

Abstract
It has been known for about seventy years that bone, in all vertebrates, contains uniquely high citrate levels. However, the role of citrate, its source, its regulation, and its implication in normal bone formation and in bone disorders have remained largely unknown. For the past thirty-five years, the relationship of citrate in bone has been a neglected area of attention and research. It has recently been discovered that citrate is critical for the structure of the apatite nanocrystal, and is required to impart the important properties of bone such as its stability, strength, and resistance to fracture. This brings to focus the need for a renewed interest and research into the relationships of citrate in bone formation. A most fundamental question that must be resolved is “What is the source of citrate in bone?”. This presentation provides a historical review of the early research to the present status of citrate implications in bone. This leads to a new concept of the role of osteoblasts as specialized citrate producing cells that provide the source of citrate in bone formation; i.e. the “osteoblast citration” process. This also brings into focus a new insight into the role of zinc in bone in relation to osteoblast citrate production. The genetic/hormonal/metabolic relationships of “net citrate production” are described. The intent of this presentation is to provide the background for a new perspective of the important implications of osteoblasts and citrate in bone formation; which, hopefully, will stimulate a renewed interest and essential research.
Keywords: Bone formation, osteoblasts, citrate, zinc, citrate metabolism, vitamin D
INTRODUCTION
In 1941, Dickens reported [1] that bone contains extremely high levels of citrate, which had been confirmed in subsequent reports [e.g. 2–6]. As revealed in Table 1, bone and teeth citrate levels are generally ~20–80 μmols/gram, which is ~100–400-fold higher than plasma and most soft tissues, with some notable exception such as prostate. The citrate comprises ~1.6 % of the bone content; and about 80% of the total body citrate resides in bone. This bone citrate relationship is conserved in all vertebrates. As it will be discussed below, the production and accumulation of high citrate levels require unique and specialized metabolic/functional relationships that do not generally exist in most mammalian tissues. These conditions dictate that citrate is a major component of bone that must be involved in some essential process and/or property required for normal bone formation and maintenance. Despite this, the role of citrate in bone, its source, its regulation, and other critical issues remain unresolved over the seventy years since first identified as a major component of bone.
Table 1.
Typical Concentrations of Citrate and Zinc in Tissues
| Citrate | Zinc | |
|---|---|---|
| (μmols/g) | ||
| Bone/teeth | ~20–80 | ~2–5 |
| Cartilage | ~1 | ------ |
| Blood plasma | ~0.2 | ~0.015 |
| Other soft tissues | ~0.2–0.4 | ~0.2–0.6 |
| Prostate Tissue | ~10–15 | ~3–5 |
| Prostate fluid | ~50–150 | ~6–8 |
This review will provide a historical background of the bone citrate relationship up to recently important reported studies of the implication of citrate in normal bone formation; followed by a description of the unique cellular genetic/metabolic requirements for citrate production; and evidence for the role of the osteoblasts as highly specialized “citrate-producing cells” for the process of “citration” as a required event in bone formation. The presentation also focuses on the important role of zinc in the metabolic pathway of osteoblast citrate production, which heretofore has never been considered or described. To the best of our knowledge, this will be the first such “comprehensive” description and review in recent decades that focuses on the citrate relationship in bone. It is intended to bring attention to the important implications of citrate in normal bone; its potential involvement in bone disorders; and its importance in bone repair and regeneration; which have been largely ignored by the contemporary clinical and biomedical research community. Hopefully, the presentation and the new concept that we propose will stimulate the support for research into the role of citrate and the osteoblast in bone formation.
HISTORICAL PERSPECTIVE
Following the initial identification in 1941 until ~1975, significant interest and reported research studies were prevalent concerning the source of citrate, the metabolism of citrate, and the role of hormones (particularly parathyroid hormone, calcitonin and vitamin D) in the regulation of citrate. Indeed, this early focus on bone citrate and citrate homeostasis provided one of the authors (LCC) the opportunity to receive NIH grant awards and publication of several papers ~1965–1978 concerning citrate homeostasis and hormonal regulation relationships. However, during the following period of ~1980–2010, interest and research concerning the implications of citrate in bone markedly diminished and essentially disappeared. This caused Schwartz et al. [2] in 2010 to state “After about 1975, interest in the role of citrate in bone waned, and many recent texts on bone do not identify citrate as a major constituent…” Hu et al. [6] also commented that “citrate is no longer even mentioned in most of the prominent literature on the bone nanocomposite published during the last thirty years”.
Despite the early research efforts and activities, the important issues such as the role of citrate in bone and the source of citrate were not resolved. At that time, the research was limited to studies with “crude” bone preparations such as bone slices and homogenates, which included mixed populations of various cell types and acellular components of bone. The advantages of isolated purified cell preparations, the availability of cell lines, and genetic/proteomic manipulation and identification capabilities did not exist. Methodology and technology advances for cellular genetic/metabolic studies that now exist were not available. These contemporary resources have since provided the capability to address the issues of the implications of citrate in bone. However, missing over the past three decades has been the interest, and even the recognition, of the citrate relationship in bone.
