

Abstract
Background
To determine (1) whether age-standardized cognitive declines and brain morphometric change differ between Young-Old (YOAD) and Very-Old (VOAD) patients with Alzheimer’s disease (AD) and (2) whether apolipoprotein E (APOE) genotype modifies these neuropsychological and morphometric changes.
Methods
Baseline and 12-month follow up neuropsychological and morphometric measures were examined for healthy control and AD individuals. The two AD groups were further divided into subgroups on the basis of the presence of at least one APOE ε4 allele.
Results
The YOAD showed more severe deficits andsteeper declines in cognition than the VOAD. Moreover, the presence of an APOE ε4 allele had a more deleterious effect on the YOAD than the VOAD on cognition and brain structure both cross-sectionally and longitudinally.
Conclusions
Results underscore the importance of integrating an individual’s age and genetic susceptibility—and their interaction—when examining neuropsychological and neuroimaging changes in the early stages of Alzheimer’s disease.
Keywords: Alzheimer’s disease, APOE genotype, cognition, morphometry, MRI, longitudinal
1. Introduction
Dementia incidence increases exponentially between the ages of 65 and 90 years. Although much progress has been made in identifying the typical cognitive deficits and brain morphometry changes associated with early Alzheimer’s disease (AD), the boundaries between normal age-related functional and structural changes and early signs of AD remain especially difficult to delineate in the Very-Old (i.e., over the age of 80) [1]. A few studies have shown that AD-related cognitive [2–4] and morphometric [3, 4] changes observed in Young-Old patients are less salient in the Very-Old because brain and behavioral changes observed in normal aging overlap with indicators of AD to a greater degree in the Very-Old than in the Young-Old.
Another factor that may lead to differences in the cognitive deficit profiles of AD in the Young-Old and Very-Old is an age-related change in the influence of the ε4 allele of the apolipoprotein E (APOE) gene. Although it is well established that the APOE ε4 allele is the most common genetic risk factor for AD, controversy exists as to whether or not APOE allelic variants are associated with different cognitive and morphometric AD phenotypes [1, 5, 6]. Several cross-sectional studies have reported that AD ε4 carriers have greater impairment in memory and executive function than AD non-ε4 carriers [5]. Longitudinal studies have shown mixed results with AD ε4 carriers demonstrating slower [7, 8], faster [9–11], or equivalent [12, 13] rates of cognitive decline as their non-ε4 counterparts. Several studies have shown that the effects of APOE ε4 genotype on cognition appear to wane with advancing age in AD and in at-risk individuals [2, 5, 14]. This latter effect suggests that the cognitive phenotypic expression of the APOE ε4 allele may vary as a function of the age of patients. However, most of these studies were cross-sectional in design and incapable of ruling out cohort effects such as differences in subjects’ age or severity of dementia. Furthermore, it is still largely unknown whether morphometric profiles differ by the patient’s age at onset of disease and APOE status.
The present study compared baseline and longitudinal patterns of cognitive decline and regional brain atrophy in Young-Old and Very-Old patients with AD and sought to determine if APOE genotype differentially affects these patterns in the two cohorts. Based upon previous results [2–4], we predicted that, relative to Young-Old AD patients, Very-Old AD patients would exhibit less severe cognitive deficits and less atrophic regional brain changes at baseline, and the rate of cognitive and morphometric changes over time would be slower when using age-standardized cognitive and brain morphometric measures. We further predicted interactive effects of age and APOE genotype—with the presence of an APOE ε4 allele having a more deleterious effect on cognition and morphometry in the Young-Old with AD than in the Very-Old AD group. Specifically, we expected that interactive effects of age and APOE genotype would be limited to memory and executive function given previous reports of the rather specific effect of APOE ε4 genotype on these cognitive functions [5]. With respect to morphometry, we predicted that interactive effects of age and APOE genotype would be apparent in medial temporal lobe, temporoparietal, and frontal regions given their well-documented roles in the pathology of early Alzheimer’s disease.
2. Methods
The raw data used in the current study were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (www.loni.ucla.edu/ADNI). ADNI was launched in 2003 by the National Institute on Aging (NIA), the National Institute of Biomedical Imaging and Bioengineering (NIBIB), the Food and Drug Administration (FDA), private pharmaceutical companies, and non-profit organizations as a $60 million, 5-year public-private partnership. The primary goal of ADNI is to test whether serial MRI, PET, other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment (MCI) and early AD. ADNI is the result of efforts of co-investigators from a broad range of academic institutions and private corporations. Subjects have been recruited from over 50 sites across the U.S. and Canada (see www.adni-info.org). The ADNI study was approved by an ethical standards committee on human experimentation at each institution. Written informed consent was obtained from all study participants, or their authorized representatives, after the procedures of the study were fully explained.
2.1 Participants
ADNI general eligibility criteria have been described elsewhere[15]. Participants included in the present study were Healthy Control (HC) participants or mildly demented patients with AD, 60–92 years old, non-depressed, with a modified Hachinski score of 4 or less, and with a study partner able to provide an independent evaluation of functioning. Participants with a history of stroke or neurological disorders other than AD were excluded from ADNI. HC had a Mini-Mental State Exam (MMSE) score between 24–30 (inclusive), a global Clinical Dementia Rating [16] score of 0, and did not meet criteria for MCI [17]. Mild AD participants had MMSE scores between 20–26, global CDR of 0.5 or 1.0, and met NINCDS/ADRDA criteria for probable AD [18]. The CDR-Sum of Boxes (CDR-SB) score was calculated for all participants to further estimate level of clinical impairment.
HC participants in ADNI were followed for 3 years, with assessments at 0, 6, 12, 24, and 36 months. AD patients were followed for 2 years, with assessments at 0, 6, 12, and 24 months. Participants who completed the baseline visit and at least the 1 year follow-up visit were included in this study. Seven HC participants (five in the Young-Old and two in the Very-Old) who converted to a diagnosis of MCI or AD during any of the ADNI follow-up visits were excluded from the analysis.
The HC and AD participants were divided into subgroups on the basis of their age at baseline testing: (1) Young-Old groups comprised individuals aged 75 years or younger (range 60–75), and (2) Very-Old groups comprised individuals aged 80 years or greater (ranged 80–91). The five-year age gap (i.e., participants were not studied if they fell between the ages of 76–80) resulted in an approximately 12-year age difference between the Young-Old and Very-Old groups. The subject pool was further restricted to those participants for whom adequately processed and quality checked MR baseline data existed. The final sample consisted of 227 participants: 83 Young-Old HC participants, 64 Young-Old AD patients, 40 Very-Old HC participants, and 40 Very-Old AD patients (see upper portion of Table 1). All participants were genotyped for apolipoprotein E (APOE) allele status using DNA extracted from peripheral blood cells. Participants with APOE genotype ε4/ε2 were excluded (n = 4) from the present study. To examine the impact of APOE ε4 allele on rate of decline in Young-Old and Very-Old AD patients, the patient groups were divided into subgroups on the basis of the presence of at least one APOE ε4 allele. The Young-Old ε4 AD group consisted of 49 participants; the Young-Old non-ε4 AD group consisted 15 participants; the Very-Old ε4 AD group consisted of 20 participants; the Very-Old non-ε4 AD group consisted 20 participants (see lower portion of Table 1).
