

Abstract
Podocytes are highly specialized epithelial cells with complex actin cytoskeletal architecture crucial for maintenance of the glomerular filtration barrier. The mammalian Rho GTPases Rac1 and Cdc42 are molecular switches that control many cellular processes, but are best known for their roles in the regulation of actin cytoskeleton dynamics. Here we employed podocyte-specific Cre-lox technology and found that mice with deletion of Rac1 display normal podocyte morphology without glomerular dysfunction well into adulthood. Using the protamine sulfate model of acute podocyte injury, podocyte-specific deletion of Rac1 prevented foot process effacement. In a long-term model of chronic hypertensive glomerular damage, however, loss of Rac1 led to an exacerbation of albuminuria and glomerulosclerosis. In contrast, mice with podocyte-specific deletion of Cdc42 had severe proteinuria, podocyte foot process effacement, and glomerulosclerosis beginning as early as 10 days of age. In addition, slit diaphragm proteins nephrin and podocin were redistributed and cofilin was de-phosphorylated. Cdc42 is necessary for the maintenance of podocyte structure and function, but Rac1 is entirely dispensable in physiologic steady state. However, Rac1 has either beneficial or deleterious effects depending on the context of podocyte impairment. Thus, our study highlights the divergent roles of Rac1 and Cdc42 function in podocyte maintenance and injury.
Introduction
The podocyte is a highly differentiated epithelial cell essential for a functional glomerular filtration barrier. Located on the outside of the glomerulus, covering the capillary wall and in the urinary space, the podocyte adopts an intricate and polarized cellular organization consisting of a cell body, major processes, and foot processes that interdigitate with foot processes from neighboring podocytes. The unique shape derives from an abundantly rich actin cytoskeleton that is key to podocyte morphology and function and crucial for establishing stability between the cell-cell and the cell-matrix contacts.1, 2 Regulation of the podocyte cytoskeleton is dynamic, and dysregulation, morphologically identified as foot process effacement, is closely associated with proteinuria, the clinical signature of podocyte injury.1
Mammalian Rho GTPases comprise a family of more than 20 intracellular signaling molecules that regulate diverse biological processes, but are best known for their important roles in regulating the actin cytoskeleton.3, 4 The GTPases of the Rho subfamily, of which Rac1 and Cdc42 are two of the best studied, are likely to play key roles in regulation of the podocyte cytoskeleton. Each GTPase acts as a molecular switch, cycling between an active GTP-bound state and an inactive GDP-bound state. Once activated, Rho GTPases bind to a range of effectors to regulate downstream signaling pathways in addition to those linked to the actin cytoskeleton including cell polarity, cell-extracellular matrix adhesion, microtubule dynamics, membrane trafficking, and gene transcription.5-7
In this study we demonstrate that mice with podocyte-specific deletion of Rac1 show no kidney dysfunction and have morphologically normal podocytes well into adulthood. When acutely injured by protamine sulfate perfusion, Rac1 deletion prevents foot process effacement in podocytes. However, mice with podocyte-specific Rac1 deletion display exacerbated albuminuria and glomerulosclerosis in a chronic model of progressive glomerular failure secondary to uninephrectomy and deoxycorticosterone acetate - high salt (UNX/DOCA-salt) induced hypertension. In sharp contrast, podocyte-specific deletion of Cdc42 results in heavy proteinuria, kidney failure, and death. This was accompanied by foot process effacement, glomerulosclerosis, and eventually end-stage kidneys. Our findings demonstrate Cdc42 has a crucial role in podocyte cell maintenance. Rac1, however, is dispensable for preservation of the glomerular filtration barrier in the unchallenged setting, but has complex and divergent roles in acute and chronic podocyte injury.
Results
Podocyte-specific deletion of Rac1 and Cdc42
To define the function of Rac1 and Cdc42 in podocytes in vivo, we used mice that express Cre-recombinase under control of a podocyte-specific promoter and crossed them with mice with floxed exon 3 of the Rac1 gene, or mice with floxed exon 2 of the Cdc42 gene, resulting in targeted inactivation of either Rac1 (podoRac1−/−) or Cdc42 (podoCdc42−/−) (Figure 1A). Mouse genotype containing Cre-recombinase expressing construct and homozygous for either floxed Rac1 or Cdc42 was confirmed by PCR analysis of tail genomic DNA (Figure 1B). Western blot analysis of protein lysates obtained from glomeruli isolated from podoRac1−/− and podoCdc42−/− mice demonstrated profound reduction of Rac1 and Cdc42 protein expression compared to glomerular lysates from floxed controls (Rac1-fl/fl and Cdc42-fl/fl) (Figure 1C). As podocytes constitute a fraction of the glomerular cell population, endothelial and mesangial cells contribute to the remaining signals in podoRac1−/− and podoCdc42−/− glomeruli.
Figure 1.

Targeted inactivation of Rac1 and Cdc42 in podocytes. (A) The diagram demonstrates the strategy for generation of podocyte-specific Rac1 and Cdc42 knockout mice. Mice expressing Cre-recombinase under control of the podocyte promoter (2.5P-Cre) were bred with mice carrying floxed Rac1 locus (exon 3) and floxed Cdc42 locus (exon 2). (B) PCR analysis of genomic DNA from tail clippings. The PCR product band of floxed (280 bp) and wild-type (200 bp) Rac1 as well as floxed (300 bp) and wild-type (200 bp) Cdc42 are shown. In addition, the 2.5P-Cre PCR product band (268 bp) is indicated. (C) Western blot analysis of Rac1 and Cdc42 in isolated glomeruli from podoRac1−/− and podoCdc42−/− mice with antibodies against Rac1, Cdc42, and β-actin reveals strong reduction of specific protein signal. (D) Survival curve for podoCdc42−/− and podoRac1−/− mice shows 100% mortality with loss of podocyte-specific Cdc42 by day 60. (E) SDS-PAGE analysis of urine samples demonstrates variable selective proteinuria by 10 days of age in podoCdc42−/− mice and heavy nonspecific proteinuria by 16 days of age compared to floxed control. Loss of podocyte-specific Rac1 has no effect on urine protein at 6 months. (F) Urine albumin-to-creatinine ratios in podoCdc42−/− mice were significantly increased versus floxed controls (n=5 per group). (G) Whole kidneys from podoCdc42−/− mouse at 3 weeks of age are pale yellow and display a granular surface compared to floxed control.