Beginning ~1975 until now, we (authors LCC and RBF) diverted our experience, interest and focus to the importance of citrate metabolism relationships in normal prostate and prostate cancer. As shown in Table 1, the prostate gland and bone are similar in regard to the unique characteristic of extremely high citrate and zinc levels. So, one can reasonably inquire as to the reason at this time for our revisiting and addressing the unresolved issues of the implications of citrate in bone formation and maintenance. For us, the issue re-surfaces as a result of recent exciting studies [6–9] that have identified an indispensible role of citrate in the apatite nanocrystal structure; which is responsible for imparting the important properties of bone such as its stability, strength, and resistance to fracture. The implications of this relationship in normal bone formation and maintenance; in normal growth and development, in bone repair and regeneration processes; and in the etiology, treatment and management of bone disorders, dictate the need for accelerated and extensive research to elucidate the unresolved issues as described above.
WHAT IS THE ROLE OF CITRATE IN BONE?
An early prevailing view of the role of citrate in bone was in the possible formation of a calcium-phosphate-citrate complex that occurs in bone. It was proposed that the presence of citrate in this complex would serve to increase the solubility of the calcium-phosphate complex. Dixon and Perkins in 1953 [10] concluded that “The facts make it seem likely that calcification mechanisms may be influenced by the amount of citrate present in the circulating fluids and at the site of calcification… The mechanism for the production of a local high concentration of citric acid therefore exists in bone. Such citric acid may be co-precipitated with calcium phosphate during its deposition, in which case it may influence the amount of deposition by holding up calcium in a complex form.” This prevailing view for nearly 60 years was neither substantially advanced nor refuted due to the absence of methodology and needed research to address the issue; so it remained dormant and forgotten until the recent studies beginning ~2010.
A new insight into the role of citrate has now been provided by the recent discovery of the incorporation and relationship of citrate in the apatite nanocrystal structure [6–9]. The studies demonstrate that citrate is a strongly bound, integral part of the apatite nanocrystal structure. The bound citrate covers about 1/6 of the available apatite surface area in bone and accounts for 5.5 wt% of the organic matter in bone. Citrate provides more carboxylate for calcium binding in bone than that provided by all of the non-collagen proteins combined. Most importantly, the incorporation of citrate into the nanocrystal structure is a determinant of the crystal thickening. This is critical for imparting the important properties of bone such as its stability, strength, and resistance to fracture. These newly discovered relationships establish an indispensible role of citrate that has critical implications in virtually every aspect of bone; such as skeletal growth and development, injury and repair, bone disorders, and more. A new era of rejuvenated interest, focus, and research is now essential. As Hu et al emphasized [6] “The discovery of apatite-bound citrate in the bone nanocomposite leads to various intriguing research questions. What is the source of citrate in bone? At what stage of bone development does citrate appear? How is the abundance of citrate on the nanocrystal surface controlled? Is the citrate concentration abnormal in diseased bone?”
THE SOURCE OF CITRATE IN BONE FORMATION
The most fundamental, yet unresolved, question is “what is the source of citrate in bone formation?” The resolution of this question is the antecedent for addressing the other important issues of the implications of citrate in bone. Early metabolic studies [e.g. 4, 10, 11] demonstrated that the concentration of citrate increased in “crude” bone preparations over time. This resulted from the de novo synthesis of citrate from precursor substrates coupled to the limited oxidation of citrate. The difficulty with these early studies with crude bone preparations was the complexity of the preparations in which the net results obtained was influenced by the various bone components in the reaction system. Consequently the specific cellular source of citrate and the metabolic relationships associated with citrate production were not identified; and have remained unidentified as of this time. There have been no reported studies (to the best of our knowledge) since ~1970 that have addressed this issue. So the first impending questions are: 1) What cell type is responsible for the production of citrate in bone?; and 2) What is the metabolic pathway associated with the production of high citrate levels, which we refer to as “net citrate production?”
“NET CITRATE PRODUCTION”: WHAT IS IT?
The high citrate content in bone occurs and exists as a result of citrate produced by the cellular metabolism of some unidentified specific bone cell type (likely to be the osteoblasts as described below). Consequently, the cellular metabolic pathway for high citrate production must be described. All mammalian cells produce citrate; which is the key intermediate in the operation of the Krebs cycle (also called “the citric acid cycle”) as shown in Fig. (1). It is synthesized in the mitochondria from acetyl CoA; which is derived from glycolysis leading to pyruvate formation as the substrate for acetyl CoA production. In some cases, mitochondrial beta oxidation of fatty acids can supply acetyl CoA. The acetyl CoA condenses (citrate synthase reaction) with oxalacetate (OAA) to produce citrate. The mitochondrial citrate is typically utilized predominantly by its oxidation via the Krebs cycle; which, by coupled phosphorylation via terminal oxidation, provides the major source of cellular ATP production. In some special instances, such as in highly proliferating cells, the mitochondrial citrate is utilized as the cytosolic source of acetyl CoA; which is required for de novo lipid biosynthesis.
Fig. 1.
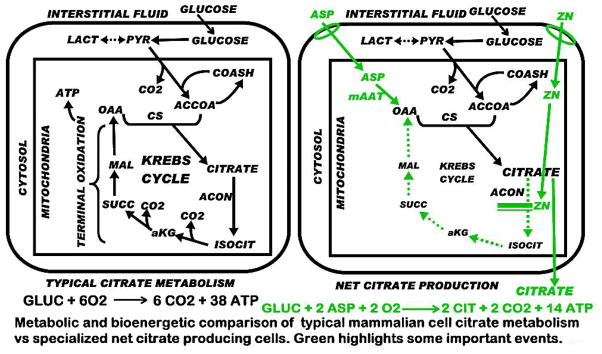
Comparison of typical mammalian cell citrate metabolism and the metabolic pathway for net citrate production. Green highlighting represents important changes associated with net citrate production.