Table 1.
Demographic, global cognitive, clinical, and MRI morphometric characteristics of Young-Old and Very-Old healthy control and Alzheimer’s disease groups (upper portion) as well as of the four AD subgroups separated by apolipoprotein E genotype (bottom portion) at baseline.
| Young-Old HC (n=83) | Very-Old HC (n=40) | Young-Old AD (n=64) | Very-Old AD (n=40) | |
|---|---|---|---|---|
| Age | 71.86 (2.30) | 83.88 (2.55) | 70.77 (3.21) | 84.01 (2.66) |
| Education | 15.37 (2.39) | 15.95 (2.97) | 14.69 (2.63) | 14.80 (3.25) |
| Gender (% men) | 51% | 54% | 44% | 56% |
| % APOE ε4 carriers | 25% | 28% | 77% * | 50% ** |
| MMSE | 29.00 (1.08) | 29.02 (1.06) | 23.52 (2.05) | 23.20 (2.19) |
| CDR sum of boxes | 0.02 (0.11) | 0.07 (0.18) | 4.07 (1.31) | 4.43 (1.92) |
| Modified HIS scores | 0.51 (0.72) | 0.45 (0.55) | 0.59 (0.66) | 0.73 (0.72) |
| Disease duration (years) | -- | -- | 3.16 (2.29) | 3.23 (2.20) |
| WMH volume (mm3) | -- | -- | 6852 (662) | 11970 (1765) § |
| Blood homocysteine, μM/L | 9.31 (2.91) † | 11.25 (3.47) | 9.99 (2.44) | 10.92 (2.64) |
| % HTN meds users | 28% | 33% | 36% | 38% |
| Loss to follow-up | 10 (12%) | 4 (10%) | 12 (18.8%) | 6 (15%) |
| Young-Old AD non-ε4 (n=15) | Young-Old AD ε4 (n=49) | Very-Old AD non-ε4 (n=20) | Very-Old AD ε4 (n=20) | |
|---|---|---|---|---|
| Age | 70.53 (3.44) | 70.84 (3.17) | 84.16 (3.10) | 83.70 (2.18) |
| Education | 16.07 (2.19) | 14.27 (2.63) | 14.90 (3.32) | 14.80 (3.32) |
| Gender (% men) | 47% | 43% | 40% | 75% |
| MMSE | 23.27 (2.34) | 23.59 (1.97) | 23.00 (2.03) | 23.50 (2.37) |
| CDR sum of boxes | 3.90 (1.35) | 4.12 (1.31) | 4.23 (1.97) | 4.60 (1.94) |
| Modified HIS scores | 0.53 (0.52) | 0.61 (0.70) | 0.65 (0.75) | 0.80 (0.70) |
| Disease duration (years) | 3.21 (2.61) | 3.14 (2.22) | 2.85 (1.97) | 3.58 (2.44) |
| WMH volume (mm3) | 7216.20 (1014.35) | 6740.65 (811.35) | 11441 (2437) | 8820.63 (824)‡ |
| Blood homocysteine, μM/L | 10.59 (2.13) | 9.81 (2.52) | 10.63 (2.46) | 11.21 (2.85) |
| % HTN meds users | 40% | 35% | 30% | 45% |
| Loss to follow-up | 2 (13.3%) | 10 (20.4%) | 3 (15%) | 3 (15%) |
Abbreviations: CDR, Clinical Dementia Rating Scale; HIS, Hachinski Ischemia Scale; HTN, hypertension; WMH, white matter hypointensity (estimated from T1 image)
indicates significant difference between the Young-Old HC group versus the Very-Old HC group and the Very-Old AD group
indicates significant difference between the Young-Old AD group and the two HC groups
indicates significant differences between the Very-Old AD group and the other three group
indicates significant difference between the Young-Old AD group and the Very-Old AD group
indicates significant difference between the Young-Old AD ε4 group and the Very-Old AD ε4 group
2.2 Neuropsychological measures
The Participants were tested with a standardized battery of neuropsychological tests [15]. We assessed performance at baseline and at the one-year follow-up. Six neuropsychological domains were assessed with the following tests: (1) Language: 30-item Boston Naming Test and Category Fluency (animal and vegetable categories); (2) Attention/ Psychomotor Processing Speed: Trail Making Test Part A, and Wechsler Adult Intelligence Scale-Revised (WAIS-R) Digit Span Forward and Digit Symbol subtests; (3) Executive Function: Trail Making Test Part B and WAIS-R Digit Span Backward; (4) Immediate Recall: Rey Auditory Verbal Learning Test (RAVLT) Trials 1–5 Total Recall and Wechsler Memory Scale-Revised (WMS-R) Logical Memory (LM) Immediate Recall; (5) Delayed Recall: RAVLT Long-Delay Recall and WMS-R Logical Memory Delayed Recall; (6) Recall Savings: RAVLT Percent Long-Delay Savings and WMS-R Logical Memory Percent Delayed Recall Savings. The test scores achieved by the Young-Old and Very-Old AD patients on each measure were converted to z-scores based upon the mean and standard deviations of their respective HC group and summed to create the six neuropsychological domain composite scores. To ease interpretation, z-scores were modified to ensure that negative scores represented poorer performance.
2.3 MRI acquisition and analysis
Image acquisition and analysis methods were developed within the NIH/NCRR sponsored Morphometry Biomedical Informatics Research Network (mBIRN) and ADNI [19–22]. Data were collected across a variety of 1.5 T scanners. Protocols are described in detail at http://adni.loni.ucla.edu/research/protocols/mri-protocols/. Two T1-weighted volumes were acquired for each participant, one at baseline and one at follow-up approximately 1 year later. These raw DICOM MRI scans were downloaded from the public ADNI site (http://adni.loni.ucla.edu/about-data-samples/how-to-apply/). Locally, images were reviewed for quality, automatically corrected for spatial distortion due to gradient nonlinearity [21] and B1 field inhomogeneity [23], registered, and averaged to improve signal-to-noise. Volumetric segmentation [24, 25] and cortical surface reconstruction [25–28] used methods based on FreeSurfer software optimized for use on large, multi-site datasets. To measure thickness, the cortical surface was reconstructed [26, 27] and parcellated into distinct regions of interest (ROIs) [25, 29]. FreeSurfer also provides estimates of white matter hypointensity (WMH) based on the automatic segmentation of the T1 weighted images. Details of the application of these methods to the ADNI data have been described in full elsewhere [30]. To limit the number of multiple comparisons, only regions assumed to be involved in early AD pathology [19–22] were included in the present analyses. These included volumetric measures of bilateral hippocampal formation, including dentate gyrus, CA fields, subiculum/ parasubiculum and the fimbria [31], and thickness measures of frontal, temporal, and parietal lobe cortical areas and bilateral cingulate cortex regions (see ROIs listed in Fig 3). To decrease the number of comparisons, the caudal and rostral anterior cingulate regions were combined as anterior cingulate cortex (ACC); the isthmus and posterior cingulate regions were combined as posterior cingulate cortex (PCC); and the pars opercularis and pars triangularis were combined as the frontal operculum. Following Buckner and colleagues [32], baseline volumetric data were corrected for individual differences in head size by regressing the estimated total intracranial volume (eTIV).