Podocyte-specific deletion of Cdc42, but not Rac1, results in decreased survival and heavy proteinuria
Survival was severely limited in podoCdc42−/− animals, whereas podoRac1−/− mice demonstrated no difference in lifespan compared to floxed controls (Figure 1D). The majority of podoCdc42−/− mice died by the age of 4 weeks, and no podoCdc42−/− mice survived past day 50 of life. Though born at a normal Mendelian distribution and normal in appearance at birth, podoCdc42−/− animals began to display significant morbidity with growth retardation at approximately two weeks postnatal (body weight 10.4±0.8 vs. 18.0±1.6 of controls at sacrifice, P<0.01, n=5 per group), most likely the result of heavy proteinuria (Figure1E). By 10 days of age, SDS-PAGE analysis of urine samples revealed significant selective proteinuria (albuminuria) in some podoCdc42−/− mice. By 16 days of age, the proteinuria had progressed dramatically and was nonselective in nature, as evidenced by the presence of proteins of varying molecular weight (Figure 1E). Quantitation of the albumin-to-creatinine ratio revealed an increase of several orders of magnitude in podoCdc42−/− mice compared to controls (Figure 1F, n=5 per group). Mice heterozygous for podocyte-specific deletion of Cdc42 (podocinCre/+, Cdc42fl/+) displayed no phenotype up to 12 months of age (data not shown). Kidneys from 3 to 4-week-old podoCdc42−/− mice were grossly pale yellow, firm, and with a granular surface, consistent with end-stage kidneys (Figure 1G). In contrast, podoRac1−/− mice remained alive and healthy up to 1 year (Figure 1D), demonstrated no proteinuria at 6 months of age (Figure 1E), and showed no gross renal pathology.
Podocyte-specific deletion of Cdc42, but not Rac1, results in severe glomerular disease and disruption of podocyte foot process architecture
Kidney morphology at 4 weeks of age was examined by light microscopy (Figure 2). Compared to floxed Cdc42 or Rac1 controls (Figure 2, A and B), podoRac1−/− kidneys (Figure 2, C and D) showed no alteration in glomerular or tubulo-interstitial morphology for up to 12 months of age. In contrast, podoCdc42−/− mice displayed progressive focal and global glomerulosclerosis accompanied by diffuse tubular dilatation with protein casts and tubular injury (Figure 2, E and F). Podocytes often appeared prominent and vacuolated. Segmentally sclerotic portions contained abundant extracellular matrix and adhered to Bowman's capsule. Additional tubulo-interstitial lesions (not shown in Figure 2) included focal atrophy and fibrosis.
Figure 2.
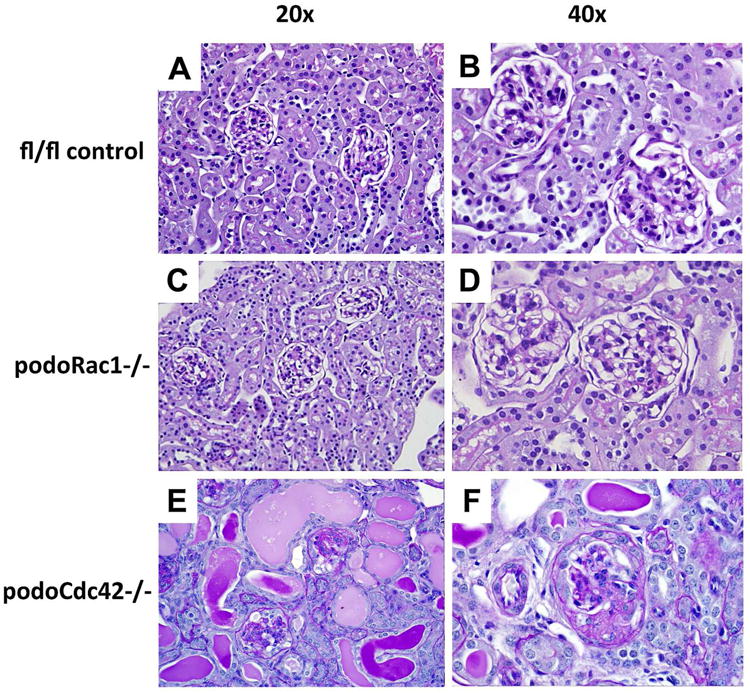
Progressive glomerulosclerosis in podoCdc42−/− but not podoRac1−/− mice by light microscopy (PAS staining at 20× and 40× magnification). (A, B) Floxed control mice demonstrate normal tubulointerstitial and glomerular morphology at 3-4 weeks of age. (C, D) podoRac1−/− tubulointerstitial and glomerular morphology at 12 months of age is indistinguishable from control. Podocyte-specific loss of Cdc42 results in tubular protein casts and injury (E) as well as focal segmental glomerulosclerosis in a minority of glomeruli (F) at 3-4 weeks of age.
Transmission electron microscopy (TEM) (Figure 3, A, C, and E) and scanning electron microscopy (Figure 3, B, D, and F) were performed on 4-week-old podoRac1−/−, podoCdc42−/− and floxed controls. Control mice displayed a normal, intact arrangement of interdigitating foot processes and preserved filtration slits (Figure 3, A and B). Ultrastructural examination of podoRac1−/− podocytes revealed morphology indistinguishable from control podocytes (Figure 3, C and D). In contrast, well-formed foot processes were replaced by broad cellular extensions (effacement) covering glomerular capillaries in podoCdc42−/− mice (Figure 3, E and F).
Figure 3.

Severe podocyte foot process effacement in podoCdc42−/− but not podoRac1−/− mice by electron microscopy (14600× transmission and 7000× scanning magnification). (A, B) Transmission and scanning electron micrographs of control mouse glomeruli show an intact arrangement of podocyte foot processes and normal filtration slits. (C, D) podoRac1−/− podocytes demonstrate no aberrant morphology and are indistinguishable from control. In contrast, podoCdc42−/− mouse podocytes show total effacement of foot processes with near absence of filtration slits. (E, F)
Expression and distribution of slit diaphragm molecules are altered in podoCdc42−/− mice
To define molecular alterations of the podocyte slit diaphragm, we measured glomerular gene expression and examined the cellular distribution of nephrin, podocin, and synaptopodin by quantitative RT-PCR (Figure 4A) and confocal laser microscopy in 4-week-old mice (Figure 4B and C ). Analysis of mRNA obtained from isolated glomeruli revealed a reduction of nephrin and podocin transcript levels in podoCdc42−/− mice (P<0.05), but no significant change in the expression of synaptopodin mRNA, compared to floxed controls. In contrast, nephrin and podocin expression in podoRac1−/− mice did not change (Figure 4A). Through an analysis of nephrin, podocin, and synaptopodin distribution in podocytes at the protein level by immunofluorescence, we observed continuous distribution along the glomerular capillary wall for each marker in wild-type animals. In contrast, nephrin and podocin immunofluorescence staining appeared discontinuous and granular in glomeruli from podoCdc42−/− mice, whereas synaptopodin remained unchanged. The intensity and pattern of these markers in podoRac1−/− mice, however, was identical to floxed controls (Figure 4B and C). These observations were consistent with the morphologic findings and lack of proteinuria described above.