Under these typical metabolic relationships, the existing cellular concentration of citrate is not a measurement of the production of citrate. The total amount of citrate produced is the existing citrate concentration plus the citrate that was utilized by the cell; which would be the “gross citrate production”. We define the “net citrate production” as the component of the synthesized citrate that is not utilized by the cells, and remains as accumulated citrate. In typical cell metabolism, citrate that is synthesized is essentially utilized; so that high citrate accumulation does not occur.
NET PRODUCTION OF HIGH LEVELS OF CITRATE IS A UNIQUE SPECIALIZED METABOLIC/FUNCTION RELATIONSHIP
There are serious metabolic and energetic alterations and consequences of net citrate production (Fig. 1). In typical cell metabolism the complete oxidation of 1 mole of glucose produces 6CO2 and 38ATP. Of this, 24ATP is due to the oxidation of citrate via the Krebs cycle. In net citrate production, in which citrate is accumulated rather than oxidized, the utilization of glucose results in 14ATP. Thus, the cell sacrifices ~70% ATP production from glucose (Fig. 1) to achieve net citrate production; which demonstrates the unique specialization of citrate-producing cells. To compensate for this loss of ATP production, the citrate-producing cells will exhibit an increased aerobic glycolysis for the accelerated increase in production of ATP (as in prostate cells); as well as an increase in pyruvate availability for acetyl CoA production for continued citrate synthesis.
Also, in typical cell metabolism, when citrate is oxidized via the Krebs cycle, OAA is regenerated; which then reacts with acetyl CoA from glucose (via pyruvate) to synthesize citrate. In this scheme, citrate oxidation involves the loss of 2 carbons (the acetyl CoA component) from the metabolic pool; but the 4 carbon component (OAA) is regenerated, and used for new synthesis of citrate. In net citrate production, the entire 6 carbons of citrate are lost. Therefore, continued production of citrate requires the 2-carbon acetyl CoA produced from glucose, plus a 4-carbon source for OAA production. Consequently, citrate producing cells require a unique metabolic alteration that will provide a continuing 4-carbon source for OAA production.
The key altered metabolic reaction in net citrate producing cells is the inhibition of citrate oxidation via the Krebs cycle. The maximal accumulation of citrate will be achieved by the prevention of citrate entry into the oxidative phase of the Krebs cycle; which is the m-aconitase reaction. Therefore, the citrate producing cells must exhibit some mechanism that inhibits or minimizes m-aconitase activity; which essentially aborts the Krebs cycle (Fig. 1).
Thus it becomes apparent that citrate producing cells are highly specialized cells in which a unique altered metabolic pathway and bioenergetic transformation are required. The above description represents some major requirements for net citrate production; but does not describe how the cell achieves the actual metabolic requirements. There is only one citrate-producing cell-type in which the pathway of net citrate production has been identified, i.e. citrate producing prostate epithelial cells; as we elucidated over several years of research (Fig. 2).
Fig. 2.
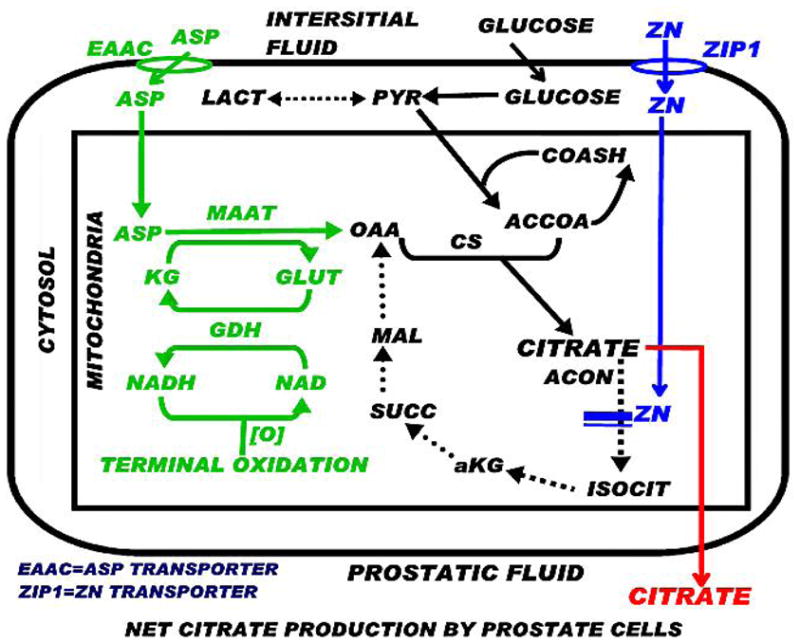
The pathway of net citrate production as exists in prostate cells. Green, blue, and red represent key events for net citrate production.
In prostate cells, aspartate transamination by mAAT provides OAA for citrate synthesis; i.e. the “glutamate-aspartate-citrate pathway” [12]. A continual source of aspartate is required, which is obtained by the transport of aspartate from circulation into the cell. Therefore, aspartate is an essential amino acid in these cells. In contrast, most mammalian cells synthesize aspartate from OAA; so aspartate is generally characterized as a non-essential amino acid. In citrate-producing cells, the mAAT reaction is in the direction of OAA production, which is opposite to its direction in most mammalian cells.