Figure 3.

Regional brain atrophy in the Young-Old and Very-Old Alzheimer’s disease (AD) patients utilizing standardized z-scores derived from their age-appropriate healthy control counterparts at baseline. Error bars denote SEM. * p < .001. Abbreviations: ACC, anterior cingulate cortex; PCC, posterior cingulate cortex; STG, superior temporal gyrus; MTG, middle temporal gyrus; ITG, inferior temporal gyrus; T pole, temporal pole; mF, middle frontal; F pole, frontal pole; SFG, superior frontal gyrus; orbitoF, orbitofrontal; SPL, superior parietal lobule; IPL, inferior parietal lobule.
2.3.1 Longitudinal analysis of MRI scans
The longitudinal data was derived through Quarc (quantitative anatomical regional change, [33]). Dual 3-D follow-up structural scans for each participant were rigid-body aligned, averaged and affine aligned to the participant’s baseline scan. Nonlinear registration of the images was then performed, where voxel centers were moved about until a good match was made between the images [33, 34]. This is achieved in the following way. The images are heavily blurred (smoothed), making them almost identical, and a merit or potential function is calculated. This merit function expresses the intensity difference between the images at each voxel, and depends on the displacement field for the voxel centers of the image being transformed; it is also regularized to keep the displacement field spatially smooth. The merit function by design will have a minimum when the displacement field induces a good match between the images. The displacement field in general will turn cubic voxels into displaced irregular hexahedra whose volumes give the volume change field. The merit function is minimized efficiently using standard numerical methods. Having found a displacement field for the heavily blurred pair of images, the blurring is reduced and the procedure repeated, thus iteratively building up a better displacement field. Two important additions to this are: 1) applying the final displacement field to the image being transformed, then nonlinearly registering the resultant image to the same target, and finally tracing back through the displacement fields thus calculated to find the net displacement field; and 2) restricting to regions of interest and zooming when tissue structures are separated by only a voxel or two. These additional features enable very precise registration involving large or subtle deformations, even at small spatial scales with low boundary contrast. Although large deformations are allowed by multiple nonlinear registration (or relaxation) steps, nonphysical deformations are precluded because at each level of blurring the image undergoing deformation is constrained to conform to the target. Note that calculating the deformation field does not depend on initially segmenting tissue. This deformation field was used to align scans at the sub-voxel level.
The follow-up aligned image underwent skull stripping and volumetric segmentations (subcortical structures, as well as hippocampus and cerebellum gray matter), with labels applied from the baseline scan. For the cortical reconstructions, surface coordinates for the white matter and pial boundaries were derived from the baseline images and mapped onto the follow-up images using the deformation field. Parcellations from the baseline image were then applied to the follow-up image. This resulted in a one-to-one correspondence between each vertex in the base image and the follow-up image. This procedure produced an estimate of the percent cortical volume loss at each vertex and within each ROI. To the extent that regional cortical areas are relatively stable across time points, the volume change is likely driven almost exclusively by changes in thickness. Atrophy rates were defined as the percent cortical volume loss over the course of one-year. These atrophy rates represent within-subject change over time and are independent of group differences in baseline measurements (e.g., eTIV). Because the procedure is fully automated, the test-retest reliability is 1. The method has been validated in model studies of complex spherical-shell geometries with low contrast and noise where a prescribed volume change is numerically estimated to accuracies of within 0.5% [33].
2.4 Statistical analyses
Group comparisons for demographic, clinical (i.e., CDR sum of boxes scores, disease duration), and global cognitive (i.e., MMSE scores) variables were performed with analyses of variance (ANOVAs) and independent samples t tests or chi-square tests, as appropriate. Repeated measure GLMs were used for analyzing the longitudinal cognitive data with group as the between-subjects variable and, in separate analyses, each of the six cognitive variables (i.e., language, attention/psychomotor processing speed, executive function, immediate recall, delayed recall, and recall savings) as a within-subjects measures. ANOVAs and/or independent samples t tests were performed for follow-up pair-wise comparisons. Based on Bonferroni correction, α level was set to p < .008 for analyses of the 6 neuropsychological composite scores. Whenever the assumption of homogeneity of variance was not met, the t-value and significance of the comparison were reported according to the assumption that variances were unequal.
To assess group difference in baseline morphometric variables, effect of gender was first regressed from all thickness and volumetric measures. Bilateral hippocampal volumes were also corrected for differences in head size by regressing the estimated total cranial vault (eTIV) volume [32]. Next, the volumetric and cortical thickness measures of the two AD groups at baseline were z-transformed relative to their respective HC group such that negative values indicated smaller volume or thinner cortex. Regional atrophy rates were used in analyses of longitudinal morphometric differences. Because the current study did not pose specific hypotheses about hemispheric effects, the volumes and cortical thickness variables were averaged across right and left hemisphere values to decrease number of comparisons. Univariate ANOVAs were performed, followed by ANOVAs or independent samples t tests for post-hoc comparisons. Based on Bonferroni correction, the α level was set to .0026 for assessment of baseline and longitudinal morphometry in the 19 ROIs. Effect sizes (Cohen’s d) were calculated for neuropsychological and morphometric variables that reached statistical significance.
To rule out possible confounding effects driven by vascular risk factors separate analyses controlling for both modified Hachinski Ischemic Scale scores and blood homocysteine levels, were performed. Effects are reported when control for these factors changed the main results.
3. Results
3.1 Demographic and clinical data
The demographic and clinical characteristics for the two HC subgroups and the two AD subgroups are presented in Table 1. Consistent with the design of the study, the two Very-Old groups were older than the two younger groups (F(3,223) = 376.27, p < .001), although no significant age difference was observed between the two younger groups (p= .17) or the two older groups (p= .98). The four groups did not differ in educational attainment (F(3,223) = 2.28, p = .08), gender distribution (χ2(3) = 2.01; p = .57), modified Hachinski Ischemic Scale scores (F(3,223) = 1.38, p = .25), or percentage of people currently taking hypertension medications (F(3,227) = 1.67, p = .65). The four groups differed significantly on the blood homocysteine level (F(3,223) = 5.41, p <.005). Post-hoc analyses revealed that the Young-Old HC showed significantly lower homocysteine level compared to the Very-Old HC (p < .005) and the Very-Old AD (p = .05) groups, while the two AD groups showed comparable level of homocysteine (p = .07).
As expected, AD patients scored lower than HC participants on the MMSE (F(3,223) = 230.13, p < .001) and CDR-SB (F(3,222) = 280.07, p < .001). The two HC groups (MMSE: p= .81; CDR-SB: p= .11) and the two AD groups (MMSE: p= .53; CDR-SB: p= .33) did not differ significantly on the MMSE or the CDR-SB scores. The two AD groups did not differ significantly in the estimated years of disease duration (t 101 = −.12, p = .91), but the Very-Old AD group showed greater volume of WMH than the Young-Old AD group (t 102 = −3.15, p = .002). Both AD groups showed a higher frequency of APOE ε4 carriers compared to the two HC groups (all p-values < .005). The Young-Old AD group had a higher frequency of APOE ε4 carriers than their Very-Old AD counterparts (χ2(1) = 7.78; p = .005). The percentage of subjects lost to follow-up did not significantly differ between the four groups (χ2(3) = 2.01; p = .57).