Figure 4.

Glomerular mRNA and protein expression of podocyte markers from one-month-old podoRac1−/− and podoCdc42−/− mice. (A) Gene expression for podocyte markers nephrin, podocin, and synaptopodin in podoRac1−/− glomeruli does not differ from floxed control. In contrast, isolated glomeruli from podoCdc42−/− mice display a significant reduction in transcript levels of nephrin and podocin compared to floxed control mice (*p<0.05). Synaptopodin gene expression is not significantly different. (B) Immunofluorescence studies from kidney sections stained with antibodies against nephrin, podocin, and synaptopodin demonstrate continuous distribution in podocytes from floxed control and podoRac1−/− mice. In contrast, immunofluorescence for nephrin and podocin in podocytes from podoCdc42−/− mice exhibit a significantly impaired, granular distribution along the basement membrane.
Loss of Cdc42 reduces phosphorylation of the downstream effector molecule cofilin
Cofilin is a tightly regulated effector molecule for both Cdc42 and Rac1 that severs actin filaments and promotes disassembly which are essential for actin remodeling and productive membrane protrusions.8, 9 Cofilin inactivation occurs through phosphorylation by LIM kinases, which are activated by the PAK family of Rac/Cdc42-dependent kinases. Western blot analysis of phospho-cofilin to cofilin ratio from isolated glomeruli revealed a near total loss of phospho-cofilin in podoCdc42−/− mice compared to all floxed controls (0.005±0.005 versus 0.62±0.14, P<0.05), but no significant change in the phosphorylation status in podoRac1−/− mice (0.56±0.16 versus 0.62±0.14, P=0.78) (Figure 5). Thus loss of Cdc42 in podocytes results in a significant imbalance in cofilin activation.
Figure 5.

Total cofilin and phospho-cofilin (p-cofilin), the active and inactive states respectively, in isolated glomeruli from podoRac1−/−, podoCdc42−/−, and floxed control mice by Western and densitometry analysis. (A and C) Compared to all floxed controls, protein expression of p-cofilin/total cofilin in glomeruli from podoRac1−/− appear similar. In stark contrast, (B and D) podoCdc42−/− glomeruli display no appreciable p-cofilin, highlighting a significant imbalance in cofilin phosphorylation.
Podocyte-specific Cdc42−/− mice appear normal at 1 week of age
At 10 days of age, some, but not all, podoCdc42−/− mice display albuminuria (Figure 1E). In order to determine if dysfunction of the glomerular filtration barrier can be detected earlier, 1-week-old podoCdc42−/− and floxed controls were examined. Semiquantitative SDS-PAGE analysis of urine samples revealed no significant albuminuria in any animal (Figure 6A). Both control and podoCdc42−/− kidneys displayed an immature nephrogenic zone and primitive tubules at low power (Figure 6B, left). Closer inspection revealed immature appearing glomeruli with prominent podocytes in all animals as expected at this age, with no evidence of glomerulosclerosis (Figure 6B, right). Equally well-formed podocyte foot processes and slit diaphragms could be identified in podoCdc42−/− and control glomeruli by transmission electron microscopy (Figure 6C and inset). Thus podoCdc42−/− mice appear to form a functional glomerular filtration barrier in utero and through the perinatal period.
Figure 6.

Normal phenotype in 1-week-old podoCdc42−/− mice. (A) SDS-PAGE shows no albumin bands in urine from podocyte-specific Cdc42 knockout mice. (B) Tubulointerstitial and glomerular morphology demonstrates an immature appearance in both podoCdc42−/− and floxed control mice (PAS staining at 10× and 40× magnification). (C) Ultrastructural examination reveals no difference in glomerular capillary or podocyte foot process (inset) morphology between podoCdc42−/− and floxed control mice.
PodoRac1−/− mice are resistant to protamine sulfate perfusion-mediated foot process effacement
Protamine sulfate in rodent models results in alterations in podocyte shape characterized by foot process effacement within minutes of perfusion. This is thought to be an actin dependent process triggered through neutralization of anionic charge and/or disruption of podocyte-basement membrane interactions.10 Infusing control mice with protamine sulfate resulted in morphologically distinct foot process effacement (Figure 7A, upper panels). Calculation of the podocyte filtration slit frequency (FSF) revealed a significant reduction by approximately 27% (Figure 7B). In contrast, the podocyte foot process morphology appeared unchanged by protamine sulfate perfusion in podoRac1−/− mice (Figure 7A, lower panels), and FSF was not significantly reduced after 15 min. of perfusion (Figure 7B).
Figure 7.

Loss of podocyte-specific Rac1 protects against induction of podocyte foot process effacement by protamine sulfate. (A) Transmission EM of glomerular capillary walls of floxed control mice after perfusion with HBSS control (top left) and protamine sulfate (top right) demonstrates partial foot process effacement (arrowheads). Glomerular capillary walls from podoRac1−/− mice after perfusion with HBSS control (bottom left) and protamine sulfate (bottom right) shows no qualitative difference in foot process morphology. Results are representative of 6-8 mice per group. ×7900 magnification (B) Filtration slit frequency per micron as seen by transmission EM reflects the morphologic interpretation. *P<0.05
Albuminuria and glomerulosclerosis are exacerbated by UNX/DOCA-salt-induced hypertension in podoRac1−/− mice
Albuminuria and progressive glomerulosclerosis are recognized as hallmarks of podocyte injury in the UNX/DOCA-salt-hypertensive rodent model.11 To investigate the effect of podocyte-specific Rac1 deletion in a chronic model of podocyte injury, we employed this injury model in podoRac1−/− and Rac1-fl/fl controls and compared them to sham treatment. Four weeks after UNX/DOCA-salt treatment, the average body weights of all four groups were not significantly different (Figure 8A). As expected, systolic blood pressure, whole kidney weight, and left ventricular heart weight were elevated by treatment in both Rac1 fl/fl and podoRac1−/− mice compared to sham control, with no difference observed between UNX/DOCA-salt treated Rac1 fl/fl and podoRac1−/− (Figure 8B-D).
