

Abstract
On the surface heat shock protein 90 (Hsp90) is an unlikely drug target for the treatment of any disease, let alone cancer. Hsp90 is highly conserved and ubiquitously expressed in all cells. There are two major isoforms α and β encoded by distinct genes and together they may constitute 1%–3% of the cellular protein. Deletion of the protein is embryonic lethal and there are no recognized polymorphisms suggesting an association or causal relationship with any human disease. With respect to cancer, the proteins absence from two recent high profile articles, ‘Hallmarks of cancer: the next generation’ [Hanahan & Weinberg (2011) Cell 144, 646–674] and ‘Comprehensive molecular portraits of human breast tumours’ [Koboldt et al. (2012) Nature] underlines the perception that it is an unlikely bona fide target to treat this disease. Yet, to date, there are 17 distinct Hsp90 inhibitors in clinical trials for multiple indications in cancer. The protein has been championed for over 20 years by the National Cancer Institute (Bethesda, MD, USA) as a cancer target since the discovery of the antitumor activity of the natural product geldanamycin. This review aims to look at the conundrum of why Hsp90 can even be considered a druggable target for the treatment of cancer. We propose that in contrast to the majority of chemotherapeutics our growing armamentarium of investigational Hsp90 drugs represents an elegant choice that offers real hope in the long-term treatment of certain cancers.
Keywords: cancer, chaperone, combination therapy, drug discovery, extracellular Hsp90, Hsp90, protein activation
Introduction
Undoubtedly, any protein or gene of interest that has relevance to cancer will receive superfluous recommendations to pursue it pharmacologically. Heat shock protein 90 (Hsp90) is no exception. With each new interaction and publication about Hsp90 there is augmenting enthusiasm to pursue clinical applications. The fact is that Hsp90 inhibitors are being pursued and advancing through clinical trials and their applications toward multiple cancers are expanding. Despite these successes, a few recent high profile articles on the mechanisms of cancer [1,2] have overlooked this already established therapeutic target, and it does not appear to be an anomaly with the authors of these cancer reviews. Cancer pharmacologists within our own institution are often blindsided by the clinical trial data or even the notion that one could target an abundant chaperone protein, such as Hsp90, that has no known disease-linked polymorphisms [3,4]. On the surface this seems reasonable: how could one pharmacologically target a protein that is essential for normal cell viability and has a documented interactome that involves over 400 putative clients and over a dozen cellular pathways and processes?
Because it was hard to envision any clinical success at first, the application of Hsp90 inhibitors was relinquished to purely academic exercises as pharmaceutical companies considered it inconsequential. However, despite the high abundance and pleiotropic effects of Hsp90 protein–protein interactions, Hsp90 inhibitors have advanced into clinical trials. Counterintuitively, the aspects that at first made Hsp90 inhibitors seem inappropriate are now demonstrating advantages over single target therapies, and the potential of combination therapies that include Hsp90 inhibitors should effectively prevent or prolong the development of cancer drug resistance that is commonly seen with tyrosine kinase inhibitors and other target-based therapies [5–7].
To underline the improved successes of Hsp90 inhibitors, we highlight results published for ganetespib, an Hsp90 inhibitor developed by Synta Pharmaceuticals (Lexington, MA, USA). In in vitro tumor cytotoxicity studies, it was shown that a 5-min exposure to ganetespib at 1 μM (a readily achievable plasma level in vivo) was sufficient to reduce cell death within 72 h. This suggests that a brief exposure to Hsp90 inhibitors results in a permanent and lasting effect on the cell. These observations extend to animal studies. A single weekly dose of 100–150 mg·kg−1 for 3 weeks resulted in 50–90% tumor reduction in solid and hematological xenograft models [8]. Most importantly, in recent clinical studies it was also shown that once-weekly ganetespib treatment in patients with non-small-cell lung carcinoma resulted in a 50% overall response rate [9,10]. Development of ganetespib in many ways is therefore a good example of how second generation synthetic Hsp90 inhibitors are surpassing their first generation geldanamycin based counterparts. Ganetespib not only shows increased efficacy and improved formulation over the geldanamycins, but also significantly reduces cardiovascular and hepatotoxicity side effects [8]. In our view such consistent advances are a testament to the validity of Hsp90 as a druggable target. While safety for off target effects is being improved and desired tumor effects continue to progress, the mechanisms for Hsp90 targeting in tumors over Hsp90 found in normal tissue is less understood.
Hsp90, the basics
To frame the review we offer a brief overview of the basics of Hsp90 and its known biology and then address the distinguishing characteristics of Hsp90 between cancer cells and normal cells. Hsp90 plays an essential role in maintaining cellular protein homeostasis by acting as a molecular chaperone to aid in folding as well as in intracellular trafficking of its protein clients [11,12]. It is expressed as a 90 kDa protein (Hsp90α 732 amino acids; Hsp90β 724 amino acids) in the cytosol and the nucleus and contains an N-terminal ATP-binding domain that is essential for most of its cellular functions [13]. ATP hydrolysis is thought to drive various conformational changes within Hsp90 and this process is highly regulated by interactions with co-chaperones and possibly a variety of posttranslational modifications (Table 1) [12,14]. Through these mechanisms the apparently ubiquitous cellular chaperone functions of Hsp90 are thought to be acutely regulated.
Table 1.
Post-translational modifications of Hsp90. p300, histone acetyl transferase; CK2, casein kinase II; PKA, protein kinase A; DNA-PK, DNA-dependent protein kinase; eNOS, endothelial nitric oxide synthase.
| Modification | Hsp90 residue (α) | Enzyme | Result | Reference |
|---|---|---|---|---|
| 4-HNE, 4-ONE | C572 | Decreased client association | 99 | |
| Acetylation | K294 | HDAC6 | Decreased client association | 100 |
| Acetylation | K69, K100, K292, K327, K546, K558 | p300, HDAC6 | Increased inhibitor binding, increased extracellular expression | 74,101–104 |
| Dephosphorylation | Ppt1/PP5 | Decreased chaperone function | 105 | |
| Phosphorylation | Y309 | SRC | VEGFR2-induced angiogenesis | 106 |
| Phosphorylation | S231, S263 | CK2 | Increased apoptosome formation, decreased client association | 107,108 |
| Phosphorylation | Y38 | Swe1/Wee1 | Increased client association, decreased inhibitor binding | 109,110 |
| Phosphorylation | T36 | CK2 | Decreased ATPase activity, increased inhibitor binding | 111,112 |
| Phosphorylation | Y197 | Yes | Decreased co-chaperone association | 113 |
| Phosphorylation | Y309 | Yes | Increased co-chaperone association | 113 |
| Phosphorylation | T5, T7 | DNA-PK | Increased extracellular expression | 75,114 |
| Phosphorylation | T | P2X7 receptor repressor | 115 | |
| Phosphorylation | S263 | B-Raf | Unknown | 116 |
| Phosphorylation | S460 | PKA | Unknown | 117 |
| Phosphorylation | T90 | PKA | Increased extracellular expression, decreased client association | 84,118 |
| S-nitrosylation | C597 | eNOS | Decreased chaperone function, decreased ATPase activity | 119,120 |
| Ubiquitination | Decreased client association | 121 |
Much effort has been seen in the past two decades to characterize the specific protein interactions with other proteins termed co-chaperones. Co-chaperones assist Hsp90 throughout its conformational cycling that is required for its normal function, act as substrate recognition proteins and even provide additional enzymatic activity. The predominant class of co-chaperones are the tetratricopeptide repeat (TPR) domain containing proteins, which bind the MEEVD motif found in the C-terminus of Hsp90 [15–18]. Among the co-chaperones with a TPR domain are C-terminus of Hsp70-interacting protein, Hsp70–Hsp90 organizing protein (Hop), cyclophilin 40, FK506-binding protein and protein phosphatase 5 (PP5). While most co-chaperones facilitate the recruitment of other substrate proteins, some of these co-chaperones add enzymatic functionality to the chaperone complex as is the case for the isomerases, phosphatases and ligases [19–21]. Other co-chaperones that interact with Hsp90 via alternative domains are activator of Hsp90 ATPase homolog 1 (Aha1), which enhances the function of Hsp90 by stimulating its ATPase activity [22,23]. The co-chaperone that is most implicated in facilitating tumorigenesis is cell division cycle 37 (Cdc37) because it associates with mutant kinases that drive cancer progression [24]. Recently it was shown that the Hsp90–Cdc37 complex binds two-thirds of the kinome to varying degrees, while at the same time demonstrating negligible interactions with ligases and transcription factors [25]. Another co-chaperone that is responsible for the complexing of nuclear hormone receptors and Hsp90 is p23. However, its interactions are not as limited as Cdc37, as p23 has been found in a broad range of Hsp90–client complexes [26–29]. Unlike the activating co-chaperone Aha1, the co-chaperones Cdc37, p23 and Hop inhibit ATPase activity. These co-chaperones therefore add another layer of regulation of this multifaceted master chaperone, underlining the complexity of the Hsp90 chaperone cycle and its folding functions. Hsp90 is also regulated transcriptionally, primarily through direct interactions with the transcription factor HSF [30]. Despite its high level of expression in normal cells, cellular stresses such as heat shock (37–42 °C) have been reported to induce Hsp90 levels by as much as twofold [31].