Regarding the issue of the mechanism for inhibition of citrate oxidation, this is achieved in prostate cells by the accumulation of high cellular mitochondrial zinc; which we identified to be a specific inhibitor of m-aconitase activity (discussed below). These metabolic relationships as identified in prostate cells should be applicable and likely exists (perhaps with some variation) in all citrate producing cells. As shown in Fig. (2), net citrate production requires alterations of key enzymes and transporters; which, in turn, requires altered expression of their respective “metabolic” genes. Thus, cells that function for “net citrate production” are highly specialized cells in which genetic/metabolic transformation has occurred.
THE IMPLICATIONS OF ZINC IN NET CITRATE PRODUCTION
It is notable that bone also contains high zinc levels [13–17] along with high citrate, as it exists in prostate cells. Some early studies had recognized that impaired oxidation of citrate was involved in bone citrate production; but the prevailing view [such as 4, 10, 11] was that limited isocitrate dehydrogenase was the cause of decreased citrate oxidation although this was never established. Our studies established that inhibited m-aconitase activity is the reaction that limits citrate oxidation and permits the accumulation of citrate in prostate cells. We identified that the high cellular zinc level, particularly the high mitochondrial zinc concentration, is a specific inhibitor of m-aconitase activity [18].
The accumulation of zinc in mammalian cells is firstly dependent upon the uptake of zinc from the extracellular fluid; i.e. the interstitial fluid derived from blood plasma. Cellular zinc uptake requires a zinc transport process, which is generally provided by a ZIP-family zinc uptake transporter. ZIP1 (SLC39A1) has been identified as the important functional zinc uptake transporter associated with the accumulation of zinc in prostate cells; and also in osteoblasts [19]. A similar corresponding relationship probably exists for all specialized citrate producing cells; which are also likely to be zinc-accumulating cells. This expectation is supported by the citrate relationship in vitamin D-deficient rachitic bone. Citrate is decreased in vitamin D-deficient rickets, and treatment with vitamin D restores bone citrate levels. Vitamin D has been shown to increase citrate predominantly due to inhibition of mitochondrial citrate oxidation [20–24]; and not due to increased citrate synthesis by bone slices. Also, vitamin D inhibition of citrate oxidation by bone preparation is mimicked by fluoroacetate treatment [20]; and fluoroacetate is converted to fluorocitrate, which is a specific inhibitor of m-aconitase activity as is zinc.
The importance of zinc in bone is well described by Vidica-Gurban and Mederle [13] who state “Zn2+ is the most abundant trace element in bone, being present at a concentration of up to 300 μg/g bone, and it has been considered an important factor in bone metabolism… Zinc ions (Zn2+) are stimulating for osteoblasts proliferation and differentiation.” This is echoed in the excellent review of Yamaguchi [14] and in other reports that demonstrate the importance of zinc in bone formation, and the importance of zinc deficiency in osteoporosis. It is now important to consider and investigate the possible involvement of these zinc relationships in regard to osteoblast citrate production.
ARE OSTEOBLASTS THE SPECIALIZED “CITRATE-PRODUCING” CELLS IN BONE?
To our knowledge, there are no reported studies regarding citrate production by osteoblasts. Recent reported studies of osteoblast intermediary energy metabolism [such as 25–30] have not included any involvement of the specialized metabolic relationships of citrate and/or zinc in osteoblasts. Chen et al in their 1999 report of zinc effects on bone formation [31] emphasized that “Studies of bone in tissue culture typically do not include addition of zinc specifically… Interpretation of data from such studies should take this variable (zinc) into account.” The important relationship of zinc accumulation on cellular citrate metabolism as described above (Fig. 2), and its inhibition of terminal oxidation and respiration [32, 33], indicate that osteoblast metabolic studies need to be conducted in the presence of physiological levels of zinc in order to determine metabolic/bioenergetic relationships that likely represent the in situ osteoblast metabolic/functional role in bone formation.
What is the currently available evidence that supports the contention that the osteoblasts are the specialized citrate-producing cells in bone? The recent discovery of the essential citrate involvement in the apatite nanocrystal structure makes it evident that this is an important role in bone formation. Therefore, the production of citrate must be associated with the cells responsible for the process of bone formation; i.e., most likely the osteoblasts. It has been shown that zinc uptake and accumulation is increased during the differentiation of mesenchyme stem cells to osteoblasts and the onset of mineralization; due to the up regulation of ZIP1 [19]. Such relationships would suggest that the increased uptake and accumulation of zinc could result in increased citrate production as a result of its inhibition of m-aconitase activity.
Aside from the speculation based on the above information, there exist no reported studies of citrate production by osteoblasts. Such evidence is necessary to support the concept that the osteoblasts are the specialized -producing cells in bone. To obtain initial supporting evidence, we conducted some preliminary experiments to determine if osteoblasts might exhibit the metabolic capability of net citrate production. We have been able to show that differentiated osteoblasts are capable of net citrate production in contrast to their undifferentiated MC3T3 cells (Table 2). In addition, in the presence of zinc-supplemented medium, citrate production by the differentiated osteoblasts was increased four-fold (Table 3). While these results must be confirmed and extended by additional studies, they serve the purpose to provide supporting evidence for the “concept” that is being proposed.
Table 2.
Citrate Production of Undifferentiated MC3T3 Cells Versus Differentiate Osteoblasts (nmols Produced)
| Days | 1–3 | 4–6 |
|---|---|---|
| Undiff cells | nil | nil |
| Diff OSBs | 16+/−5 | 18+/−3 |
Table 3.