We further compared the demographic and clinical variables for the four AD subgroups (APOE genotype x age). The four AD groups did not differ in educational attainment (F(3,100) = 1.55, p = .21), gender distribution (χ2(3) = 6.83; p = .08), CDR-SB scores (F(3,100) = .64, p = .59), MMSE scores (F(3,100) = .41, p = .75), modified Hachinski Ischemic Scale scores (F(3,100) = 0.51, p = .68), disease duration (F(3,99) = .35, p = .79), percentage of participants currently taking hypertension medications (F(3,104) = 1.14, p = .77), or blood homocysteine level (F(3,100) = 1.65, p = .18). As expected, the two Very-Old AD groups were older than the two younger AD groups (F(3,100) = 154.47, p < .001) whereas no significant age difference was observed between the two younger groups (p= .75) or the two older groups (p= .60). The Very-Old AD ε4 group had greater volume of white matter hypointensity than the Young-Old AD ε4 group (F(3,100) = 3.31, p = .023)
3.2 Neuropsychological assessment
Age main effects
Main effects for age using the age-normalized z-scores revealed significantly poorer performance in the Young-Old AD group than in the Very-Old AD group in the domains of executive function (t 92 = −3.83, p < .001; Cohen’s d = 0.78), attention/psychomotor processing speed (t 97 = −3.21, p= .002; Cohen’s d = 0.68), and immediate memory (t 101 = −2.77, p =.007; Cohen’s d = 0.58). The two AD groups had comparable performances on language (t 95 = 1.29, p = .20), delayed memory (t 102 = 0.69, p = .50), and recall savings (t 92 = −.82, p = .42) (see Fig 1).
Figure 1.

Baseline performances of Young-Old AD vs. Very-Old AD by cognitive domains computed by age-normalized z-scores.
APOE genotype polymorphism effects
There were no significant main effects of APOE genotype on any of the cognitive domains in the AD groups.
Time main effects
Significant main effects for time were found on all six cognitive composite scores. Relative to their age-appropriate HC group, the AD patients as a whole demonstrated decline in performance over one year in all cognitive domains, including language (F(1,79) = 27.98, p < .001), executive function (F(1,70) = 49.31, p < .001), attention/psychomotor processing speed (F(1,81) = 8.14, p = .005), immediate memory (F(1,81) = 35.26, p < .001), delayed memory (F(1,80) = 9.43, p = .003), and recall savings (F(1,66) = 8.42, p = .005).
Age x APOE genotype interaction effects
Significant age x APOE genotype interaction effects were obtained for three of the baseline cognitive composite scores. Specifically, the presence of an APOE ε4 allele had a more deleterious effect on performance in the Young-Old AD group than in the Very-Old AD group on executive function (t 54 = −3.26, p = .002; Cohen’s d = 0.82), attention/psychomotor processing speed (t 61= −3.74, p < .001; Cohen’s d = 0.88), and marginally for recall savings (t 52 = −2.32, p = .02; Cohen’s d = 0.50). In contrast, there were no significant differences on any of the cognitive domains between the two AD age groups for the non-ε4 carriers (all p-values > .05).
Age x time interactions effects
Significant age x time interaction effects were observed for four of the cognitive composite scores. The Young-Old AD group showed steeper declines over time than the Very-Old AD group on language (F(1,81) = 21.07, p < .001, ηp2 = 0.21), executive function (F(1,72) = 29.64, p < .001, ηp2 = .29), immediate recall (F(1,83) = 21.23, p < .001, ηp2 = .20), and delayed recall (F(1,82) = 33.43, p < .001, ηp2 = .29) composite scores.
APOE genotype x time interaction effects
Significant APOE genotype x time interactions were obtained only for the language domain (F(1,81) = 7.58, p = .007, ηp2 = .09) with the AD ε4 group, as a whole, showing steeper decline than the AD non-ε4 group over the one year follow-up interval.
Age x APOE genotype x time interaction effects
Significant age group x APOE genotype x time interaction effects were obtained on immediate recall. The Young-Old ε4 AD group showed steeper declines on immediate recall over one year than did the Very-Old ε4 AD group (F(3,81) = 7.80, p < .001, ηp2 = .22), whereas the two non-ε4 AD groups showed comparable rates of decline. The presence of an APOE ε4 allele also had a more deleterious effect on language function for the Young-Old AD group than for the Very-Old AD group (F(3,79) = 8.25, p < .001, ηp2 = .24) (Fig 2).
Figure 2.

Baseline and 12-month follow-up performances of the four AD age x APOE genotype subgroups on the age-normalized z-scores for the different cognitive domains.
3.3 Brain morphometry
Age main effects
The analyses were based on the age-normalized z-scores that also controlled for gender effects. The Young-Old AD group showed thinner baseline cortex than the Very-Old AD group in inferior parietal regions (t 102 = −3.89, p < .001; Cohen’s d = 0.80) (see Figure 3).
APOE genotype polymorphism effects
There were no significant main effects of APOE genotype on baseline morphometric measures in any of the ROIs in the AD groups.
Age x APOE genotype interaction effects
A significant age x APOE genotype interaction was obtained for the inferior parietal region (F(3,100) = 5.34, p = .002, ηp2 = .14). The Young-Old ε4 AD group showed more cortical thinning than the Very-Old ε4 AD group in this cortical region, whereas the two non-ε4 AD groups showed comparable thickness (p >. 05).
Age x time interaction effects
Significant age x time (annual brain atrophy rate) interaction effects were found in both AD and HC groups. There was a greater rate of atrophy in the Young-Old AD group than in the Very-Old AD group for superior temporal (t 64= −5.04, p < .001; Cohen’s d = 1.15), middle temporal (t 64= −6.70, p < .001; Cohen’s d = 1.57), inferior temporal (t 68= −6.42, p < .001; Cohen’s d = 1.48), caudal middle frontal (t 60= −3.18, p = .002; Cohen’s d = 0.78), inferior parietal lobule (t 68= −5.64, p < .001; Cohen’s d = 1.42), precuneus (t 68= −4.25, p < .001; Cohen’s d = 0.99), superior parietal lobule (t 64= −4.31, p < .001; Cohen’s d = 1.05), supramarginal (t 68= −5.69, p < .001; Cohen’s d = 1.31), and posterior cingulate (t 68= −3.10, p = .002; Cohen’s d = 0.72) regions. The two AD groups showed comparable rates of atrophy in hippocampus, but a marginally significant effect in entorhinal (p = .054) and parahippocampal (p = .04) regions with a greater rate of atrophy in the Young-Old AD group than in the Very-Old AD group. After controlling for both modified Hachinski Ischemic Scale scores and blood homocysteine level, the previously observed age x time effect on caudal middle frontal and posterior cingulate were no longer significant.