Two other immediate family members, glucose regulated protein 94 (Grp94) and tumor necrosis factor receptor associated protein 1 (Trap1), share sequence similarity in the ATP-binding domain and are also thought to act as cellular chaperones to promote protein stability and folding either in the mitochondria (Trap1) or endoplasmic reticulum (Grp94), although they do not exemplify the same complexity and vast interactions that Hsp90 exhibits [32]. Like Hsp90, Grp94 and Trap1 contain an N-terminal ATP-binding domain and ATPase activity that is also necessary for cellular function [33–36]. Unlike Hsp90, the list of client proteins interacting with either Grp94 or Trap1 is much more limited and less well defined. Several groups have proposed that Grp94 and Trap1 are additional potential targets that can be exploited as cancer chemotherapies, although selective inhibitors of these proteins have yet to be defined.
A surprising and common finding with most Hsp90 inhibitors is their selectivity for certain tumor cells and not other cells. With few exceptions in vivo, most cancer cells are more sensitive to Hsp90 inhibition than non-transformed cells and non-toxic doses demonstrate anti-cancer activity. In animals and humans, Hsp90 inhibitors consistently accumulate in tumors whereas they are rapidly cleared from plasma and do not appear to enter most tissues [37–43]. One notable exception may be the retina. Although idiosyncratic in nature, a reversible ocular toxicity has been observed in some patients with some but not all Hsp90 inhibitors [44]. Generally this occurs at higher dosing or after prolonged exposure and manifests as a loss of night vision. Drug withdrawal or dose reduction usually reverses the symptoms. However, the idiosyncratic nature of the phenomenon suggests that the mechanisms of drug accumulation in the retina may be different from tumor cells and perhaps related to some underlying eye pathology in patients that are most susceptible. Alternatively, the structural variations of different Hsp90 inhibitors permit varying degrees of cellular uptake, with the retina being more susceptible than most other tissues. The fact that certain tumor cells preferentially absorb Hsp90 inhibitors strongly suggests that specific mechanisms exist within these cells that contribute to druggability of Hsp90. One early hypothesis suggested that in tumor cells Hsp90 preferentially exists in chaperone complexes and these multiprotein complexes were reported to exhibit a higher affinity for Hsp90 inhibitors as well as higher ATPase activity [45]. Experiments with affinity resins based on immobilized Hsp90 inhibitors suggest a large imbalance of Hsp90 to its co-chaperones in both normal and transformed cells [46,47]. A simple mechanism that could explain the drug accumulation may be that tumor cells simply express more Hsp90 than normal cells. Certainly, transformation has been shown to induce expression of Hsp90 at the protein level and this can also be measured in tumors isolated from patients. However, even in the most extreme case the fold inductions are 2–3-fold at best at the protein level. As discussed, in normal cells Hsp90 is thought to constitute 1–2% of the cellular protein and many believe that high expression of Hsp90 is evolutionarily conserved. Indeed, normal Hsp90 expression levels beg the question as to why tumor cells would need to induce more expression of the protein. We align our theory with others that the abundance of Hsp90 is evolutionarily conserved for a purpose and not wastefully guarded to bewilder scientists. If we can come to an understanding of the mechanisms that accentuate the tumor differences, Hsp90 inhibitors could be universally employed to fight the constantly evolving battle on cancer.
In this review we discuss the mechanisms that differentiate tumor Hsp90 from the Hsp90 that is abundantly expressed in normal tissues, thereby making it a druggable cancer target. The three mechanisms, not mutually exclusive, that are the most important in distinguishing Hsp90 in cancer cells from Hsp90 in non-transformed cells are (a) the induction of Hsp90 mRNA and protein, (b) the activation of the protein through either client association or post-translational modifications and (c) the localization to ectopic cellular compartments (Fig. 1). As we understand more clearly the mechanistic differences in tumor biology and normal cellular biology regarding Hsp90, the remaining hurdles for Hsp90 inhibitors to enter routine clinical practice will be overcome. For reviews on Hsp90 inhibitors see [37,48–53].
Fig. 1.

Hsp90 differences between tumor cells and adjacent normal cells. Hsp90 is elevated in tumor cells, induction. Hsp90 is activated (red) in tumor cells and localizes to the cell surface, whereas in normal cells Hsp90 resides exclusively in the cytosol.
Induction
First we look at the differential expression of Hsp90. Few will argue that Hsp90 is overexpressed in tumors 2–3-fold higher than corresponding non-tumorigenic tissue, but even under basal conditions Hsp90 is abundant, comprising 1–3% of the total cellular protein [46,51,54,55]. Hsp90 is upregulated in response to cellular stress imposed by heat, hypoxia and nutrient deprivation, which are commonly associated with the tumor microenvironment. Thus it has been proposed that Hsp90 upregulation in tumors is essential to surviving the harsh microenvironment by allowing unstable permutations to persist that drive tumor malignancy [56].
In one of the strongest cases for clinical applications, hormone and protein kinase dependent breast cancer, Hsp90 expression levels have been well characterized and correlated to patient outcome of survival. First, Hsp90 expression in the breast has been recently evaluated immunohistochemically for various types of tissue: normal, pre-malignant and malignant. Hsp90 was shown to be expressed at higher levels in cancer tissues compared with non-cancer tissues [57]. Also Pick and colleagues performed immunohistochemical analysis of breast cancer cell lines and 655 primary breast cancers, including 331 estrogen receptor positive (ER+) and 324 ER− tumors, and found detectable Hsp90 expression in all of the breast cancer cell lines and in 90% of primary breast cancers. In addition, they reported that high expression of Hsp90 is associated with poor prognosis [58]. Because of the recognition of specific molecular subtypes of breast cancer, biostatisticians evaluated Hsp90 gene expression from profiles of over 4000 breast cancer patients from 23 publicly available gene expression databases, annotated with overall survival data from over 1000 patients. They found a normal distribution of Hsp90 expression, and confirmed that high expression of Hsp90 was associated with a poor overall survival [59]. It has also been shown in melanomas, leukemia and human colonic carcinoma that Hsp90 is elevated in the transformed and even more malignant tumors [60–62]. Elevated levels are also being detected in the serum of patients with non-small-cell lung cancer and prostate cancer [63,64].