Zinc Effect on Murine Differentiated Osteoblast Citrate Production
| Zn-uM | Cit-nmols |
|---|---|
| 0 | 24 ± 8 |
| 5 | 102 ± 22 |
“OSTEOBLAST CITRATION” AS AN ESSENTIAL PROCESS IN BONE FORMATION
The discovery of the relationship of citrate in the nanocrystal structure leads us to coin a new terminology; i.e. “citration” as a process or event in bone formation. We couple this to the expectation that the osteoblasts are the specialized citrate-producing cells; which provides the new concept of “osteoblast citration” in bone formation. Moreover, when considering the mineralization role of osteoblast in bone formation, it now becomes evident that “citration” must be included in the process. Mineralization without “citration” will not result in formation of normal bone; i.e. bone that exhibits its important properties such as stability, strength and resistance to fracture.
OSTEOGENIC DIFFERENTIATION AND DEVELOPMENT OF SPECIALIZED CITRATE-PRODUCING OSTEOBLASTS
The citrate relationship brings to focus the need to assess the prevailing view of the osteogenic differentiation of mesenchymal stem cells leading to the development of osteoblasts for bone formation. Fig. (3) is a representation of the course of events in the transformation of the mesenchymal cells to osteoblasts. The current hallmark for determination of the functional osteoblasts is the achievement of mineralization and production of hydroxyapatite for normal bone formation. This capability determines the optimal conditions and factors that are required to achieve the osteogenic differentiation to bone-forming osteoblasts.
Fig. 3.
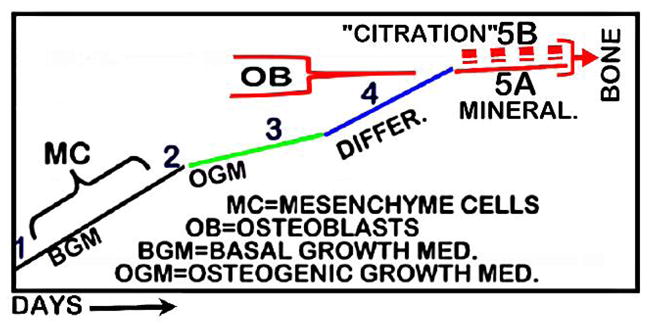
Representation of the current concept of MC cell differentiation to OB cell; with modification for inclusion of the concept of OB “citration” (5B) required for normal bone formation and its properties.
Omitted from this view is consideration of any role or involvement of osteoblast production of citrate; the absence of which will not result in the formation of normal bone. Thus, those conditions that are employed to optimize the mineralization process might not facilitate, and might even suppress, the genetic/metabolic transformation of mesenchymal cells to differentiated citrate-producing osteoblasts (Figs. 1 and 2). For example, we suggest that dexamethasone is likely to suppress osteoblast citrate production; although its inclusion in osteogenic medium facilitates cell proliferation and the mineralization process. The reason for our expectation is the gluconeogenic actions of glucocorticoids; which include inhibition of glycolysis and glucose utilization, prevention of citrate synthesis, increased fatty acid beta oxidation and citrate oxidation. Considering the importance of citrate in bone strength and resistance to fracture, this relationship likely contributes to the fracture manifestations of hypercortisolism as in the osteoporotic association in Cushing’s disease, and in sustained use of glucocorticoids. In regard to other adverse effects of dexamethasone, Zhou et al. [34] determined that vitamin D is a preferable substitute for dexamethasone in achieving proliferation and mineralization in developing osteoblasts. The established effect of vitamin D in promoting increased citrate production would indicate its advantageous inclusion as an important factor in optimizing the development of osteoblast citration in concert with mineralization as shown in Fig. (3).
THE REGULATION OF NET CITRATE PRODUCTION
Compared to the citrate metabolism that typifies most normal mammalian cells, the development of specialized cells with the capability of net citrate production requires metabolic alterations involving key enzymes and transporters similar to those shown in Figs. (1 and 2). These metabolic alterations are achieved by changes in the expression of the respective “metabolic” genes; especially under the influence of hormonal regulation; as well as the influence of cellular environment conditions on the activity of the key reaction. The events that have been identified for prostate cells (Fig. 2) are likely to be applicable to osteoblasts; and the hormonal regulation is also likely to be applicable, with some cell-specific modifications. While there are many metabolic alterations associated with net citrate production, we have focused on those key reactions that are directly involved with the synthesis of citrate and its oxidation. These are represented by ZIP1 zinc transporter, EAAC1 aspartate transporter, mAAT enzyme reaction, and m-aconitase reaction. It is important to note that citrate production by prostate cells is regulated by testosterone and prolactin; and in both cases the hormones stimulate citrate production [35–37]. This is achieved, at least in part, by the effects of testosterone and prolactin in increasing the gene expression for ZIP1, EAAC1, and mAAT. Testosterone and prolactin also increase the expression of PDHE1α, which increases PDH activity and the formation of acetyl CoA for citrate synthesis [37]. In concert with this regulation for increased synthesis of citrate; both hormones also increase the expression of ZIP1, which increases the uptake and accumulation of zinc, and inhibits m-aconitase activity and citrate oxidation; thereby resulting in the accumulation of citrate. These collective events demonstrate the coordinated genetic/hormonal/metabolic events that are altered and regulated to achieve net citrate production in specialized cells.
In relation to the osteoblasts and bone formation, there is little information regarding the regulation of citrate production. As we described above, it seems evident that vitamin D stimulates citrate levels in bone by its inhibition of citrate oxidation; which appears to be due to inhibition of m-aconitase. How this is achieved and which cell type is involved remain unknown. ZIP1 has been identified as a functional transporter for the uptake and accumulation of zinc in osteoblasts as in prostate cells (Fig. 2); and its up-regulation is essential for the differentiation of mesenchyme cells to osteoblasts [19]. This is likely to be associated with corresponding citrate production.