Figure 8.

Body weight, systolic blood pressure, and organ weights at 4 weeks following UNX/DOCA-salt treatment. (A) No difference in average total body weight among the four groups. (B-D) UNX/DOCA-salt treatment resulted in increased systolic blood pressure, kidney/body weight ratio, and left ventricle/body weight similarly in both podoRac1−/− and Rac1 fl/fl control mice. *P<0.01, **P<0.05
UNX/DOCA-salt treated mice of either group displayed elevated albuminuria at 2 and 4 weeks, however, urine albumin in UNX/DOCA-salt treated podoRac1−/− mice was nearly twice that of Rac1-fl/fl at 2 weeks (P<0.05) and 4 weeks (Figure 9A), although the difference was not significant at the latter time point (P=0.19). The percentage of glomeruli with segmental and global sclerosis was likewise doubled in treated podoRac1−/− mice at 4 weeks versus treated controls, (Figure 9, B-D) indicating an exacerbation of podocyte injury, rather than protection, in podoRac1−/− mice. Though increased in frequency, the morphologic appearance of segmental sclerosis in UNX/DOCA-salt treated podoRac1−/− mice was indistinguishable from Rac1-fl/fl controls. A qualitative ultrastructural examination by TEM revealed focal and segmental podocyte foot process effacement in both UNX/DOCA-salt treated podoRac1−/− and Rac1-fl/fl mice (Figure 10, A and B, and Supplementary Figure 1), which is consistent with the focal nature of effacement in glomerular hyperfiltration injury akin to secondary focal segmental glomerulosclerosis (FSGS), such as seen in hypertensive or obese humans.12 Interestingly, we observed focal foot process effacement in UNX/DOCA-salt treated podoRac1−/−mice, indicating that Rac1-independent mechanisms of effacement exist.
Figure 9.

Albuminuria and percent glomerulosclerosis in UNX/DOCA-salt mice. (A) Albumin-to-creatinine ratio at 1 week before, and 2 and 4 weeks after uninephrectomy and DOCA treatment demonstrate significantly more albuminuria in podoRac1−/− mice than Rac1 fl/fl control mice. (B) UNX/DOCA-salt treated podoRac1−/− demonstrates twice the glomerulosclerosis than Rac1 fl/fl control mice. Sham treated mice of both genotypes showed no glomerulosclerosis (data not shown). (C and D) Representative PAS stained glomerulus at 20× and 40× magnification from UNX/DOCA-salt treated podoRac1−/− mouse demonstrating segmental sclerosis. *P<0.05, #P=0.19
Figure 10.

Transmission EM of glomerular capillary walls of UNX/DOCA-salt-treated Rac1-fl/fl (A) and podoRac1−/− (B) mice displaying focal and segmental foot process effacement. Sham-treated podoRac1−/− and Rac1 fl/fl (C) exhibit only normal appearing, regularly interdigitating podocyte foot processes.
Discussion
Podocytes are polarized cells with an abundantly rich and highly dynamic actin-based cytoskeleton vital to proper podocyte function and glomerular filtration.1, 13 Furthermore, dysregulation of the podocyte cytoskeleton, seen as foot process effacement, is invariably seen in podocyte injury. Each cell membrane domain of the foot process (slit diaphragm, basal, and apical) has the ability to regulate actin dynamics through Rho GTPase activation.1 Nephrin phosphorylation increases Rac1 activity through phosphoinositide 3-kinase 14 and nephrin directly interacts with IQGAP1, an effector protein that binds and maintains Rac1 and Cdc42 in an active state.15 Synaptopodin, an actin-associated protein, induces RhoA stabilization and cell migration while preventing Cdc42-mediated filopodia formation in mouse podocytes.16 In addition, podocalyxin, an apical membrane domain protein, has been shown to activate RhoA and induce actin reorganization in MDCK cells.17 Bidirectional signaling between Rho GTPases and integrins to modulate cell-basement membrane adhesion has xalso been described.3 Evidence suggests current immunosuppressive strategies to reduce proteinuria and treat FSGS, such as calcineurin inhibitors and glucocorticoids, have the ability to directly target the podocyte cytoskeleton,18-20 implicating cytoskeleton as a therapeutic goal to abrogate podocyte injury. Thus elucidation of Rho GTPase contribution in podocyte function is important.
Rho GTPases, including Rac1, are important for proper neuron21 and vascular development.22 However, mice with podocytes lacking Rac1 develop normal glomeruli and display no renal dysfunction well into adulthood. Thus it appears Rac1 is not required for the development or maintenance of podocyte architecture and the glomerular filtration barrier. It should be noted that Rac1 and its isoforms Rac2 and Rac3 share a high degree of homology. Studies have revealed a redundant role between Rac1 and Rac223 and Rac1 and Rac324 in T-cell and neuronal development respectively. Therefore we cannot yet rule out a compensatory effect of Rac2 and Rac3 in podoRac1−/− mice as an explanation for the normal phenotype. However, we believe this to be unlikely because microarray analysis of normal mouse glomeruli reveal levels of Rac2 and Rac3 transcripts to be approximately 1% of Rac1 transcript abundance (data available at Gene Expression Omnibus under GEO no. GSE33744). Additional experiments will be needed to test our hypothesis.
Interestingly, we find that podoRac1−/− mice do not develop protamine sulfate-induced foot process effacement, a model of acute podocyte injury that alters anionic charge and results in the disruption of podocyte foot process architecture.10 Alterations in podocyte motility in response to injury are considered to underlie foot process effacement and recent studies have highlighted the importance of podocyte actin cytoskeleton dynamics and reorganization in these processes.25, 26 Since Rac1 is known to induce lamellipodia formation at the leading edge of motile cells,27 it would be expected to have a central role in podocyte motility, which is supported by several recent studies. In vitro, angiotensin II induces a phenotypic shift in human podocytes from being dynamically stable to adaptively migratory through regulation of the cytoskeleton involving signaling pathways dominated by Rac1.28 In addition, the urokinase receptor uPAR is activated in murine podocytes in response to lipopolysaccharide and puromycin aminoglycoside treatment in vitro and in vivo, which leads to foot process effacement and proteinuria. uPAR complexes with and activates β3-integrin in podocytes, which activates Rac1 and Cdc42, and promotes cell motility.29
Surprisingly, we found that in long-term injury by UNX/DOCA-salt treatment podoctye specific loss of Rac1 exacerbates proteinuria and glomerulosclerosis. Podocyte injury is a critical step in the development of glomerulosclerosis in models of DOCA-salt induced hypertension, likely due to an inability of the podocyte to adapt to cover increased glomerular capillary surfaces of hypertrophic glomeruli,11 Recently, Fukuda et al.30 demonstrated that a failure of podocytes to match glomerular tuft growth triggers proteinuria and glomerulosclerosis. This suggests podocyte-specific deletion of Rac1 impairs the podocyte's ability to respond to hypertrophic stress in the UNX/DOCA-salt model. This could be due to an impairment of hypertrophic signaling pathways, or a direct consequence of impaired mobility of podocyte foot processes.