Age x time interaction effects were also obtained between the two HC groups, but these were in opposite direction to effects observed in the two AD groups. Specifically, the Very-Old HC group had a greater annual atrophy rate than the Young-Old HC group in hippocampal (t 88= −3.23, p = .002; Cohen’s d = 0.75), entorhinal (t 89= 4.43, p < .001; Cohen’s d = 1.09), supramarginal (t 89= 3.29, p = .001; Cohen’s d = 0.76), and operculum (t 89= 3.19, p = .002; Cohen’s d = 0.72) regions.
APOE genotype x time interaction effects
There were no significant APOE genotype effects on annual atrophy rate in the two AD groups in any of the ROIs.
Age x APOE genotype x time interaction effects
The presence of an APOE ε4 allele was associated with a greater rate of atrophy in the Young-Old AD group than in the Very-Old AD group on several lateral temporal and parietal ROIs. Specifically, the Young-Old ε4 AD group showed a greater annual rate of atrophy in superior (F(3,65) = 6.69, p = .001, ηp2 = .24), middle (F(3,65) = 11.71, p < .001, ηp2 = .35), and inferior (F(3,65) = 11.43, p < .001, ηp2 = .35) temporal; superior (F(3,65) = 5.94, p = .001, ηp2 = .08) and inferior (F(3,65) = 10.21, p < .001, ηp2 = .17) parietal; precuneus (F(3,65) = 5.71, p = .002, ηp2 = .12), and supramarginal (F(3,65) = 9.71, p = .001, ηp2 = .14) regions than any of the other three AD groups. In contrast, the Very-Old ε4 AD group showed annual atrophy rates that were similar to those of the Very-Old non-ε4 AD and Young-Old non-ε4 AD groups in all ROIs, including medial temporal regions (all p-values > .05) (Fig 5).
Figure 5.

Annual neocortical atrophy rates (as measured by percent volume change) in regions of interest for each of the four early Alzheimer’s disease (AD) groups: Young-Old non-ε4, Young-Old ε4, Very-Old non-ε4, and Very-Old ε4 AD groups.
4. Discussion
The present results extend our previous findings [3, 4] by demonstrating an interaction between APOE genotype and age in cross-sectional and longitudinal cognitive and morphometric manifestations of AD. We previously showed that when Young-Old and Very-Old patients with AD are compared to their respective age-appropriate HC subjects, the Very-Old AD patients exhibit less severe cognitive impairment and less regional brain atrophy than the Young-Old AD patients in a cross-sectional sample [3]. In the current study, we further found that the Very-Old AD patients also show a slower rate of cognitive decline in memory, executive function, and language, as well as a slower rate of atrophy in multiple temporal, parietal, and cingulate brain regions over time. These effects were partially explained by age-related decreases in cognitive performance and cortical thickness of the respective healthy control participants, which made the age-appropriate standard scores of the Very-Old AD patients less “abnormal” than those of the Young-Old AD patients.
In the present study, we also found that Young-Old AD patients with at least one APOE ε4 allele were more impaired (relative to age-appropriate HC participants) than Young-Old AD patients without an ε4 allele, or Very-Old AD patients with or without an ε4 allele, on baseline measures of executive function, attention, and psychomotor processing speed. Furthermore, the Young-Old ε4 AD patients had steeper declines in memory and language over a one-year interval than Young-Old non-ε4 or Very-Old ε4 and Very-Old non-ε4 AD patients. These results suggest that the effect of APOE genotype on cognition and rate of decline depend upon the patient’s age at onset of disease. Variability in the results of previous studies that examined APOE-related differences in the cognitive phenotype of AD may be explained, in part, by this phenomenon. Previous studies that found APOE genotype effects on cognition or rate of cognitive decline typically studied patients in younger age ranges (i.e., mean ages less than 85)[10, 35], whereas studies that failed to find these effects may have included samples with wider or older age ranges[13].
The observed interaction between the effects of age and APOE genotype on cognition was also apparent in cortical thickness measures. Young-Old ε4 AD patients had greater thinning than Young-Old non-ε4 AD patients in inferior parietal cortex, whereas the Very-Old ε4 and non-ε4 AD patients did not differ. In addition, Young-Old ε4 AD patients had greater atrophy over one-year than Young-Old non-ε4 AD and Very-Old ε4 and non-ε4 AD patients in a number of lateral temporal lobe and parietal cortical regions including precuneus cortex and the supramarginal gyrus. These findings are consistent with our hypothesis that decreased cortical thickness or accelerated rates of brain atrophy related to age and APOE ε4 genotype would be found in regions particularly susceptible to deposition of neurofibrillary tangles and neuronal loss such as the supramarginal gyrus.
However, not all brain regions usually affected by AD showed differential levels of volumetric or cortical thickness abnormality in the AD groups that were stratified by age and APOE genotype. For example, Young-Old ε4 AD patients showed only marginally more atrophy over the one-year interval than Young-Old non-ε4 AD and Very-Old ε4 and non-ε4 AD patients in hippocampus, entorhinal cortex and parahippocampal gyrus, and this was due largely to the fact that Very-Old HC participants showed more atrophy than Young-Old HC participants in these regions which skewed the AD patients age-adjusted atrophy scores. It should be noted, however, that while atrophy of the hippocampus and medial temporal gyrus increased with age and the presence of an APOE ε4 allele in the HC participants, the degree of difference between the HC and AD groups remained large. Thus, our findings do not contradict the position that early and severe atrophy of the hippocampus and medial temporal lobe cortex due to AD greatly eclipses normal age-related changes and allows atrophy in these regions to be a salient marker of AD. It should also be noted that, as with cognition, our findings of interactions between age and APOE genotype on morphometric brain changes related to AD may partially account for inconsistent findings across studies examining the effect of the APOE ε4 allele on brain atrophy in patients with AD.
The present findings have potential clinical implications because they imply that there are age and APOE genotype-related decrements in the sensitivity of cognitive and imaging measures for detecting AD. Because cognitive impairment (relative to age-appropriate HC) in Very-Old AD patients is less apparent than in Young-Old AD patients, the likelihood of false negative diagnostic errors is increased in very elderly patients. This could have important ramifications for diagnosis under the proposed Diagnostic and Statistical Manual of Mental Disorders fifth edition (DSM-5) scheme, given that much of the distinction between ‘major’ and ‘minor’ neurocognitive disorder (i.e., analogous to dementia vs. mild cognitive impairment, respectively) rests on the severity of cognitive impairment. In this proposed scheme, −1 to −2 SDs below appropriate norms on cognitive testing defines minor neurocognitive disorder, whereas −2 or more SDs defines major neurocognitive disorder. Our findings suggest that application of this approach would likely give rise to greater numbers of false negative diagnostic errors (e.g., mis-assigning those who are demented as having a ‘minor’ neurocognitive disorder) in very elderly individuals than in younger elderly individuals. The common use of a −1.5 SD cutoff on memory testing for the identification of MCI may also need adjustment upward if it is to retain sensitivity for the detection of MCI in the Very-Old [2].