Estrogen is a known osteogenic factor that exerts direct effects on bone mineralization by osteoblasts [38, 39], and its effects are enhanced by zinc. This, along with the relationship of zinc deficiency and estrogen deficiency in osteoporosis [13, 14] would suggest a possible targeted role of estrogen at the production of citrate by osteoblasts.
The important role and actions of testosterone in stimulating prostate citrate production could also have similar implications in the regulation of osteoblast citrate production. Low testosterone levels as found in elderly men are associated with an increased risk of osteoporosis and fracture; and testosterone treatment reportedly increases their bone mineral density and reduces fracture [40]. Although still inconclusive, antiosteoporotic testosterone treatment reportedly increases bone formation [41, 42]. The complexity of confounding factors in such studies prevents the identification of possible direct testosterone effects on osteoblast bone formation, including citrate production. This is also confounded by the dual role of testosterone effects on bone cells being mediated directly by androgen receptor and/or by aromatization to estrogen [43]. The fact that testosterone is the major regulator of prostate citrate production and osteoblasts exhibit androgen receptor and direct responses to DHT (5OH-testosterone) makes it plausible to expect that it contributes to the regulation of osteoblast citrate production.
Obviously, the above description is highly speculative; and does not include other potential hormonal regulators of osteoblast citrate production. The presentation reveals the very limited information regarding the regulation of citrate in bone formation. The fact is that these important issues of factors involved and their mechanisms in the regulation of citrate production by osteoblasts during bone formation cannot be established until the metabolic pathway for osteoblast net citrate production is identified.
OTHER ASPECTS OF CITRATE RELATIONSHIPS IN BONE
This review has focused on the important implications of the role of citrate and the osteoblasts as citrate-producing cells in the process of bone formation. However, other citrate relationships in bone need to be considered and addressed. For example, the resorption of bone, such as in bone turnover, involves the localized loss of citrate via its entry into circulation. This seems evident from the hyperciticemic effect associated with bone resorption, as with the action of parathyroid hormone; although other tissues contribute to this effect [44–46]. Consequently, there is a citrate turnover process involved at sites where remodeling exists. The mechanism and cellular involvement associated with the citrate relationship during bone resorption remains largely unknown. This too was an issue of considerable early interest and research; particularly in regard to the early studies of the mechanism of actions of parathyroid hormone and calcitonin. It is likely that the osteoclasts play an important role with specific actions regarding the bone resorption citrate relationship; however this association has not received attention in recent years. The broader issue is the role of bone citrate relationships in the physiological homeostatic regulation of citrate. It is important to recognize that the plasma citrate concentration is tightly regulated to maintain a constant concentration, which in humans is 112+/−6 uM. Bone citrate and its regulation is an important component of the homeostatic maintenance of citrate. These are important issues to address; but are beyond the scope of this review.
CONCLUSIONS
There now exists compelling evidence that citrate is indispensible and essential for the formation of normal bone that exhibits the important properties of stability, strength, and resistance to fracture. In answer to the important question, “What is the source of citrate in bone formation?”, we present the concept with supporting evidence that the osteoblasts are the specialized citrate-producing cells for bone formation. This new insight provides the basis to pursue the critical issues of the implications of the citrate relationship in bone. What are the genetic/metabolic changes required for the osteogenic differentiation of mesenchymal stem cells to citrate-producing osteoblasts? What are the hormonal factors that regulate this capability, and when during the differentiation process does this occur? How is osteoblast “citration” synchronized in concert with mineralization? What are the implications of the citrate relationship in bone disorders such as osteoporosis? What are the implications in bone regeneration and repair; and in the formulation of synthetic bone materials? These and other relevant issues need to be addressed in order to provide a new understanding of bone biology and pathophysiology.
Footnotes
Send Orders of Reprints at reprints@benthamscience.org
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.
This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/), which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
References
- 1.Dickens F. The citric acid content of animal tissues with reference to its occurrence in bone and tumour. Biochem J. 1941;35:1011–23. doi: 10.1042/bj0351011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schwarcz HP, Agur K, Jantz LM. A new method for determination of postmortem interval: citrate content of bone. J Forensic Sci. 2010;55:1516–22. doi: 10.1111/j.1556-4029.2010.01511.x. [DOI] [PubMed] [Google Scholar]
- 3.Knuuttilla M, Lappalainen R, Alakuijala P, Lammi S. Statistical evidence for the relation between citrate and carbonate in human cortical bone. Calcif Tiss Int. 1985;37:363–6. doi: 10.1007/BF02553702. [DOI] [PubMed] [Google Scholar]
- 4.Kenny A, Draskczy P, Goldhaber P. Citric acid production by resorbing bone in tissue culture. Am J Physiol. 1959;197:502–4. doi: 10.1152/ajplegacy.1959.197.2.502. [DOI] [PubMed] [Google Scholar]
- 5.Taylor TG. The nature of bone citrate. Biochim Biophys Acta. 1960;39:148–9. doi: 10.1016/0006-3002(60)90131-1. [DOI] [PubMed] [Google Scholar]
- 6.Hu YY, Rawal A, Schmidt-Rohr K. Strongly bound citrate stabilizes the apatite nanocrystals in bone. Proc Natl Acad Sci USA. 2010;107:22425–9. doi: 10.1073/pnas.1009219107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu Y-Y, Liu XP, Ma X, et al. Biomimetic self-assembling copolymer_hydroxyapatite nanocomposites with the nanocrystal size controlled by citrate. Chem Mater. 2011;23:2481–90. [Google Scholar]
- 8.Schmidt-Rohr K. Citrate key in bone’s nanostructure. Science Daily. 2011 Available from http://www.sciencedaily.com/releases/-2011/06/110608153548.htm.
