubMed] [Google Scholar]
- 59.Hackeng TM, Griffin JH, Dawson PE. Protein synthesis by native chemical ligation: expanded scope by using straightforward methodology. Proc Natl Acad Sci U S A. 1999;96:10068–73. doi: 10.1073/pnas.96.18.10068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Luger K, Rechsteiner TJ, Richmond TJ. Preparation of nucleosome core particle from recombinant histones. Methods Enzymol. 1999;304:3–19. doi: 10.1016/s0076-6879(99)04003-3. [DOI] [PubMed] [Google Scholar]
- 61.Little JW, Kim B, Roland KL, Smith MH, Lin LL, Slilaty SN. Cleavage of LexA repressor. Methods Enzymol. 1994;244:266–84. doi: 10.1016/0076-6879(94)44022-0. [DOI] [PubMed] [Google Scholar]
- 62.Thastrom A, Lowary PT, Widom J. Measurement of histone-DNA interaction free energy in nucleosomes. Methods. 2004;33:33–44. doi: 10.1016/j.ymeth.2003.10.018. [DOI] [PubMed] [Google Scholar]
- 63.Clegg RM. Fluorescence resonance energy transfer and nucleic acids. Methods Enzymol. 1992;211:353–88. doi: 10.1016/0076-6879(92)11020-j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.