Based on these studies it is believed that high expression of Hsp90 represents an important oncogenic signaling node for malignant behavior in cancer and detecting upregulation or activation of Hsp90 in cancer cells could be an early indicator of malignant behavior. However, increased Hsp90 expression is not sufficient to explain the mechanisms that drive tumorigenicity. One discrepancy is that even though Hsp90 is elevated in breast tumors compared with normal breast tissue, there are other normal tissues such as the bladder, spleen and brain that express higher Hsp90 in the ratio of total Hsp90 : total protein (Fig. 2) [55,65]. This leads to the argument that there is something unique about the protein in cancer cells that distinguishes it from Hsp90 found in normal tissue. With hundreds of putative client proteins and over 30 sites where post-translational modifications can occur, one can easily envision a scenario where tumor Hsp90 proteins are poised to behave differently from normal Hsp90.
Fig. 2.
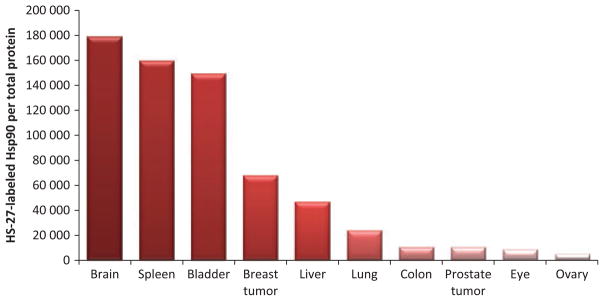
Labeled Hsp90 in various lysed mouse tissues normalized to total protein. The fluorescently labeled small molecule inhibitor HS-27 detects active Hsp90, and surprisingly two xenograft tumor samples did not exhibit the highest active Hsp90 to total protein ratio.
Activation
Hsp90 is implicated in cancer cell survival and growth through its transient interactions with client proteins. Over 400 clients have been thought to be identified thus far and many of these are involved in mediating signal transduction pathways that govern cellular growth, apoptotic evasion, differentiation and metastasis [25,66–68]. For a current and comprehensive list of reported Hsp90 protein interactions visit the Picard laboratory’s website at http://www.picard.ch/Hsp90Int/index.php.
Due to the fact that Hsp90 inhibitors preferentially target tumors, one may infer that these drugs target a subset pool of Hsp90, and this subset exhibits a high affinity conformation of Hsp90 for the inhibitors. One possibility is that Hsp90 exists in a multi-chaperone complex with transformation-specific oncoproteins, such as classical mutant kinases, human epidermal growth factor receptor 2 (Her2) and Bcr-Abl. These proteins may represent a small fraction of the client proteins regulating the transformed phenotype. The latent Hsp90 complexes regulating normal misfolding processes and comprising at any time the majority of total cellular Hsp90 may not be effectively inhibited by these drugs at the relatively non-toxic doses used. Such an interpretation leads to the hypothesis that, under normal conditions, Hsp90 interacts with client proteins in a dynamic, low affinity manner regulated by low affinity binding and release of ATP and ADP. Upon mutation or deregulation, which is characteristic of the cancer phenotype, many of these client proteins may display unusually stable association with Hsp90, representing the active and druggable state.
A 2003 Nature paper by Kamal et al. [45] claimed that Hsp90 in tumors exists entirely in multi-chaperone complexes and that when Hsp90 is in these specific complexes it has higher ATPase activity and a 100-fold higher affinity for the inhibitor 17-AAG. However, one incorrect assumption was that all Hsp90 has an equal opportunity of binding ATP or its mimetics that are immobilized to a bead. We and others have shown that only a fraction (20–30%) of Hsp90 binds to ATP or its ligands. Radiolabeled PU-H71 also only labeled 30% of the Hsp90 in MDA-MB-468 cells and only half that in CML cells [46]. As far as co-chaperone involvement, Kamal et al. demonstrated that when Hsp90 was reconstituted in vitro with Hsp70, Hsp40, Hop and p23, the highest ATPase activity was observed. Moulick et al. also showed that Hsp90 recognized by immobilized ligand precipitated the co-chaperones Hsp70, Hsp40, Hop and Hip and that these co-chaperones were not found in the fraction of the antibody-isolated Hsp90, but they were found in the flow-through [45,46]. It is thus hypothesized that the population of Hsp90 that binds to the ligand also exists in complex with several co-chaperones, but the ‘inactive’ pool does not exist with co-chaperones. In their in vivo analysis they found that mouse tumors compared with non-corresponding normal tissue do not differ that much in total Hsp90 levels as determined by western blotting. However, their ATPase activity was higher and their affinity for Hsp90 inhibitors was more [45], thus supporting that transformation and malignancy cannot be explained solely by the elevated expression of Hsp90. On the other hand, efforts to replicate this work have failed to show the exclusive complex of Hsp90 found in cancer. With regard to the complex having a higher affinity for Hsp90 inhibitors is believed to be an artifact of non-specific binding to the affinity resin. Our laboratory has shown that non-specific binding to an Hsp90 affinity resin decreases upon extending the ligand away from the immobilized bead. Hsp90 was cleanly and competitively eluted from the affinity resin [47], suggesting an alternative hypothesis that when Hsp90 is in complex with an inhibitor that targets the ATP-binding domain, co-chaperones that should be in stoichiometric abundance are displaced and not recovered.
The studies to elucidate the client–chaperone interactions for Hsp90 are incomplete and provide little rationale for these interactions. For example, Hsp90 does not recognize an amino acid sequence that is common among the vast array of putative client proteins, nor do proteins within the same family that are structurally similar interact with Hsp90 in a comparable manner, such as is the case with epidermal growth factor receptor and Her2. Due to the many criticisms that have been offered for the various approaches of identifying the Hsp90–client interaction, whether by immunoprecipitation, yeast two-hybrid assays or mass spectrometry analysis, a recent study attempted to circumvent previous obstacles by expressing tagged potential client proteins (i.e. kinases, ligases and transcription factors) with essential co-chaperones in order to study the interactions in a quantifiable manner. While no specific recognition sequence or structure was determined, the researchers concluded that a co-chaperone, Cdc37 in this case, provided a recognition of an as yet undefined fold and the thermal and conformational stability determined the extent of the interaction of Hsp90 with many of its kinase clients [25]. Cynically, one could also conclude from this study that any denatured protein is more likely to interact with Hsp90 than properly folded ones. Certainly the observation that inclusion of protein kinase inhibitors generally reduced binding to Hsp90 supports such a notion. Expressed protein kinases are generally stabilized in the presence of ATP competitive inhibitors as reflected by an increase in thermal stability. In our experience, studies that use assays relying on affinity pull-downs to study protein–protein interactions are fraught with artifacts and designing the appropriate controls to prove that interactions are real is not easy. Our own experience in the development of affinity resins targeting Hsp90 revealed the importance of carrying out appropriate controls in discriminating proteins that bind directly to Hsp90 versus those that are non-specific (Fig. 3). Figure 3 shows that changing the linker that is used to immobilize an Hsp90 inhibitor can dramatically affect the patterns of proteins recovered from a cell extract. One could conclude for example from the resin shown in the decane linker lane that this resin recovers a large amount of Hsp90 clients in addition to Hsp90. However, by blocking binding of Hsp90 itself to the affinity medium by including a free Hsp90 inhibitor in the cell extract prior to mixing with the affinity resin shows that, although recovery of Hsp90 is blocked, none of the other proteins are affected, showing that these are artifacts and have nothing to do with Hsp90 [47]. Ultimately this has led us to develop a medium that clearly recovers only Hsp90 and very little else including co-chaperones. We are somewhat perplexed by the absence of the Hsp90 interactome with this medium, despite the observation that it clearly recovers Hsp90 in a competitive manner. Our own conclusions from these studies is that the Hsp90 interactome should be revisited using more rigorous controls such as including Hsp90 inhibitors in control extracts to eliminate non-specific binding to the medium surface. We feel that such studies are likely to shorten the stable interactome considerably.
Fig. 3.

Directed chemical evolution of a selective affinity medium for Hsp90. SDS/PAGE silver stain showing the effects of different side chain modifications on Hsp90 recovery and recovery of nonspecifically bound proteins. Selectivity towards Hsp90 was demonstrated by inclusion of 1 mM of the free ligand (+) in the tissue extract prior to mixing with affinity resin. Mass spectrometry was used to define all of the bound protein. In the azo-6-PEG resin bound proteins were eluted with 25 mM sodium dithionite in NaCl/Pi. PEG, polyethylene glycol. Figure reproduced from [47].