- 9.Xie B, Nancollas GH. How to control the size and morphology of apatite nanocrystals in bone. Proc Natl Acad Sci USA. 2010;107:22369–70. doi: 10.1073/pnas.1017493108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dixon TF, Perkins HR. Citric acid and bone metabolism. Biochem J. 1952;52:260–5. doi: 10.1042/bj0520260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seifter E, Lavine LS. Aspects of citric acid chemistry related to bone. Bull N Y Acad Med. 1961;37:156–66. [PMC free article] [PubMed] [Google Scholar]
- 12.Franklin RB, Costello LC. Glutamate dehydrogenase in rat ventral prostate and a proposed aspartate-glutamate pathway of citrate synthesis. J Urol. 1984;132:1239–43. doi: 10.1016/s0022-5347(17)50113-5. [DOI] [PubMed] [Google Scholar]
- 13.Vidica-Gurban C, Mederle O. The OPG÷RANKL system and zinc ions are promoters of bone remodeling by osteoblast proliferation in postmenopausal osteoporosis. Rom J Morphol Embryol. 2011;52:1113–9. [PubMed] [Google Scholar]
- 14.Yamaguchi M. Role of nutritional zinc in the prevention of osteoporosis. Mol Cell Biochem. 2010;338:241–54. doi: 10.1007/s11010-009-0358-0. [DOI] [PubMed] [Google Scholar]
- 15.Takasugi S, Matsui T, Omori H, Yano H. Excess calcium increases bone zinc concentration without affecting zinc absorption in rats. Biol Trace Elem Res. 2007;116:311–20. doi: 10.1007/BF02698015. [DOI] [PubMed] [Google Scholar]
- 16.Walravens PA. Zinc metabolism and its implications in clinical medicine. West J Med. 1979;130:133–42. [PMC free article] [PubMed] [Google Scholar]
- 17.Gaubeur I, Avila-Terra LH, Masini JC, Suárez-Iha ME. Spectrophotometric flow injection methods for zinc determination in pharmaceutical and biological samples. Anal Sci. 2007;23:1227–31. doi: 10.2116/analsci.23.1227. [DOI] [PubMed] [Google Scholar]
- 18.Costello LC, Liu Y, Franklin RB, Kennedy MC. Zinc inhibition of mitochondrial aconitase and its importance in citrate metabolism of prostate epithelial cells. J Biol Chem. 1997;272:28875–81. doi: 10.1074/jbc.272.46.28875. [DOI] [PubMed] [Google Scholar]
- 19.Tang Z, Sahu SN, Khadeer MA, Bai G, Franklin RB, Gupta A. Overexpression of the ZIP1 zinc transporter induces an osteogenic phenotype in mesenchymal stem cells. Bone. 2006;38:181–98. doi: 10.1016/j.bone.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 20.Steenbock H, Bellin SA. Vitamin D and tissue citrate. J Biol Chem. 1953;205:985–91. [PubMed] [Google Scholar]
- 21.Carlson A, Hollunger G. The effect of vitamin D on the citric acid metabolism. Acta Physiol Scand. 954(31):317–33. doi: 10.1111/j.1748-1716.1954.tb01143.x. [DOI] [PubMed] [Google Scholar]
- 22.Norman AW, DeLuca HF. Vitamin D and the incorporation of [1-14C] acetate into the organic acids of bone. Biochem J. 1964;91:124–30. doi: 10.1042/bj0910124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Luca HF, Gran FC, Steenbock H. Vitamin D and citrate oxidation. J Biol Chem. 1957;224:201–8. [PubMed] [Google Scholar]
- 24.De Luca HF, Gran FC, Steenbock H, Reiser S. Vitamin D and citrate oxidation by kidney mitochondria. J Biol Chem. 1957;228:469–74. [PubMed] [Google Scholar]
- 25.Pattappa G, Heywood HK, de Bruijn JD, Lee DA. The metabolism of human mesenchymal stem cells during proliferation and differentiation. J Cell Physiol. 2011;226:2562–70. doi: 10.1002/jcp.22605. [DOI] [PubMed] [Google Scholar]
- 26.Komarova SV, Ataullakhanov FI, Globus RK. Bioenergetics and mitochondrial transmembrane potential during differentiation of cultured osteoblasts. Am J Physiol. 2000;279:C1220–9. doi: 10.1152/ajpcell.2000.279.4.C1220. [DOI] [PubMed] [Google Scholar]
- 27.Mischen BT, Follmar KE, Moyer KE, et al. Metabolic and functional characterization of human adipose-derived stem cells in tissue engineering. Plast Reconstr Surg. 2008;122:725–38. doi: 10.1097/PRS.0b013e318180ec9f. [DOI] [PubMed] [Google Scholar]
- 28.Malladi P, Xu Y, Chiou M, Giaccia AJ, Longaker MT. Effect of reduced oxygen tension on chondrogenesis and osteogenesis in adipose-derived mesenchymal cells. Am J Physiol. 2006;290:C1139–46. doi: 10.1152/ajpcell.00415.2005. [DOI] [PubMed] [Google Scholar]
- 29.Grayson WL, Zhao F, Bunnell B, Ma T. Hypoxia enhances proliferation and tissue formation of human mesenchymal stem cells. Biochem Biophys Res Commun. 2007;358:948–53. doi: 10.1016/j.bbrc.2007.05.054. [DOI] [PubMed] [Google Scholar]