With respect to MR imaging, there may be less volumetric integrity (and more variability) in temporal lobe and other brain regions in the Very-Old. Consequently, imaging approaches that measure change in these structures as a diagnostic sign of AD may also be less useful in this cohort because the change occurs against a backdrop of age-related change and increased variability in hippocampal and other regional atrophy [36]. Future empirical studies that define age-specific cutoff values for morphometric measures will be useful.
Although APOE genotype per se did not necessarily influence the rate of cognitive decline or brain morphometric change in patients with AD, the age by APOE genotype interaction effect is likely an important factor in determining and modulating decline. It is important to understand the role of APOE genotype and its interaction with age of onset in the progression of neurodegeneration to optimize treatment regimens, including therapies that target APOE function (see also [37]). Our results suggest, however, that less additional prognostic information would be provided by knowledge of APOE genotype in very elderly patients with AD.
Some limitations of the present study should be noted. First, histopathological verification of disease is not available so it is possible that some participants have a disorder other than AD or have AD with co-morbid pathologies (e.g., infarcts, Lewy bodies, etc.) that contribute to the cognitive and neuroimaging presentations. For example, the impact of white matter changes on the pattern of cognitive and regional brain changes in AD across different age groups may relate to some of the observed differences in cognitive profiles, as we noted a significantly greater amount of WMH in Very-Old AD relative to the Young-Old AD. However, ADNI exclusionary criteria ensure a low prevalence of vascular risk factors. Moreover, after controlling for the effects of select vascular risk factors (i.e., modified Hachinski Scale scores and blood homocysteine level), our findings of the distinct patterns in Young-Old versus Very-Old and their interaction with APOE status on the cognition and morphometry were retained. It suggested that the vascular factors were, for the most part, not driving these effects. Second, despite having a larger sample size than some previous studies, a sample size of 227 participants is relatively small, particularly once diagnostic groups were stratified on the basis of age and APOE status. This raises the issue of generalizability of the results. Thus, the study warrants replication in a larger and preferably autopsy-confirmed sample. Third, despite an effort made to ensure that none of the healthy control participants progressed to MCI or AD within three years after baseline evaluation, it is still possible that some participants with sub-clinical AD were misclassified into control groups (given the relatively short follow-up duration in the present study) potentially obscuring diagnostic group differences. Fourth, the relatively high number of men in the Very-Old group, though statistically non-significant, is inconsistent with studies that show female survival advantages [38]. Thus, the present sample may be biased towards a more physically healthy sample which limits the generalizability of results showing cognitive and morphometric differences between groups.
Despite these limitations, our results clearly argue against the simple application of our understanding of neuropsychological and neuroimaging changes in AD in the Young-Old to the detection of the disease in the Very-Old. Because there are normal age-related changes in cognitive performance and age-related changes in the influence of the APOE ε4 allele on cognition and morphometry, a multi-faceted approach that integrates neuropsychological assessment, APOE genotyping, and neuroimaging technologies [39, 40] may be needed to characterize the early and preclinical stages of AD in this fastest growing and most vulnerable segment of our population.
Figure 4.
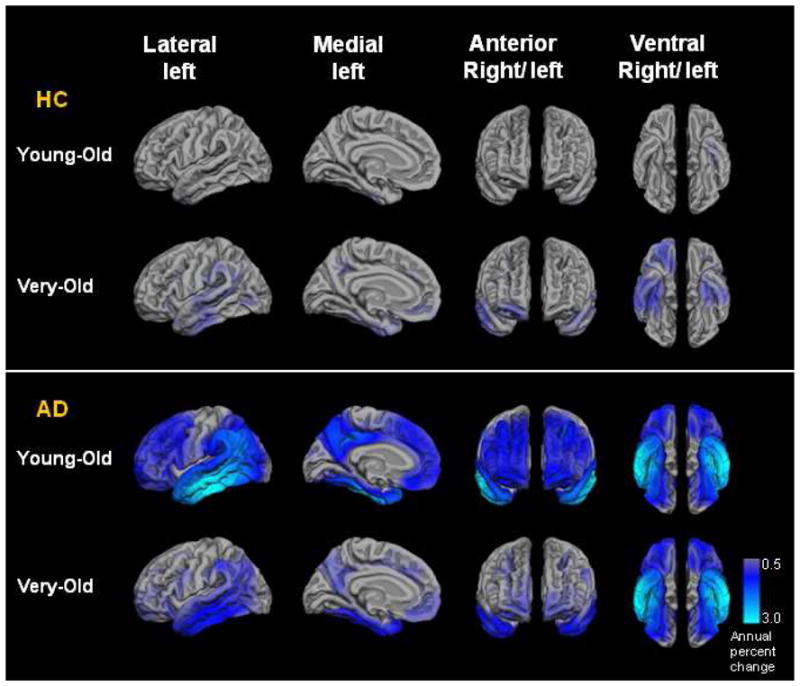
Annual neocortical atrophy rates (as measured by percent volume change) in regions of interest in the Young-Old healthy control (HC), Very-Old HC, Young-Old Alzheimer’s disease (AD), and Very-Old AD groups.
Research in Context.
Systematic review
The cognitive phenotypic expression of the APOE ε4 allele may vary as a function of the age of patients. However, most of these studies were cross-sectional in design and it is still largely unknown whether morphometric profiles differ by the patient’s age at onset of disease and APOE status.
Interpretation
Our results clearly argue against the simple application of our understanding of neuropsychological and neuroimaging changes in AD in the Young-Old to the detection of the disease in the Very-Old. A multi-faceted approach that integrates neuropsychological assessment, APOE genotyping, and neuroimaging technologies may be needed to characterize the early and preclinical stages of AD.
Future directions
Future studies should explore the role of APOE genotype and its interaction with age of onset in the progression of neurodegeneration to optimize treatment regimens, including therapies that target APOE function.
Acknowledgments
Data used in the preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (www.loni.ucla.edu\ADNI). As such, the investigators within ADNI contributed to the design and implementation of ADNI and/or provided data, but did not participate in data analysis or writing this report. A complete listing of the ADNI investigators is available at: http://www.loni.ucla.edu/ADNI/Data/ADNI_Authorship_List.pdf. We thank Alain Koyama, Michele Perry, Chris Pung and Elaine Wu for downloading and preprocessing the ADNI MRI data. We also thank the ADNI participants.
This research was supported by grants from the Taiwan National Science Council (99-2410-H-002-262-MY2, 101-2628-H-002-003-MY3) and National Institute of Aging (K24 AG026431, R01 AG012674, R01 AG05131, R01 AG031224, and K01AG029218). Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Abbott, AstraZeneca AB, Bayer Schering Pharma AG, Bristol-Myers Squibb, Eisai Global Clinical Development, Elan Corporation, Genentech, GE Healthcare, GlaxoSmithKline, Innogenetics, Johnson and Johnson, Eli Lilly and Co., Medpace, Inc., Merck and Co., Inc., Novartis AG, Pfizer Inc, F. Hoffman-La Roche, Schering-Plough, Synarc, Inc., and Wyeth, as well as non-profit partners the Alzheimer’s Association and Alzheimer’s Drug Discovery Foundation, with participation from the U.S. Food and Drug Administration. Private sector contributions to ADNI are facilitated by the Foundation for the National Institutes of Health (www.fnih.org <http://www.fnih.org/> <http://www.fnih.org <http://www.fnih.org/≫). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for NeuroImaging at the University of California, Los Angeles. This research was also supported by NIH grants P30 AG010129, K01 AG030514, and the Dana Foundation.