In fact, a role for Rac1-dependent hypertrophic signaling pathways has been known for some time in the myocardium through enhancement of hypertrophic gene expression via reactive oxygen species signaling and direct effects on transcription factors.31 In addition, the balance between activation of GTPases Rac1 and RhoA may be important in the podocyte. It is known that Rac1 stimulation results in down-modulation of RhoA activity32, 33 and that excessive RhoA activation in podocytes induces FSGS.34 Thus, loss of Rac1 may allow excessive RhoA signaling and induce significant podocyte dysfunction and loss. Understanding the mechanisms driving, or abrogating, these process may lead to new opportunities for targeted therapeutic interventions in glomerular disease.
Additional evidence indicates excess Rac1 activity is harmful to the mature podocyte. Recently, a mutant form of the ARHGAP24 gene that impairs Arhgap24 Rac1-GAP activity was found to be associated with FSGS in humans.35 Ahgap24 inactivates Rac1 and suppresses lamellipodia formation downstream of RhoA signaling. Arhgap24 knockdown studies in mouse podocytes revealed increased motility and increased Rac1 as well as Cdc42 activity.35 In another study, mice lacking the Rho GDP dissociation inhibitor RhoGDIα,a RhoGDP dissociation inhibitor that binds Rho family members in an inactive state, display massive albuminuria, foot process effacement, and FSGS.36, 37 These mice demonstrate enhanced Rac1 activity, but not RhoA activity, in the kidney. Repression of Rac1 activity with a specific inhibitor significantly reduced albuminuria and histological damage,36 however these studies do not rule out dysregulation of Cdc42 as a contributing factor.
Rac1 is also known to regulate multiple signaling pathways apart from cytoskeleton organization including transcription, reactive oxygen species generation, and cell proliferation.38 In studies using RhoGDI-α knockout mice and Dahl salt-sensitive rats by Shibata et al.,36, 39 a cross-talk between Rac1 and the mineralocorticoid receptor was observed. Shibata reported a protective effect of subcutaneous infusion of a Rac1 inhibitor against hypertension-induced glomerular injury through prevention of mineralocorticoid overactivation in the kidney. The reasons for the discrepant findings in our UNX/DOCA-salt experiments in podoRac1−/− mice could be a consequence of the cell specificity of the cre-lox technology employed in our study compared to the effect of pharmacological inhibition with potentially competing local and systemic effects. With pharmacological and genetic tools to modulate Rac1 activity available, further detailed experiments will be able to elucidate the complexities of Rac1 activity in the glomerulus.
Mice with podocytes lacking Cdc42 developed prodigious proteinuria at approximately 2 weeks of age, associated with the loss of normal podocyte foot process architecture and an aberrant distribution of nephrin and podocin. This is followed quickly by segmental and global glomerulosclerosis, severe tubulointerstitial injury, renal failure and death. Podocyte-specific loss of Cdc42, but not Rac1, left nearly all cofilin in its nonphosphorylated active state. Cdc42 activates LIM kinase to stimulate cofilin phosphorylation and inactivation in a well-characterized pathway, and cofilin regulates actin dynamics by stimulating actin-filament severing and depolymerization.8 Because the complex podocyte morphology in vivo requires a dynamic regulation of actin polymerization and remodeling, we hypothesize that such an imbalance in cofilin activation results in a less stable actin cytoskeleton architecture. This is supported by increased cofilin activation following podocyte injury in a rat model and the requirement for cofilin activation in lamellipodia formation and directed motility.9, 40 In addition, recent functional analysis of INF2 gene mutations in patients with Charcot-Marie-Tooth neuropathy and FSGS demonstrate an enhanced binding of mutated INF2 protein to Cdc42 in human embryonic kidney (HEK-293T) cells. This causes mislocalization of Cdc42 and subsequent cytoskeleton disorganization.41 Though this has yet to be demonstrated in human podocytes, loss of normal Cdc42 activity may be a major causative factor leading to podocyte injury and FSGS in patients with INF2 mutations.
In the podocyte, Cdc42 may have a role in polarity and polarity signaling, which is crucial for maintenance of the glomerular filtration barrier. Recent studies have demonstrated the importance of the PAR complex [PAR6 (partitioning defective 6)-PAR3 (partitioning defective 3)-aPKC (atypical protein kinase C)], a regulator of cell polarity, as an essential determinant of podocyte morphology. The PAR complex has been identified as a component of the slit diaphragm that interacts with nephrin and Neph1.42-44 In mice, pharmacologic inhibition of aPKC,42 or podocyte-specific genetic deletion of aPKC,43, 44 results in severe proteinuria and renal failure. Cdc42 may regulate cell polarity by binding to PAR6 and inducing a conformational change that activates aPKC.5 In fact, cre-lox mouse conditional knock-out methodology has revealed that loss of Cdc42 in neural progenitors45 and telencephalon46 abolishes the apical localization of the PAR complex and induces apical basal polarity defects. The severe renal phenotype we find in podoCdc42−/− mice is nearly identical to that reported in podocyte-specific knockouts of aPKC by Hirose et al.43 and Huber et al.,44 and indicates Cdc42 regulates polarity signaling in podocytes in vivo.
Our findings in unchallenged podocyte-specific Rac1 knockout mice are in agreement with Scott et al.47 However, using podoRac1−/− mice, we extend our understanding of the effects of podocyte-specific Rac1 deletion by describing distinct and divergent effects in acute and chronic models of podocyte injury. In addition, Scott et al.47 describe a congenital phenotype of massive proteinuria with loss of foot process architecture, glomerulosclerosis, and tubular injury with protein casts at birth in mice lacking podocyte-specific Cdc42. In contrast, we find intact glomerular filtration in podoCdc42−/− mice up to 1 week of age. The reasons for this discrepancy are not clear. Both studies used mice that express Cre recombinase under control of the NPHS2 promoter and the same floxed Cdc42 gene. However, differences in environment and genetic heterogeneity are important considerations well known to influence phenotype in experiments with mice. Both studies employed mice on a mixed genetic background, thus small changes in protein stability or unrecognized functional genetic complementation could account for a delay in podocyte injury. Nevertheless, our data indicate Cdc42 is dispensable for early stages of podocyte development and not required to form a functional glomerular filtration barrier, but rather key to maintenance of normal podocyte architecture.