Having suggested that affinity based approaches might lead to misidentification of clients, other approaches examining client fate in the presence of an Hsp90 inhibitor may be more informative. Many studies that try to establish Hsp90 client associations evaluate the fate of the putative client after Hsp90 inhibition. Most often cited is the degradation of a given client, while those proteins that persist are thought not to be chaperoned by Hsp90. The best example of this phenomenon is the degradation of Her2 in breast cancer cell lines treated with Hsp90 inhibitors. By western blot we can confirm recovery of Her2 with Hsp90 on our most selective Hsp90 resin [47]. Importantly, this recovery is blocked by the inclusion of an Hsp90 inhibitor in the cell extracts prior to mixing with the affinity medium. We note, however, that the ratio of active drug bound Hsp90 recovered relative to Her2 is at least two orders of magnitude higher (Hsp90 : Her2). As suggested in studies by Lindquist and colleagues, Hsp90 interacts with its clients transiently or with low affinity, at least in the drug bound state [25]. Client degradation approaches also have their caveats; for example it is common knowledge that the fate of unfolded proteins varies–some are degraded with differing kinetics and others form more stable aggregates when improperly folded. Thus, the lack of sensitivity to Hsp90 inhibition cannot fully explain the interactions or discredit client interactions. Additional caveats in establishing clients is that only a fraction of Hsp90 is considered active due to its ability to bind inhibitors in its ATP-binding pocket leaving the remaining Hsp90 pool to continue its chaperoning functions.
With many uncertainties in the reported clients and no clear way to establish a transient interaction for a protein that is otherwise stable without the chaperoning functions of Hsp90, the nature of defining a general activated state of Hsp90 in cancer will continue to be difficult to attain. However, much progress is being made in the differences in cancer Hsp90 compared with normal tissue Hsp90 with regard to its post-translational modifications. These modifications are now demonstrating their effects in ATPase activity, as well as localization, which in turn are affecting the association Hsp90 has with other proteins. For a comprehensive list of reported modifications see Table 1. Hsp90 modifications that influence localization are highlighting some of the stronger differences that are being observed in cancer cells over normal tissue.
Localization
The emphasis of distinguishing tumorigenic Hsp90 from normal Hsp90 is being placed in the field of client association. Post-translational modifications are thought to influence the association of Hsp90 with its clients, and in the past decade the community of Hsp90 has begun to reveal how post-translational modifications affect the localization of Hsp90. More importantly, they have shown that ectopic localization can lead to the progression of a more malignant phenotype of most cancers.
No longer is Hsp90 being considered to reside exclusively in the intracellular milieu, but it can be found on the surface membrane of a variety of cancer cells, as well as being secreted into the extracellular space [60,69]. The fact that cell surface Hsp90 is higher in some cancer cells than normal cells makes it an even more attractive target to destabilize metastatic pathways that are dependent on surface Hsp90 for invasion and migration. Since the initial screen in 2004 that implicated Hsp90 in cell invasion and migration, many researchers have shown that blocking or neutralizing secreted Hsp90 has an inhibitory effect on these metastatic behaviors [70,71]. While we still do not understand the mechanisms responsible for Hsp90 extracellular expression, certain environmental stresses and growth factors have been shown to stimulate its secretory pathways [72,73]. The secretion of Hsp90 also appears to be influenced by post-translational modifications, such as acetylation and phosphorylation [74,75].
The presence of Hsp90 outside the cell was first implicated in 1986 when a mouse tumor-specific antigen was found to be a heat shock protein, now recognized as Hsp90 [76]. In 2004, it was published that Hsp90 was discovered in a functional screen looking at cell surface proteins that are necessary for cell invasion, but due to the cellular abundance of Hsp90 it was first viewed as an artifact. However, the screen was validated and Hsp90 was determined to be biologically important in the mechanisms of cell invasion as it was established that it interacts with and activates matrix metalloproteinase-2 (MMP2) [70]. Also in 2004, it was observed that in flow analysis of malignant melanoma isolated tumor cells exhibited surface expressed Hsp90 [60]. In the same year it was discovered that surface Hsp90 plays a role in the migration of developmental neurons. Both Hsp90 α and β are expressed on the surface of rat primary neural cells and antibodies against Hsp90 inhibited cell motility and lamellapodia formation. In this study crude measurements were made to determine the relative expression of surface Hsp90. Ratios of surface to total levels of Hsp90 were obtained and surface Hsp90 represented < 10% of total cellular Hsp90 [77]. Others have shown that tumor cell lines excrete Hsp90 into the conditioned medium and the lack of other classical intracellular proteins such as actin and tubulin argue that the Hsp90 does not come from lysed cells. In one study of dying cells, acrylamide-induced necrosis led to extracellular release of Hsp90 and increased HSF1 activity. Extracellular Hsp90 could be considered a danger signal to the immune system that something is detrimental to the cells [78]. However, it is unknown what external cues trigger the excretion of Hsp90 in necrotic cells.
Because intracellular Hsp90 plays a key role in proteomic homeostasis, it begs the question, are its chaperoning functions required outside the cell or does it play a different role altogether? It is known that intracellular misfolded proteins have three possible fates: chaperoning, proteolysis or aggregation. It appears that extracellular proteins require chaperoning as well. Compared with intracellular fluid, extracellular fluids have a lower protein concentration, 6% in plasma and 2% in interstitial fluid, as opposed to 30% in cytosol [79]. It is thought that extracellular Hsp90α functions with the co-chaperones Hsp70, Hsp40, Hip, Hop and p23 to assist in the cleavable activation of MMP-2 and can do it independently of ATP, an important feature for performing its function in an environment where ATP is drastically reduced [80].
Normal cells secrete Hsp90 but only when they are subjected to a compromised environment, such as heat, reactive oxygen species, γ-irradiation and injury released growth factors, whereas tumor cells constitutively secrete Hsp90 [72]. Early researchers proposed that Hsp90 is secreted by a non-canonical secretory pathway because Hsp90 lacks the conventional signal peptides in secretory proteins as well as post-translational modifications such as N-glycosylation. Also Hsp90 is not localized to the endoplasmic reticulum and Golgi apparatus and Hsp90 secretion is resistant to Brefeldin A treatment, a classical inhibitor of endoplasmic reticulum/Golgi dependent secretion [81]. Indeed it has been demonstrated that Hsp90 is excreted through the exosome pathway [72,82]. McCready et al. showed that when exosomes harvested from MDA-MB-231 cells were applied to other invasive cell lines in a wound healing assay both exogenous exosomes and recombinant Hsp90 stimulated faster migration into the scratched area. Conditioned medium was immunoprecipitated for Hsp90α and 10 client proteins were identified by mass spectrometry. The complex of interest was Hsp90 : AnnexinII : tPA : plasminogen, which results in the activation of plasmin, a key protein in tumor invasion and cancer metastasis [82]. Why, however, is some Hsp90 targeted for exosomal secretion while the majority remains in the cell? Additionally, secretion of Hsp90 does not appear to be isoform specific as first speculated, with the recent finding of Stellas et al. who showed that breast cancer cells also secrete the β isoform [70,72,73,83].