Footnotes
Conflict of Interest Disclosure: Dr. Salmon serves as a consultant for CHDI Foundation, Novartis, and Bristol-Meyers Squibb. Dr. Dale is a founder of, holds equity in, and serves on the scientific advisory board for CorTechs Labs, Inc. The terms of this arrangement have been reviewed and approved by UCSD in accordance with its conflict of interest policies. Dr. Bondi serves as an Associate Editor for the Journal of the International Neuropsychological Society. Dr. McEvoy’s spouse is president of CorTechs Labs Inc. The other authors report no disclosures.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Salmon DP, Bondi MW. Neuropsychological assessment of dementia. Annu Rev Psychol. 2009;60:257–82. doi: 10.1146/annurev.psych.57.102904.190024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bondi MW, Houston WS, Salmon DP, Corey-Bloom J, Katzman R, Thal LJ, Delis DC. Neuropsychological deficits associated with Alzheimer’s disease in the very-old: discrepancies in raw vs. standardized scores. J Int Neuropsychol Soc. 2003;9(5):783–95. doi: 10.1017/S1355617703950119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stricker NH, Chang YL, Fennema-Notestine C, Delano-Wood L, Salmon DP, Bondi MW, Dale AM. Distinct profiles of brain and cognitive changes in the very old with Alzheimer disease. Neurology. 2011;77(8):713–21. doi: 10.1212/WNL.0b013e31822b0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holland D, Desikan RS, Dale AM, McEvoy LK. Rates of Decline in Alzheimer Disease Decrease with Age. PLoS One. 2012;7(8):e42325. doi: 10.1371/journal.pone.0042325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Small BJ, Rosnick CB, Fratiglioni L, Backman L. Apolipoprotein E and cognitive performance: a meta-analysis. Psychol Aging. 2004;19(4):592–600. doi: 10.1037/0882-7974.19.4.592. [DOI] [PubMed] [Google Scholar]
- 6.Fennema-Notestine C, Panizzon MS, Thompson WR, Chen CH, Eyler LT, Fischl B, Franz CE, Grant MD, Jak AJ, Jernigan TL, Lyons MJ, Neale MC, Seidman LJ, Tsuang MT, Xian H, Dale AM, Kremen WS. Presence of ApoE epsilon4 allele associated with thinner frontal cortex in middle age. J Alzheimers Dis. 2011;26(Suppl 3):49–60. doi: 10.3233/JAD-2011-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoyt BD, Massman PJ, Schatschneider C, Cooke N, Doody RS. Individual growth curve analysis of APOE epsilon 4-associated cognitive decline in Alzheimer disease. Arch Neurol. 2005;62(3):454–9. doi: 10.1001/archneur.62.3.454. [DOI] [PubMed] [Google Scholar]
- 8.Stern Y, Brandt J, Albert M, Jacobs DM, Liu X, Bell K, Marder K, Sano M, Albert S, Del-Castillo Castenada C, Bylsma F, Tycko B, Mayeux R. The absence of an apolipoprotein epsilon4 allele is associated with a more aggressive form of Alzheimer’s disease. Ann Neurol. 1997;41(5):615–20. doi: 10.1002/ana.410410510. [DOI] [PubMed] [Google Scholar]
- 9.Martins CA, Oulhaj A, de Jager CA, Williams JH. APOE alleles predict the rate of cognitive decline in Alzheimer disease: a nonlinear model. Neurology. 2005;65(12):1888–93. doi: 10.1212/01.wnl.0000188871.74093.12. [DOI] [PubMed] [Google Scholar]
- 10.Hirono N, Hashimoto M, Yasuda M, Kazui H, Mori E. Accelerated memory decline in Alzheimer’s disease with apolipoprotein epsilon4 allele. J Neuropsychiatry Clin Neurosci. 2003;15(3):354–8. doi: 10.1176/jnp.15.3.354. [DOI] [PubMed] [Google Scholar]
- 11.Cosentino S, Scarmeas N, Helzner E, Glymour MM, Brandt J, Albert M, Blacker D, Stern Y. APOE epsilon 4 allele predicts faster cognitive decline in mild Alzheimer disease. Neurology. 2008;70(19 Pt 2):1842–9. doi: 10.1212/01.wnl.0000304038.37421.cc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Farlow MR, Cyrus PA, Nadel A, Lahiri DK, Brashear A, Gulanski B. Metrifonate treatment of AD: influence of APOE genotype. Neurology. 1999;53(9):2010–6. doi: 10.1212/wnl.53.9.2010. [DOI] [PubMed] [Google Scholar]
- 13.Kleiman T, Zdanys K, Black B, Rightmer T, Grey M, Garman K, Macavoy M, Gelernter J, van Dyck C. Apolipoprotein E epsilon4 allele is unrelated to cognitive or functional decline in Alzheimer’s disease: retrospective and prospective analysis. Dement Geriatr Cogn Disord. 2006;22(1):73–82. doi: 10.1159/000093316. [DOI] [PubMed] [Google Scholar]
- 14.Negash S, Greenwood PM, Sunderland T, Parasuraman R, Geda YE, Knopman DS, Boeve BF, Ivnik RJ, Petersen RC, Smith GE. The influence of apolipoprotein E genotype on visuospatial attention dissipates after age 80. Neuropsychology. 2009;23(1):81–9. doi: 10.1037/a0014014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petersen RC, Aisen PS, Beckett LA, Donohue MC, Gamst AC, Harvey DJ, Jack CR, Jr, Jagust WJ, Shaw LM, Toga AW, Trojanowski JQ, Weiner MW. Alzheimer’s Disease Neuroimaging Initiative (ADNI): clinical characterization. Neurology. 74(3):201–9. doi: 10.1212/WNL.0b013e3181cb3e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL. A new clinical scale for the staging of dementia. Br J Psychiatry. 1982;140:566–72. doi: 10.1192/bjp.140.6.566. [DOI] [PubMed] [Google Scholar]
- 17.Petersen RC, Doody R, Kurz A, Mohs RC, Morris JC, Rabins PV, Ritchie K, Rossor M, Thal L, Winblad B. Current concepts in mild cognitive impairment. Arch Neurol. 2001;58(12):1985–92. doi: 10.1001/archneur.58.12.1985. [DOI] [PubMed] [Google Scholar]
- 18.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34(7):939–44. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 19.Fennema-Notestine C, Ozyurt IB, Clark CP, Morris S, Bischoff-Grethe A, Bondi MW, Jernigan TL, Fischl B, Segonne F, Shattuck DW, Leahy RM, Rex DE, Toga AW, Zou KH, Brown GG. Quantitative evaluation of automated skull-stripping methods applied to contemporary and legacy images: effects of diagnosis, bias correction, and slice location. Hum Brain Mapp. 2006;27(2):99–113. doi: 10.1002/hbm.20161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han X, Jovicich J, Salat D, van der Kouwe A, Quinn B, Czanner S, Busa E, Pacheco J, Albert M, Killiany R, Maguire P, Rosas D, Makris N, Dale A, Dickerson B, Fischl B. Reliability of MRI-derived measurements of human cerebral cortical thickness: the effects of field strength, scanner upgrade and manufacturer. Neuroimage. 2006;32(1):180–94. doi: 10.1016/j.neuroimage.2006.02.051. [DOI] [PubMed] [Google Scholar]