Knowledge of the signaling functions of Rac1 and Cdc42 in mammalian cells has come primarily from studies using pharmacological agents or dominant-negative or constitutively active mutant overexpression approach, which imposes experimental limitations related to specificity and dosage.48 By employing a cell lineage specific transgene technology in mice, our study demonstrates the importance of Cdc42 in the maintenance of podocyte architecture and function in glomerular filtration in vivo, and reveals both potentially beneficial and deleterious roles for the inhibition of Rac1 dependent foot process dynamics in acute and chronic forms of podocyte injury.
Materials and Methods
Targeted inactivation of Rac1 and Cdc42 in podocytes
For selective deletion of Rac1 and Cdc42, transgenic mice that express Cre-recombinase specifically in podocytes were crossed with “floxed” mice (Figure 1), which contain loxP sites downstream of exon 2 and upstream of exon 4 of the Rac1 gene,49 and downstream of exon 1 and upstream of exon 3 of the Cdc42 gene.50 2.5P-Cre mice (podocinCre/Cre) contain the Cre-recombinase cassette under the regulation the human NPHS2 promoter that leads to podocyte-specific expression of the Cre-recombinase.51 2.5P-Cre mice were crossed with floxed Rac1 and Cdc42 mice to generate bitransgenic heterozygous offspring (podocinCre/+,Rac1fl/+ and podocinCre/+,Cdc42fl/+). These were intercrossed in such a manner to produce podocyte-specific knockout mice on a mixed C57BL/6 and 129 genetic background for Rac1 (podocinCre/+,Rac1fl/fl) and Cdc42 (podocinCre/+,Cdc42fl/fl), hereafter referred to simply as podoRac1−/− and podoCdc42−/− mice. Floxed control mice are referred to as Rac1-fl/fl and Cdc42-fl/fl. Genotyping was performed by PCR as previously described 49-51 using the following oligonucleotide primers: Cre-Recombinase (Fwd: 5′-GCATAACCAGTGAAACAGCATTGCTG-3′, Rev: 5′-GGA CATGTTCAGGGATCGCCAGGCG-3′), Rac1 (Fwd: 5′-GTCTTGAGTTACATCT CTGG-3′, Rev: 5′-CTGACGCCAACAACTATGC-3′) and Cdc42 (Fwd: 5′-ATGT AGTGTCTGTCCATTGG-3′, Rev: 5′-TCTGCCATCTACACATACAC-3′). For the UNX/DOCA-salt-induced hypertension experiments, podocinCre/+ and Rac1fl/+ were backcrossed eight generations (>99%) to 129S6/SvEvTaconic. All animal experiments were approved by the University Committee on the Use and Care of Animals Institutional Review Board at the University of Michigan Medical School and conducted in accord with the principles and procedures outlined in the National Institutes of Health Guidelines for the Care and Use of Experimental Animals.
Experimental procedures
The following experimental procedures were performed as previously described.52-54 (1) Isolation of glomeruli for molecular analysis using magnetic beads, (2) Western blot analysis and densitometry using NIH ImageJ software, version 1.42q (http://rsb.info.nih.gov/ij/index.html), (3) Urine protein analysis by SDS-PAGE and albumin-to-creatinine ration by ELISA, (4) histology and light microscopy, (5) electron microscopy and calculation of podocyte filtration slit frequency, (5) real-time quantitative PCR, (6) immunofluorescence, and (7) protamine sulfate induced podocyte injury model. Additional details are provided in the Supplementary Section.
Uninephrectomy, deoxycorticosterone acetate (DOCA), and high salt induced hypertension model
Two genotypic groups of adult male mice, Rac1-fl/fl and podoRac1−/− on 129S6/SvEvTaconic background, age 6 to 7 weeks, were weighed and underwent left unilateral nephrectomy or sham operation under isofluorane anesthesia. A DOCA-impregnated Silastic implant (2.5g DOCA/kg body weight) or Silastic implant alone, was placed subcutaneously. After surgery, DOCA mice were maintained on salt water (1% NaCl plus 0.2% KCl), sham treated mice received tap water. One week before, and at two and four weeks after surgery and treatment, mice were placed in metabolic cages for 24 hours and urine collected for albumin and creatinine measurement. Urine albumin and creatinine was determined with the Albuwell M Test and Creatinine Companion kit (Exocel, Philadelphia, PA) according to manufacturer's instructions. At four weeks, systolic blood pressure was measured noninvasively using CODA tail-cuff system (Kent Scientific, Torrington, CT) on conscious, restrained mice. Mice were sacrificed and body and organ weights were obtained. Percent glomerulosclerosis for each mouse was calculated by counting all segmentally and globally sclerosed glomeruli in one PAS stained section from formalin-fixed, paraffin-embedded kidney (total glomeruli 120-160). Qualitative ultrastructural analysis was performed as described above.
Statistical analyses
Data are given as mean ± SE. A minimum of four mice was used for each analysis, unless otherwise stated. Statistical analysis was performed using Student's t-test with Bonferroni correction for multiple measurements. P≤0.05 was considered significant.
Supplementary Material
Supplementary Figure 1. Representative transmission EMs of injured podocytes in UNX/DOCA-salt treated floxed control (A) and podoRac1−/− (B) mice. Each displays segmental foot process widening and effacement (arrowheads) and accumulation of lysosomal and/or autophagic elements (asterisks).
Acknowledgments
We thank Andreas F. Blutke of the Institute of Veterinary Pathology, Munich, Germany, for technical assistance in the methodology to isolate glomeruli. This work was supported by grants from the National Institutes of Health, National Institute of Diabetes, Digestive, and Kidney Diseases (R24 DK082841, P30 DK081943 to MK; K08 DK-088944 to JBH).
Footnotes
Disclosure: Masashi Nishio contributed to these studies at the University of Michigan as an employee of Mitsubishi Tanabe Pharma Corporation.