Some speculated whether or not the numerous possibilities for protein modification distinguish Hsp90 for secretion. Knockdown of histone deacetylase 6 (HDAC6) results in increased acetylation which in turn decreases ATP binding and chaperone activity in Hsp90. Seven lysine residues on Hsp90α revealed hyperacetylation by mass spectrometry (Table 1), and acetylation increased 17-AAG binding to Hsp90, whereas K294 acetylation in the middle domain affects co-chaperone binding. Hyperacetylated Hsp90 was found extracellularly and promoted in vitro breast cancer cell invasion. Additionally, an antiacetyl Lys69 Hsp90α antibody markedly inhibited the cell’s invasiveness. P300 is the putative histone acetyl transferase responsible for acetylating Hsp90α. The Hsp90α K292Q mutant showed increased binding to ATP whereas the other K to Q mutations at the other sites showed decreased binding. However, all the acetylated mimetics demonstrated increased binding to biotinylated geldanamycin [74]. While acetylation appears to play a role, others suggest that it is the phosphorylation of T5 and T7 of Hsp90α and others suggest that it is a C-terminal truncation [75,84]. It was shown that the secreted Hsp90 is a C-terminal truncated form and its secretion was regulated by the C-terminal EEVD motif via interacting with proteins containing TPR domains. It was also demonstrated that the secretion of Hsp90 was determined by the phosphorylation status at residue Thr90, regulated by protein kinase A and PP5. It was further demonstrated that the secretion of Hsp90 is a prerequisite for its pro-invasive function and blocking the secreted Hsp90 resulted in significant inhibition of tumor metastasis. Meanwhile, the level of plasma Hsp90 was positively correlated with tumor malignancy in clinical cancer patients [84].
While the precise mechanisms by which Hsp90 is targeted for exosomal secretion remain unclear, it is well established that extracellular Hsp90 plays pivotal roles in driving a non-motile tumor cell to become motile and invasive. Hsp90 facilitates each step in the three-step model for tumor invasion: degradation of the extracellular matrix, adhesion and migration of the cancer cells [85]. These events most probably are coordinated with intracellular chaperoning functions that drive the cell to become more malignant. These observations were made in the context of Hsp90 inhibition and its effects on cytoskeletal architecture and remodeling during cell motility and cell invasion. Hsp90 inhibition led to decreased cell motility associated with a decrease in filopodia, lamellipodia and tough cortical actin bundles and resulted in less ruffling. The Hsp90 inhibition was also associated with a loss of Diaphanous-related formin-2 and a decrease in Ras homolog gene family member A, both necessary for the formation of lamellipodia and generation of contractile force. Hsp90 inhibition stimulated an increase in the pull-down of actin in the soluble fraction with Hsp90. These observations suggested an increased interaction of Hsp90 with the soluble form of actin (G-actin) and αB-crystallin upon Hsp90 inhibition, which might be responsible for the decreased actin treadmilling at the cell periphery. Actin–enhanced cyan fluorescent protein and actin–enhanced yellow fluorescent protein showed a 50% reduction in fluorescence resonance energy transfer when inhibited with 17-AAG, and 17-AAG treatment resulted in an 80% decrease in inverse cell invasion for MDA-MB-231-GFP cells. Inhibition of Hsp90 also leads to a decrease in surface Hsp90 expression [86]. In light of the many advances in studying extracellular Hsp90, several questions remain. How do the extracellular cues communicate with the exosomal trafficking machinery to send Hsp90 out of the cell? Do the extracellular cues selectively cause Hsp90 secretion and leave other exosome-housed proteins behind or do they simply trigger secretion of the exosome vesicles as a whole? What percentage of total intracellular Hsp90 gets secreted?
One of the most important questions that we can ask pharmacologically is whether the surface expression of Hsp90 can be exploited to increase the safety threshold of future Hsp90 inhibitors. Our laboratory is currently exploring the hypothesis that expression of surface Hsp90 may contribute to the tumor selectivity of many Hsp90 inhibitors. This hypothesis is driven by recent data using cell impermeable Hsp90 inhibitors tethered to fluorophores showing that a portion of ectopically expressed Hsp90 is re-internalized causing drug accumulation (Barrott et al., unpublished results). By extension of this observation, it is conceivable that many leading inhibitors of Hsp90 preferentially enter tumor cells through a similar mechanism and may have been fortuitously optimized to do so. Such a statement is not without possibility. From our own experience in the discovery of Hsp90 inhibitors, although our initial high throughput leads were identified by elution of native Hsp90 bound to an ATP affinity column, all molecules were subsequently evaluated for biological activity in cell-based assays [41]. These assays comprised a combination of growth inhibition and evidence of the mechanism of action including inhibition of Her2 expression, induction of Hsp70 and S6 phosphorylation. Generally at this stage compounds that caused overt cell toxicity would have been considered second tier and not pursued further. Such a path would most likely have been pursued by other groups developing Hsp90 inhibitors. It will therefore be interesting to test whether Hsp90 inhibitors that have higher c Log P values, and are thus more freely diffusible, are more toxic to normal cells compared with our current armamentarium of clinically active Hsp90 inhibitors. This finding would have great significance because the development of Hsp90 inhibitors that only penetrate cells through interactions with surface Hsp90 could have vastly improved safety profiles enabling the therapeutic window of this class of drugs to be extended.
Combination therapy
Typically when Hsp90 inhibitors are successively dosed to animals bearing human tumors, the tumors stop growing. However, when the inhibitor is withdrawn the tumors tend to start growing again (Fig. 4). Generally the same phenomenon occurs in patients with solid tumors being treated with a variety of structurally unrelated Hsp90 inhibitors. These observations are consistent with the role of the protein in maintaining tumor growth and suppression of these pathways by Hsp90 inhibitors. For these reasons Hsp90 inhibitors may have limited use as a mono-therapy, although more recent observations suggest that certain tumor types may be more responsive to Hsp90 inhibition than others and mono-therapy may be sufficient to promote tumor reduction and even cellular death. In recent phase II studies with ganetespib alone, an encouraging response rate of 50% in non-small-cell lung carcinoma patients whose tumors contained ALK translocations was observed [87]. Instead, however, there is great excitement in the field for using Hsp90 inhibitors in combination with either existing chemotherapeutics or more cutting-edge drugs like Herceptin, lapatinib or Gleevec. Figure 4 shows that a combination of the drug SNX5422 and Herceptin not only shrinks the tumors dramatically and persistently (even after withdrawal), but it is also synergistic. The argument for combination therapy is in the evidence that genome sequencing of human cancers reveals numerous driver mutations within any one individual cancer [88,89]. Intratumor heterogeneity may foster tumor evolution and adaptation and hinder personalized-medicine strategies that depend on results from single tumor-biopsy samples [90]. Hsp90 inhibitors can play a unique role in preventing drug resistance in tumors because oncogenes rely heavily on Hsp90 to chaperone their otherwise unstable conformation due to their mutations. This dependence has been termed oncogene addiction. Because numerous mutant oncoproteins are ‘addicted’ to Hsp90 activity, an inhibitor to Hsp90 has the ability to affect multiple targets and pathways, which can prevent oncogene switching, a major mechanism for developing resistance [91,92].
Fig. 4.

In vivo pharmacology of SNX-5422. Effects on BT-474 tumor-bearing nude mice (n = 10) dosed with vehicle or (1) 10 mg·kg−1 SNX-5422 (SNX) by mouth three times per week, (2) trastuzumab (TZB) 3 mg·kg−1 intravenously twice per week, (3) 10 mg·kg−1 SNX by mouth three times per week and TZB 3 mg·kg−1 intravenously twice per week or (4) 40 mg·kg−1 SNX three times per week. Dosing in all drug-treated groups was for only the first 3 weeks. All groups were followed for 6 months or until sacrifice due to tumor burden. Figure reproduced from [41].
Hsp90 inhibitors have the potential to circumvent or diminish the drug resistance seen in the use of target-based therapies, but because cancers evolve, resistance to Hsp90 inhibition is also a real possibility. However, the silencing of co-chaperones such as p23, Aha1 or Cdc37 has been shown to cause dramatic sensitization to Hsp90 inhibition [93–95]. Because Hsp90 inhibition allows the trimerization and activation of HSF1, and because elevated HSF1 is linked to stages of oncogenesis, there are efforts to find inhibitors to HSF1 that can be used in conjunction with Hsp90 inhibitors to minimize the negative effects of Hsp90 inhibition [96,97]. As a result of HSF1 activation, Hsp70 transcription is dramatically upregulated, which is often used as readout in cell-based assays for efficacy of an Hsp90 inhibitor. However, Hsp70 induction is another chaperone that can drive oncogenesis in many tumors. Researchers have shown a therapeutic value for targeting Hsp70 because tumors overexpress the inducible form over the constitutive form [98]. Therefore, combination therapy that inhibits both Hsp90 and the inducible Hsp70 could be a formidable method of treating cancer by eliminating the unwanted effect of Hsp70 induction.