- 21.Jovicich J, Czanner S, Greve D, Haley E, van der Kouwe A, Gollub R, Kennedy D, Schmitt F, Brown G, Macfall J, Fischl B, Dale A. Reliability in multi-site structural MRI studies: effects of gradient non-linearity correction on phantom and human data. Neuroimage. 2006;30(2):436–43. doi: 10.1016/j.neuroimage.2005.09.046. [DOI] [PubMed] [Google Scholar]
- 22.Jack CR, Jr, Bernstein MA, Fox NC, Thompson P, Alexander G, Harvey D, Borowski B, Britson PJ, JLW, Ward C, Dale AM, Felmlee JP, Gunter JL, Hill DL, Killiany R, Schuff N, Fox-Bosetti S, Lin C, Studholme C, DeCarli CS, Krueger G, Ward HA, Metzger GJ, Scott KT, Mallozzi R, Blezek D, Levy J, Debbins JP, Fleisher AS, Albert M, Green R, Bartzokis G, Glover G, Mugler J, Weiner MW. The Alzheimer’s Disease Neuroimaging Initiative (ADNI): MRI methods. J Magn Reson Imaging. 2008;27(4):685–91. doi: 10.1002/jmri.21049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sled JG, Zijdenbos AP, Evans AC. A nonparametric method for automatic correction of intensity nonuniformity in MRI data. IEEE Trans Med Imaging. 1998;17(1):87–97. doi: 10.1109/42.668698. [DOI] [PubMed] [Google Scholar]
- 24.Fischl B, Salat DH, Busa E, Albert M, Dieterich M, Haselgrove C, van der Kouwe A, Killiany R, Kennedy D, Klaveness S, Montillo A, Makris N, Rosen B, Dale AM. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron. 2002;33(3):341–55. doi: 10.1016/s0896-6273(02)00569-x. [DOI] [PubMed] [Google Scholar]
- 25.Fischl B, van der Kouwe A, Destrieux C, Halgren E, Segonne F, Salat DH, Busa E, Seidman LJ, Goldstein J, Kennedy D, Caviness V, Makris N, Rosen B, Dale AM. Automatically parcellating the human cerebral cortex. Cereb Cortex. 2004;14(1):11–22. doi: 10.1093/cercor/bhg087. [DOI] [PubMed] [Google Scholar]
- 26.Dale AM, Sereno MI. Improved Localization of Cortical Activity by Combining Eeg and Meg with Mri Cortical Surface Reconstruction - a Linear-Approach. Journal of Cognitive Neuroscience. 1993;5(2):162–176. doi: 10.1162/jocn.1993.5.2.162. [DOI] [PubMed] [Google Scholar]
- 27.Dale AM, Fischl B, Sereno MI. Cortical surface-based analysis. I. Segmentation and surface reconstruction. Neuroimage. 1999;9(2):179–94. doi: 10.1006/nimg.1998.0395. [DOI] [PubMed] [Google Scholar]
- 28.Fischl B, Sereno MI, Dale AM. Cortical surface-based analysis. II: Inflation, flattening, and a surface-based coordinate system. Neuroimage. 1999;9(2):195–207. doi: 10.1006/nimg.1998.0396. [DOI] [PubMed] [Google Scholar]
- 29.Desikan RS, Segonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, Buckner RL, Dale AM, Maguire RP, Hyman BT, Albert MS, Killiany RJ. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31(3):968–80. doi: 10.1016/j.neuroimage.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 30.Fennema-Notestine C, Hagler DJ, Jr, McEvoy LK, Fleisher AS, Wu EH, Karow DS, Dale AM. Structural MRI biomarkers for preclinical and mild Alzheimer’s disease. Hum Brain Mapp. 2009;30(10):3238–53. doi: 10.1002/hbm.20744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Makris N, Meyer JW, Bates JF, Yeterian EH, Kennedy DN, Caviness VS. MRI-Based topographic parcellation of human cerebral white matter and nuclei II. Rationale and applications with systematics of cerebral connectivity. Neuroimage. 1999;9(1):18–45. doi: 10.1006/nimg.1998.0384. [DOI] [PubMed] [Google Scholar]
- 32.Buckner RL, Head D, Parker J, Fotenos AF, Marcus D, Morris JC, Snyder AZ. A unified approach for morphometric and functional data analysis in young, old, and demented adults using automated atlas-based head size normalization: reliability and validation against manual measurement of total intracranial volume. Neuroimage. 2004;23(2):724–738. doi: 10.1016/j.neuroimage.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 33.Holland D, Dale AM. Nonlinear registration of longitudinal images and measurement of change in regions of interest. Med Image Anal. 2011;15(4):489–97. doi: 10.1016/j.media.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holland D, Brewer JB, Hagler DJ, Fennema-Notestine C, Dale AM. Subregional neuroanatomical change as a biomarker for Alzheimer’s disease. Proc Natl Acad Sci U S A. 2009;106(49):20954–9. doi: 10.1073/pnas.0906053106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gutierrez-Galve L, Lehmann M, Hobbs NZ, Clarkson MJ, Ridgway GR, Crutch S, Ourselin S, Schott JM, Fox NC, Barnes J. Patterns of cortical thickness according to APOE genotype in Alzheimer’s disease. Dement Geriatr Cogn Disord. 2009;28(5):476–85. doi: 10.1159/000258100. [DOI] [PubMed] [Google Scholar]
- 36.Jernigan TL, Archibald SL, Fennema-Notestine C, Gamst AC, Stout JC, Bonner J, Hesselink JR. Effects of age on tissues and regions of the cerebrum and cerebellum. Neurobiol Aging. 2001;22(4):581–94. doi: 10.1016/s0197-4580(01)00217-2. [DOI] [PubMed] [Google Scholar]
- 37.Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, Holtzman DM, Betsholtz C, Armulik A, Sallstrom J, Berk BC, Zlokovic BV. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485(7399):512–6. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Austad S. Why women live longer than men: sex differences in longevity. Gender Medicine. 2006;3:79–92. doi: 10.1016/s1550-8579(06)80198-1. [DOI] [PubMed] [Google Scholar]
- 39.Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, Gamst A, Holtzman DM, Jagust WJ, Petersen RC, Snyder PJ, Carrillo MC, Thies B, Phelps CH. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):270–9. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Jr, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, Mohs RC, Morris JC, Rossor MN, Scheltens P, Carrillo MC, Thies B, Weintraub S, Phelps CH. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263–9. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]