References
- 1.Faul C, Asanuma K, Yanagida-Asanuma E, et al. Actin up: regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol. 2007;17:428–437. doi: 10.1016/j.tcb.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 2.Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003;83:253–307. doi: 10.1152/physrev.00020.2002. [DOI] [PubMed] [Google Scholar]
- 3.Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9:690–701. doi: 10.1038/nrm2476. [DOI] [PubMed] [Google Scholar]
- 4.Wennerberg K, Der CJ. Rho-family GTPases: it's not only Rac and Rho (and I like it) J Cell Sci. 2004;117:1301–1312. doi: 10.1242/jcs.01118. [DOI] [PubMed] [Google Scholar]
- 5.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 6.Jaffe AB, Hall A. Rho GTPases in transformation and metastasis. Adv Cancer Res. 2002;84:57–80. doi: 10.1016/s0065-230x(02)84003-9. [DOI] [PubMed] [Google Scholar]
- 7.Schwartz M. Rho signalling at a glance. J Cell Sci. 2004;117:5457–5458. doi: 10.1242/jcs.01582. [DOI] [PubMed] [Google Scholar]
- 8.Bamburg JR. Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu Rev Cell Dev Biol. 1999;15:185–230. doi: 10.1146/annurev.cellbio.15.1.185. [DOI] [PubMed] [Google Scholar]
- 9.Garg P, Verma R, Cook L, et al. Actin-depolymerizing factor cofilin-1 is necessary in maintaining mature podocyte architecture. J Biol Chem. 2010;285:22676–22688. doi: 10.1074/jbc.M110.122929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pippin JW, Brinkkoetter PT, Cormack-Aboud FC, et al. Inducible rodent models of acquired podocyte diseases. Am J Physiol Renal Physiol. 2009;296:F213–229. doi: 10.1152/ajprenal.90421.2008. [DOI] [PubMed] [Google Scholar]
- 11.Kretzler M, Koeppen-Hagemann I, Kriz W. Podocyte damage is a critical step in the development of glomerulosclerosis in the uninephrectomised-desoxycorticosterone hypertensive rat. Virchows Arch. 1994;425:181–193. doi: 10.1007/BF00230355. [DOI] [PubMed] [Google Scholar]
- 12.D'Agati VD, Kaskel FJ, Falk RJ. Focal segmental glomerulosclerosis. N Engl J Med. 2011;365:2398–2411. doi: 10.1056/NEJMra1106556. [DOI] [PubMed] [Google Scholar]
- 13.Shankland SJ. The podocyte's response to injury: role in proteinuria and glomerulosclerosis. Kidney Int. 2006;69:2131–2147. doi: 10.1038/sj.ki.5000410. [DOI] [PubMed] [Google Scholar]
- 14.Zhu J, Sun N, Aoudjit L, et al. Nephrin mediates actin reorganization via phosphoinositide 3-kinase in podocytes. Kidney Int. 2008;73:556–566. doi: 10.1038/sj.ki.5002691. [DOI] [PubMed] [Google Scholar]
- 15.Liu XL, Kilpelainen P, Hellman U, et al. Characterization of the interactions of the nephrin intracellular domain. FEBS J. 2005;272:228–243. doi: 10.1111/j.1432-1033.2004.04408.x. [DOI] [PubMed] [Google Scholar]
- 16.Yanagida-Asanuma E, Asanuma K, Kim K, et al. Synaptopodin protects against proteinuria by disrupting Cdc42:IRSp53:Mena signaling complexes in kidney podocytes. Am J Pathol. 2007;171:415–427. doi: 10.2353/ajpath.2007.070075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmieder S, Nagai M, Orlando RA, et al. Podocalyxin activates RhoA and induces actin reorganization through NHERF1 and Ezrin in MDCK cells. J Am Soc Nephrol. 2004;15:2289–2298. doi: 10.1097/01.ASN.0000135968.49899.E8. [DOI] [PubMed] [Google Scholar]
- 18.Faul C, Donnelly M, Merscher-Gomez S, et al. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med. 2008;14:931–938. doi: 10.1038/nm.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ransom RF, Lam NG, Hallett MA, et al. Glucocorticoids protect and enhance recovery of cultured murine podocytes via actin filament stabilization. Kidney Int. 2005;68:2473–2483. doi: 10.1111/j.1523-1755.2005.00723.x. [DOI] [PubMed] [Google Scholar]
- 20.Schonenberger E, Ehrich JH, Haller H, et al. The podocyte as a direct target of immunosuppressive agents. Nephrol Dial Transplant. 2011;26:18–24. doi: 10.1093/ndt/gfq617. [DOI] [PubMed] [Google Scholar]
- 21.Govek EE, Newey SE, Van Aelst L. The role of the Rho GTPases in neuronal development. Genes Dev. 2005;19:1–49. doi: 10.1101/gad.1256405. [DOI] [PubMed] [Google Scholar]
- 22.Tan W, Palmby TR, Gavard J, et al. An essential role for Rac1 in endothelial cell function and vascular development. FASEB J. 2008;22:1829–1838. doi: 10.1096/fj.07-096438. [DOI] [PubMed] [Google Scholar]
- 23.Guo F, Cancelas JA, Hildeman D, et al. Rac GTPase isoforms Rac1 and Rac2 play a redundant and crucial role in T-cell development. Blood. 2008;112:1767–1775. doi: 10.1182/blood-2008-01-132068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Corbetta S, Gualdoni S, Ciceri G, et al. Essential role of Rac1 and Rac3 GTPases in neuronal development. FASEB J. 2009;23:1347–1357. doi: 10.1096/fj.08-121574. [DOI] [PubMed] [Google Scholar]
- 25.Mundel P, Reiser J. Proteinuria: an enzymatic disease of the podocyte? Kidney Int. 2010;77:571–580. doi: 10.1038/ki.2009.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Welsh GI, Saleem MA. The podocyte cytoskeleton-key to a functioning glomerulus in health and disease. Nat Rev Nephrol. 2011;8:14–21. doi: 10.1038/nrneph.2011.151. [DOI] [PubMed] [Google Scholar]
- 27.Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167–179. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- 28.Hsu HH, Hoffmann S, Endlich N, et al. Mechanisms of angiotensin II signaling on cytoskeleton of podocytes. J Mol Med (Berl) 2008;86:1379–1394. doi: 10.1007/s00109-008-0399-y. [DOI] [PubMed] [Google Scholar]
- 29.Wei C, Moller CC, Altintas MM, et al. Modification of kidney barrier function by the urokinase receptor. Nat Med. 2008;14:55–63. doi: 10.1038/nm1696. [DOI] [PubMed] [Google Scholar]
- 30.Fukuda A, Chowdhury MA, Venkatareddy MP, et al. Growth-dependent podocyte failure causes glomerulosclerosis. J Am Soc Nephrol. 2012;23:1351–1363. doi: 10.1681/ASN.2012030271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown JH, Del Re DP, Sussman MA. The Rac and Rho hall of fame: a decade of hypertrophic signaling hits. Circ Res. 2006;98:730–742. doi: 10.1161/01.RES.0000216039.75913.9e. [DOI] [PubMed] [Google Scholar]