Conclusion
While some are still not cognizant to the advances of Hsp90 inhibitors in the clinic, others, including members of the Hsp90 community, still question whether or not any of the now 17 Hsp90 inhibitors will successfully exit clinical trials and enter the market. We think the day will come when Hsp90 inhibitors will be commonplace in the clinic and that in the oncological application of Hsp90 inhibitors these drugs will be used as first line chemotherapeutics in combination with other potent anti-cancer therapies to prevent or prolong the development of drug resistance. In the immediate term combination therapy involving an Hsp90 inhibitor and cutting-edge targeted therapies clearly holds the greatest promise. If the consistent synergisms in efficacy and tumor elimination observed in animal combination studies with drugs such as Herceptin hold true in human trials, perhaps we are well on the way to seeing actual ‘cures’ rather thinking about merely halting disease progression over the shorter term. Beyond combination therapy, as we have discussed, other aspects of Hsp90 biology may render it even more druggable. Exploring the differences in Hsp90 between cancer cells and normal tissues may reveal additional strategies on how to target Hsp90 and how to improve its therapeutic window. One of these in our sights is the role of ectopically expressed Hsp90 in metastatic progression. Although yet to be demonstrated in human biopsy samples, clearly, if there is a correlation between expression of ectopic Hsp90 and metastatic progression, this is likely to be of both diagnostic and therapeutic importance. If expression of ectopic Hsp90 correlates with poor outcomes it could be used as an early biomarker to discriminate indolent forms from aggressive disease. We are particularly excited by the prospect of developing Hsp90 inhibitors that only target ectopically expressed Hsp90 as a means to improve the selectivity of Hsp90 inhibitors and deliver tumor-killing payloads to tumor cells. On top of all these tangible possibilities for extending the therapeutic potential of Hsp90 is the co-chaperone/ chaperone machinery of associated proteins regulating and regulated by Hsp90. Once bona fide clients are established and validated, one could envision strategies that could selectively inhibit these protein–protein interactions, thus potentially making Hsp90 therapy highly targeted and disease specific.
Finally, from a pharmacologist’s perspective, there are lessons to be learned from the development of Hsp90 as a cancer target that are pertinent to 21st century drug discovery. Clearly, new therapies to treat cancer are not always going to be obvious as has been the case in the past, i.e. one must not assume that a ‘hallmark of cancer’ involves a point mutation and that alone signifies a usable target. Not all targets are going to be ‘Gleevec type stories’. If we make the assumption that this is the case we are likely to miss the boat on some great opportunities. A second lesson is that although an Hsp90 drug has yet to be brought to market, there is clearly a ground swell of independent data that strongly indicate that this will happen soon, despite some side effect concerns such as retinal toxicities. This ground swell is self-evident in the form of the numbers of independently derived inhibitors that are currently being advanced clinically. Within these independent paths we count an extraordinary six distinct structurally diverse scaffolds upon which the current clinical armamentarium of Hsp90 inhibitors is currently derived. In future drug discovery efforts involving unconventional targets where we do not have obvious genetic data to validate a target we should look for similar trends to assure pharmaceutical companies that such targets should not be ignored.
Acknowledgments
This work was funded by 1R01-AI089526-01 and 1R01AI090644-01 to TAJH.
Abbreviations
- Aha1
activator of heat shock 90 kDa protein ATPase homolog 1
- Cdc37
cell division cycle 37
- ER±
estrogen receptor positive or negative
- Grp94
glucose regulated protein 94
- HDAC6
histone deacetylase 6
- Her2
human epidermal growth factor receptor 2
- Hip
Hsp70-interacting protein
- Hop
Hsp70–Hsp90 organizing protein
- HSF
heat shock factor
- Hsp90
heat shock protein 90
- MMP2
matrix metalloproteinase-2
- PP5
protein phosphatase 5
- tPa
tissue plasminogen activator
- TPR
tetratricopeptide repeat
- Trap1
tumor necrosis factor receptor associated protein 1
References
- 1.Koboldt D, Fulton C, McLellan RS, Schmidt MD, Kalicki-Veizer H, McMichael J, Fulton JF, Dooling LL, Ding DJ, Mardis L, et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Passarino G, Cavalleri GL, Stecconi R, Franceschi C, Altomare K, Dato S, Greco V, Luca Cavalli Sforza L, Underhill PA, de Benedictis G. Molecular variation of human HSP90alpha and HSP90beta genes in Caucasians. Hum Mutat. 2003;21:554–555. doi: 10.1002/humu.9141. [DOI] [PubMed] [Google Scholar]
- 4.Urban JD, Budinsky RA, Rowlands JC. An evaluation of single nucleotide polymorphisms in the human heat shock protein 90 kDa alpha and beta isoforms. Drug Metab Pharmacokinet. 2012;27:268–278. doi: 10.2133/dmpk.dmpk-11-sc-114. [DOI] [PubMed] [Google Scholar]
- 5.Komarova NL, Wodarz D. Drug resistance in cancer: principles of emergence and prevention. Proc Natl Acad Sci USA. 2005;102:9714–9719. doi: 10.1073/pnas.0501870102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bozic I, Allen B, Nowak MA. Dynamics of targeted cancer therapy. Trends Mol Med. 2012;18:311–316. doi: 10.1016/j.molmed.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ying W, Du Z, Sun L, Foley KP, Proia DA, Blackman RK, Zhou D, Inoue T, Tatsuta N, Sang J, et al. Ganetespib, a unique triazolone-containing Hsp90 inhibitor, exhibits potent antitumor activity and a superior safety profile for cancer therapy. Mol Cancer Ther. 2012;11:475–484. doi: 10.1158/1535-7163.MCT-11-0755. [DOI] [PubMed] [Google Scholar]
- 9.Proia DA, Sang J, He S, Smith DL, Sequeira M, Zhang C, Liu Y, Ye S, Zhou D, Blackman RK, et al. Synergistic activity of the Hsp90 inhibitor ganetespib with taxanes in non-small cell lung cancer models. Invest New Drugs. 2012;30:2201–2209. doi: 10.1007/s10637-011-9790-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Acquaviva J, Smith DL, Sang J, Friedland JC, He S, Sequeira M, Zhang C, Wada Y, Proia DA. Targeting KRAS-mutant non-small cell lung cancer with the Hsp90 inhibitor ganetespib. Mol Cancer Ther. 2012;11:2633–2643. doi: 10.1158/1535-7163.MCT-12-0615. [DOI] [PubMed] [Google Scholar]
- 11.Wiech H, Buchner J, Zimmermann R, Jakob U. Hsp90 chaperones protein folding in vitro. Nature. 1992;358:169–170. doi: 10.1038/358169a0. [DOI] [PubMed] [Google Scholar]
- 12.Pearl LH, Prodromou C. Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu Rev Biochem. 2006;75:271–294. doi: 10.1146/annurev.biochem.75.103004.142738. [DOI] [PubMed] [Google Scholar]