- 30.Chen CT, Shih YR, Kuo TK, Lee OK, Wei YH. Coordinated changes of mitochondrial biogenesis and antioxidant enzymes during osteogenic differentiation of human mesenchymal stem cells. Stem Cells. 2008;26:960–8. doi: 10.1634/stemcells.2007-0509. [DOI] [PubMed] [Google Scholar]
- 31.Chen D, Waite LC, Pierce WM., Jr In vitro effects of zinc on markers of bone formation. Biol Trace Elem Res. 1999;68:225–34. doi: 10.1007/BF02783905. [DOI] [PubMed] [Google Scholar]
- 32.Costello LC, Franklin RB, Feng P. Mitochondrial function, zinc, and intermediary metabolism relationships in normal prostate and prostate cancer. Mitochondrion. 2005;5:143–53. doi: 10.1016/j.mito.2005.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Costello LC, Guan Z, Kukoyi B, Feng P, Franklin RB. Terminal oxidation and the effects of zinc in prostate versus liver mitochondria. Mitochondrion. 2004;4:331–8. doi: 10.1016/j.mito.2004.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou YS, Liu YS, Tan JG. Is 1, 25-dihydroxyvitamin D3 an ideal substitute for dexamethasone for inducing osteogenic differentiation of human adipose tissue-derived stromal cells in vitro? Chin Med J (Engl) 2006;119:1278–86. [PubMed] [Google Scholar]
- 35.Costello LC, Franklin RB. Testosterone and prolactin regulation of metabolic genes and citrate metabolism of prostate epithelial cells. Horm Metabol Res. 2002;34:417–24. doi: 10.1055/s-2002-33598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Franklin RB, Zou J, Yu Z, Costello LC. EAAC1 is expressed in rat and human prostate epithelial cells; functions as a high-affinity L-aspartate transporter; and is regulated by prolactin and testosterone. BMC Biochem. 2006;7:10. doi: 10.1186/1471-2091-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Costello LC, Liu Y, Zou J, Franklin RB. The pyruvate dehydrogenase E1 alpha gene is testosterone and prolactin regulated in prostate epithelial cells. Endocr Res. 2000;26:23–39. doi: 10.1080/07435800009040143. [DOI] [PubMed] [Google Scholar]
- 38.Brennan O, O’Brien FJ, McNamara L. Estrogen plus estrogen receptor antagonists alter mineral production by osteoblasts in vitro. Horm Metab Res. 2012;44:47–53. doi: 10.1055/s-0031-1291358. [DOI] [PubMed] [Google Scholar]
- 39.Ikeda K, Tsukui T, Horie-Inoue K, Inoue S. Conditional expression of constitutively active estrogen receptor alpha in osteoblasts increases bone mineral density in mice. FEBS Lett. 2011;585:1303–9. doi: 10.1016/j.febslet.2011.03.038. [DOI] [PubMed] [Google Scholar]
- 39.Modder UI, Roforth MM, Hoey K, et al. Effects of estrogen on osteoprogenitor cells and cytokines/bone-regulatory factors in postmenopausal women. Bone. 2011;49:202–7. doi: 10.1016/j.bone.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Amory JK, Watts NB, Easley KA, et al. Exogenous testosterone or testosterone with finasteride increases bone mineral density in older men with low serum testosterone. J Clin Endocrinol Metab. 2004;89:503–10. doi: 10.1210/jc.2003-031110. [DOI] [PubMed] [Google Scholar]
- 41.Wang C, Swerdloff RS, Iranmanesh A, et al. Effects of transdermal testosterone gel on bone turnover markers and bone mineral density in hypogonadal men. Clin Endocrinol (Oxf) 2001;54:739–50. doi: 10.1046/j.1365-2265.2001.01271.x. [DOI] [PubMed] [Google Scholar]
- 42.Szulc P. Biochemical bone turnover markers and osteoporosis in older men: where are we? J Osteoporos. 2011;2011:240328. doi: 10.4061/2011/704015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sinnesael M, Boonen S, Claessens F, Gielen E, Vanderschueren D. Testosterone and the male skeleton: a dual mode of action. J Osteoporos. 2011;2011:240328. doi: 10.4061/2011/240328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Freeman S, Chang TS. Effect of thyroparathyroidectomy and vitamin D upon serum calcium and citric acid of normal and nephrectomized dogs. Am J Physiol. 1950;160:341–7. doi: 10.1152/ajplegacy.1950.160.2.341. [DOI] [PubMed] [Google Scholar]
- 45.Munson PL, Hirsch PF, Tashjian AH., Jr Parathyroid gland. Ann Rev Physiol. 1963;25:325–60. doi: 10.1146/annurev.ph.25.030163.001545. [DOI] [PubMed] [Google Scholar]
- 46.Costello LC, Stacey R, Stevens R. Hypocitricemic effects of calcitonin, parathyroidectomy and surgical stress. Horm Metab Res. 1971;3:120–5. doi: 10.1055/s-0028-1094163. [DOI] [PubMed] [Google Scholar]