- 32.Sander EE, ten Klooster JP, van Delft S, et al. Rac downregulates Rho activity: reciprocal balance between both GTPases determines cellular morphology and migratory behavior. J Cell Biol. 1999;147:1009–1022. doi: 10.1083/jcb.147.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu YI, Frey D, Lungu OI, et al. A genetically encoded photoactivatable Rac controls the motility of living cells. Nature. 2009;461:104–108. doi: 10.1038/nature08241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu L, Jiang R, Aoudjit L, et al. Activation of RhoA in podocytes induces focal segmental glomerulosclerosis. J Am Soc Nephrol. 2011;22:1621–1630. doi: 10.1681/ASN.2010111146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Akilesh S, Suleiman H, Yu H, et al. Arhgap24 inactivates Rac1 in mouse podocytes, and a mutant form is associated with familial focal segmental glomerulosclerosis. J Clin Invest. 2011;121:4127–4137. doi: 10.1172/JCI46458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shibata S, Nagase M, Yoshida S, et al. Modification of mineralocorticoid receptor function by Rac1 GTPase: implication in proteinuric kidney disease. Nat Med. 2008;14:1370–1376. doi: 10.1038/nm.1879. [DOI] [PubMed] [Google Scholar]
- 37.Togawa A, Miyoshi J, Ishizaki H, et al. Progressive impairment of kidneys and reproductive organs in mice lacking Rho GDIalpha. Oncogene. 1999;18:5373–5380. doi: 10.1038/sj.onc.1202921. [DOI] [PubMed] [Google Scholar]
- 38.Bosco EE, Mulloy JC, Zheng Y. Rac1 GTPase: a “Rac” of all trades. Cell Mol Life Sci. 2009;66:370–374. doi: 10.1007/s00018-008-8552-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shibata S, Mu S, Kawarazaki H, et al. Rac1 GTPase in rodent kidneys is essential for salt-sensitive hypertension via a mineralocorticoid receptor-dependent pathway. J Clin Invest. 2011;121:3233–3243. doi: 10.1172/JCI43124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nagata-Ohashi K, Ohta Y, Goto K, et al. A pathway of neuregulin-induced activation of cofilinphosphatase Slingshot and cofilin in lamellipodia. J Cell Biol. 2004;165:465–471. doi: 10.1083/jcb.200401136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boyer O, Nevo F, Plaisier E, et al. INF2 mutations in Charcot-Marie-Tooth disease with glomerulopathy. N Engl J Med. 2011;365:2377–2388. doi: 10.1056/NEJMoa1109122. [DOI] [PubMed] [Google Scholar]
- 42.Hartleben B, Schweizer H, Lubben P, et al. Neph-Nephrin proteins bind the Par3-Par6-atypical protein kinase C (aPKC) complex to regulate podocyte cell polarity. J Biol Chem. 2008;283:23033–23038. doi: 10.1074/jbc.M803143200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hirose T, Satoh D, Kurihara H, et al. An essential role of the universal polarity protein, aPKClambda, on the maintenance of podocyte slit diaphragms. PLoS One. 2009;4:e4194. doi: 10.1371/journal.pone.0004194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huber TB, Hartleben B, Winkelmann K, et al. Loss of podocyte aPKClambda/iota causes polarity defects and nephrotic syndrome. J Am Soc Nephrol. 2009;20:798–806. doi: 10.1681/ASN.2008080871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cappello S, Attardo A, Wu X, et al. The Rho-GTPase cdc42 regulates neural progenitor fate at the apical surface. Nat Neurosci. 2006;9:1099–1107. doi: 10.1038/nn1744. [DOI] [PubMed] [Google Scholar]
- 46.Chen L, Liao G, Yang L, et al. Cdc42 deficiency causes Sonic hedgehog-independent holoprosencephaly. Proc Natl Acad Sci U S A. 2006;103:16520–16525. doi: 10.1073/pnas.0603533103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scott RP, Hawley SP, Ruston J, et al. Podocyte-specific loss of cdc42 leads to congenital nephropathy. J Am Soc Nephrol. 2012;23:1149–1154. doi: 10.1681/ASN.2011121206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Melendez J, Grogg M, Zheng Y. Signaling role of Cdc42 in regulating mammalian physiology. J Biol Chem. 2011;286:2375–2381. doi: 10.1074/jbc.R110.200329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chrostek A, Wu X, Quondamatteo F, et al. Rac1 is crucial for hair follicle integrity but is not essential for maintenance of the epidermis. Mol Cell Biol. 2006;26:6957–6970. doi: 10.1128/MCB.00075-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu X, Quondamatteo F, Lefever T, et al. Cdc42 controls progenitor cell differentiation and beta-catenin turnover in skin. Genes Dev. 2006;20:571–585. doi: 10.1101/gad.361406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moeller MJ, Sanden SK, Soofi A, et al. Two gene fragments that direct podocyte-specific expression in transgenic mice. J Am Soc Nephrol. 2002;13:1561–1567. doi: 10.1097/01.asn.0000015614.68893.0b. [DOI] [PubMed] [Google Scholar]
- 52.El-Aouni C, Herbach N, Blattner SM, et al. Podocyte-specific deletion of integrin-linked kinase results in severe glomerular basement membrane alterations and progressive glomerulosclerosis. J Am Soc Nephrol. 2006;17:1334–1344. doi: 10.1681/ASN.2005090921. [DOI] [PubMed] [Google Scholar]
- 53.Takemoto M, Asker N, Gerhardt H, et al. A new method for large scale isolation of kidney glomeruli from mice. Am J Pathol. 2002;161:799–805. doi: 10.1016/S0002-9440(10)64239-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Verma R, Kovari I, Soofi A, et al. Nephrin ectodomain engagement results in Src kinase activation, nephrin phosphorylation, Nck recruitment, and actin polymerization. J Clin Invest. 2006;116:1346–1359. doi: 10.1172/JCI27414. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Representative transmission EMs of injured podocytes in UNX/DOCA-salt treated floxed control (A) and podoRac1−/− (B) mice. Each displays segmental foot process widening and effacement (arrowheads) and accumulation of lysosomal and/or autophagic elements (asterisks).