- 13.Sawarkar R, Sievers C, Paro R. Hsp90 globally targets paused RNA polymerase to regulate gene expression in response to environmental stimuli. Cell. 2012;149:807–818. doi: 10.1016/j.cell.2012.02.061. [DOI] [PubMed] [Google Scholar]
- 14.Mollapour M, Neckers L. Post-translational modifications of Hsp90 and their contributions to chaperone regulation. Biochim Biophys Acta. 2012;1823:648–655. doi: 10.1016/j.bbamcr.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Owens-Grillo JK, Stancato LF, Hoffmann K, Pratt WB, Krishna P. Binding of immunophilins to the 90 kDa heat shock protein (hsp90) via a tetratricopeptide repeat domain is a conserved protein interaction in plants. Biochemistry. 1996;35:15249–15255. doi: 10.1021/bi9615349. [DOI] [PubMed] [Google Scholar]
- 16.Ratajczak T, Carrello A. Cyclophilin 40 (CyP-40), mapping of its hsp90 binding domain and evidence that FKBP52 competes with CyP-40 for hsp90 binding. J Biol Chem. 1996;271:2961–2965. doi: 10.1074/jbc.271.6.2961. [DOI] [PubMed] [Google Scholar]
- 17.Young JC, Obermann WM, Hartl FU. Specific binding of tetratricopeptide repeat proteins to the C-terminal 12-kDa domain of hsp90. J Biol Chem. 1998;273:18007–18010. doi: 10.1074/jbc.273.29.18007. [DOI] [PubMed] [Google Scholar]
- 18.Chen S, Sullivan WP, Toft DO, Smith DF. Differential interactions of p23 and the TPR-containing proteins Hop, Cyp40, FKBP52 and FKBP51 with Hsp90 mutants. Cell Stress Chaperones. 1998;3:118–129. doi: 10.1379/1466-1268(1998)003<0118:diopat>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith DF, Baggenstoss BA, Marion TN, Rimerman RA. Two FKBP-related proteins are associated with progesterone receptor complexes. J Biol Chem. 1993;268:18365–18371. [PubMed] [Google Scholar]
- 20.Silverstein AM, Galigniana MD, Chen MS, Owens-Grillo JK, Chinkers M, Pratt WB. Protein phosphatase 5 is a major component of glucocorticoid receptor.hsp90 complexes with properties of an FK506-binding immunophilin. J Biol Chem. 1997;272:16224–16230. doi: 10.1074/jbc.272.26.16224. [DOI] [PubMed] [Google Scholar]
- 21.Jiang J, Ballinger CA, Wu Y, Dai Q, Cyr DM, Hohfeld J, Patterson C. CHIP is a U-box-dependent E3 ubiquitin ligase: identification of Hsc70 as a target for ubiquitylation. J Biol Chem. 2001;276:42938–42944. doi: 10.1074/jbc.M101968200. [DOI] [PubMed] [Google Scholar]
- 22.Panaretou B, Siligardi G, Meyer P, Maloney A, Sullivan JK, Singh S, Millson SH, Clarke PA, Naaby-Hansen S, Stein R, et al. Activation of the ATPase activity of hsp90 by the stress-regulated cochaperone aha1. Mol Cell. 2002;10:1307–1318. doi: 10.1016/s1097-2765(02)00785-2. [DOI] [PubMed] [Google Scholar]
- 23.Lotz GP, Lin H, Harst A, Obermann WM. Aha1 binds to the middle domain of Hsp90, contributes to client protein activation, and stimulates the ATPase activity of the molecular chaperone. J Biol Chem. 2003;278:17228–17235. doi: 10.1074/jbc.M212761200. [DOI] [PubMed] [Google Scholar]
- 24.Gray PJ, Jr, Prince T, Cheng J, Stevenson MA, Calderwood SK. Targeting the oncogene and kinome chaperone CDC37. Nat Rev Cancer. 2008;8:491–495. doi: 10.1038/nrc2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taipale M, Krykbaeva I, Koeva M, Kayatekin C, Westover KD, Karras GI, Lindquist S. Quantitative analysis of hsp90–client interactions reveals principles of substrate recognition. Cell. 2012;150:987–1001. doi: 10.1016/j.cell.2012.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson JL, Toft DO. A novel chaperone complex for steroid receptors involving heat shock proteins, immunophilins, and p23. J Biol Chem. 1994;269:24989–24993. [PubMed] [Google Scholar]
- 27.Johnson JL, Toft DO. Binding of p23 and hsp90 during assembly with the progesterone receptor. Mol Endocrinol. 1995;9:670–678. doi: 10.1210/mend.9.6.8592513. [DOI] [PubMed] [Google Scholar]
- 28.Cox MB, Miller CA., III Cooperation of heat shock protein 90 and p23 in aryl hydrocarbon receptor signaling. Cell Stress Chaperones. 2004;9:4–20. doi: 10.1379/460.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Forsythe HL, Jarvis JL, Turner JW, Elmore LW, Holt SE. Stable association of hsp90 and p23, but not hsp70, with active human telomerase. J Biol Chem. 2001;276:15571–15574. doi: 10.1074/jbc.C100055200. [DOI] [PubMed] [Google Scholar]
- 30.Trinklein ND, Chen WC, Kingston RE, Myers RM. Transcriptional regulation and binding of heat shock factor 1 and heat shock factor 2 to 32 human heat shock genes during thermal stress and differentiation. Cell Stress Chaperones. 2004;9:21–28. doi: 10.1379/481.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bagatell R, Paine-Murrieta GD, Taylor CW, Pulcini EJ, Akinaga S, Benjamin IJ, Whitesell L. Induction of a heat shock factor 1-dependent stress response alters the cytotoxic activity of hsp90-binding agents. Clin Cancer Res. 2000;6:3312–3318. [PubMed] [Google Scholar]
- 32.Felts SJ, Owen BA, Nguyen P, Trepel J, Donner DB, Toft DO. The hsp90-related protein TRAP1 is a mitochondrial protein with distinct functional properties. J Biol Chem. 2000;275:3305–3312. doi: 10.1074/jbc.275.5.3305. [DOI] [PubMed] [Google Scholar]
- 33.Tsutsumi S, Mollapour M, Graf C, Lee CT, Scroggins BT, Xu W, Haslerova L, Hessling M, Konstantinova AA, Trepel JB, et al. Hsp90 charged-linker truncation reverses the functional consequences of weakened hydrophobic contacts in the N domain. Nat Struct Mol Biol. 2009;16:1141–1147. doi: 10.1038/nsmb.1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dollins DE, Warren JJ, Immormino RM, Gewirth DT. Structures of GRP94-nucleotide complexes reveal mechanistic differences between the hsp90 chaperones. Mol Cell. 2007;28:41–56. doi: 10.1016/j.molcel.2007.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frey S, Leskovar A, Reinstein J, Buchner J. The ATPase cycle of the endoplasmic chaperone Grp94. J Biol Chem. 2007;282:35612–35620. doi: 10.1074/jbc.M704647200. [DOI] [PubMed] [Google Scholar]
- 36.Leskovar A, Wegele H, Werbeck ND, Buchner J, Reinstein J. The ATPase cycle of the mitochondrial Hsp90 analog Trap1. J Biol Chem. 2008;283:11677–11688. doi: 10.1074/jbc.M709516200. [DOI] [PubMed] [Google Scholar]
- 37.Chiosis G, Neckers L. Tumor selectivity of Hsp90 inhibitors: the explanation remains elusive. ACS Chem Biol. 2006;1:279–284. doi: 10.1021/cb600224w. [DOI] [PubMed] [Google Scholar]
- 38.Bachleitner-Hofmann T, Sun MY, Chen CT, Liska D, Zeng Z, Viale A, Olshen AB, Mittlboeck M, Christensen JG, Rosen N, et al. Antitumor activity of SNX-2112, a synthetic heat shock protein-90 inhibitor, in MET-amplified tumor cells with or without resistance to selective MET inhibition. Clin Cancer Res. 2011;17:122–133. doi: 10.1158/1078-0432.CCR-10-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chandarlapaty S, Sawai A, Ye Q, Scott A, Silinski M, Huang K, Fadden P, Partdrige J, Hall S, Steed P, et al. SNX2112, a synthetic heat shock protein 90 inhibitor, has potent antitumor activity against HER kinase-dependent cancers. Clin Cancer Res. 2008;14:240–248. doi: 10.1158/1078-0432.CCR-07-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang KH, Veal JM, Fadden RP, Rice JW, Eaves J, Strachan JP, Barabasz AF, Foley BE, Barta TE, Ma W, et al. Discovery of novel 2-aminobenzamide inhibitors of heat shock protein 90 as potent, selective and orally active antitumor agents. J Med Chem. 2009;52:4288–4305. doi: 10.1021/jm900230j. [DOI] [PubMed] [Google Scholar]
- 41.Fadden P, Huang KH, Veal JM, Steed PM, Barabasz AF, Foley B, Hu M, Partridge JM, Rice J, Scott A, et al. Application of chemoproteomics to drug discovery: identification of a clinical candidate targeting hsp90. Chem Biol. 2010;17:686–694. doi: 10.1016/j.chembiol.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 42.Eiseman JL, Lan J, Lagattuta TF, Hamburger DR, Joseph E, Covey JM, Egorin MJ. Pharmacokinetics and pharmacodynamics of 17-demethoxy 17-[[(2-dimethylamino)ethyl]amino] geldanamycin (17DMAG, NSC 707545) in C.B-17 SCID mice bearing MDA-MB-231 human breast cancer xenografts. Cancer Chemother Pharmacol. 2005;55:21–32. doi: 10.1007/s00280-004-0865-3. [DOI] [PubMed] [Google Scholar]
- 43.Vilenchik M, Solit D, Basso A, Huezo H, Lucas B, He H, Rosen N, Spampinato C, Modrich P, Chiosis G. Targeting wide-range oncogenic transformation via PU24FCl, a specific inhibitor of tumor Hsp90. Chem Biol. 2004;11:787–797. doi: 10.1016/j.chembiol.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 44.Zhou DTF, Liu Y, Ye J, Ying W, Shin Ogawa L, Inoue T, Lee W, Adjiri-Awere A, Kolodzieyski L, Tatsuta N, et al. Associating retinal drug exposure and retention with the ocular toxicity profiles of Hsp90 inhibitors. J Clin Oncol. 2012;30(suppl 3086) [Google Scholar]
- 45.Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Burrows LCFFJ. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–410. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- 46.Moulick K, Ahn JH, Zong H, Rodina A, Cerchietti L, Gomes DaGama EM, Caldas-Lopes E, Beebe K, Perna F, Hatzi K, et al. Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nat Chem Biol. 2011;7:818–826. doi: 10.1038/nchembio.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hughes PF, Barrott JJ, Carlson DA, Loiselle DR, Speer BL, Bodoor K, Rund LA, Haystead TA. A highly selective Hsp90 affinity chromatography resin with a cleavable linker. Bioorg Med Chem. 2012;20:3298–3305. doi: 10.1016/j.bmc.2012.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li Y, Zhang T, Schwartz SJ, Sun D. New developments in Hsp90 inhibitors as anti-cancer therapeutics: mechanisms, clinical perspective and more potential. Drug Resist Updates. 2009;12:17–27. doi: 10.1016/j.drup.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Powers MV, Workman P. Inhibitors of the heat shock response: biology and pharmacology. FEBS Lett. 2007;581:3758–3769. doi: 10.1016/j.febslet.2007.05.040. [DOI] [PubMed] [Google Scholar]
- 50.Chiosis G, Caldas Lopes E, Solit D. Heat shock protein-90 inhibitors: a chronicle from geldanamycin to today’s agents. Curr Opin Invest Drugs. 2006;7:534–541. [PubMed] [Google Scholar]
- 51.Workman P, Burrows F, Neckers L, Rosen N. Drugging the cancer chaperone HSP90: combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann NY Acad Sci. 2007;1113:202–216. doi: 10.1196/annals.1391.012. [DOI] [PubMed] [Google Scholar]
- 52.Neckers L, Workman P. Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res. 2012;18:64–76. doi: 10.1158/1078-0432.CCR-11-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer. 2010;10:537–549. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Welch WJ, Feramisco JR. Purification of the major mammalian heat shock proteins. J Biol Chem. 1982;257:14949–14959. [PubMed] [Google Scholar]
- 55.Sahu D, Zhao Z, Tsen F, Cheng CF, Park R, Situ AJ, Dai J, Eginli A, Shams S, Chen M, et al. A potentially common peptide target in secreted heat shock protein-90alpha for hypoxia-inducible factor-1alpha-positive tumors. Mol Biol Cell. 2012;23:602–613. doi: 10.1091/mbc.E11-06-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 57.Zagouri F, Sergentanis TN, Nonni A, Papadimitriou CA, Michalopoulos NV, Domeyer P, Theodoropoulos G, Lazaris A, Patsouris E, Zogafos E, et al. Hsp90 in the continuum of breast ductal carcinogenesis: evaluation in precursors, preinvasive and ductal carcinoma lesions. BMC Cancer. 2010;10:353. doi: 10.1186/1471-2407-10-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pick E, Kluger Y, Giltnane JM, Moeder C, Camp RL, Rimm DL, Kluger HM. High HSP90 expression is associated with decreased survival in breast cancer. Cancer Res. 2007;67:2932–2937. doi: 10.1158/0008-5472.CAN-06-4511. [DOI] [PubMed] [Google Scholar]
- 59.Cheng Q, Chang JT, Geradts J, Neckers LM, Haystead T, Spector NL, Lyerly HK. Amplification and high-level expression of heat shock protein 90 marks aggressive phenotypes of human epidermal growth factor receptor 2 negative breast cancer. Breast Cancer Res. 2012;14:R62. doi: 10.1186/bcr3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Becker B, Multhoff G, Farkas B, Wild PJ, Landthaler M, Stolz W, Vogt T. Induction of Hsp90 protein expression in malignant melanomas and melanoma metastases. Exp Dermatol. 2004;13:27–32. doi: 10.1111/j.0906-6705.2004.00114.x. [DOI] [PubMed] [Google Scholar]
- 61.Flandrin P, Guyotat D, Duval A, Cornillon J, Tavernier E, Nadal N, Campos L. Significance of heat-shock protein (HSP) 90 expression in acute myeloid leukemia cells. Cell Stress Chaperones. 2008;13:357–364. doi: 10.1007/s12192-008-0035-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kubota H, Yamamoto S, Itoh E, Abe Y, Nakamura A, Izumi Y, Okada H, Iida M, Nanjo H, Itoh H, et al. Increased expression of co-chaperone HOP with HSP90 and HSC70 and complex formation in human colonic carcinoma. Cell Stress Chaperones. 2010;15:1003–1011. doi: 10.1007/s12192-010-0211-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Burgess EF, Ham AJ, Tabb DL, Billheimer D, Roth BJ, Chang SS, Cookson MS, Hinton TJ, Cheek KL, Hill S, et al. Prostate cancer serum biomarker discovery through proteomic analysis of alpha-2 macroglobulin protein complexes. Proteomics Clin Appl. 2008;2:1223. doi: 10.1002/prca.200780073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhong L, Peng X, Hidalgo GE, Doherty DE, Stromberg AJ, Hirschowitz EA. Antibodies to HSP70 and HSP90 in serum in non-small cell lung cancer patients. Cancer Detect Prev. 2003;27:285–290. doi: 10.1016/s0361-090x(03)00097-7. [DOI] [PubMed] [Google Scholar]
- 65.Li W, Sahu D, Tsen F. Secreted heat shock protein-90 (Hsp90) in wound healing and cancer. Biochim Biophys Acta. 2012;1823:730–741. doi: 10.1016/j.bbamcr.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Samant RS, Clarke PA, Workman P. The expanding proteome of the molecular chaperone HSP90. Cell Cycle. 2012;11:1301–1308. doi: 10.4161/cc.19722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Prince T, Neckers L. A network of its own: the unique interactome of the Hsp90 cochaperone, Sba1/p23. Mol Cell. 2011;43:159–160. doi: 10.1016/j.molcel.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Echeverria PC, Bernthaler A, Dupuis P, Mayer B, Picard D. An interaction network predicted from public data as a discovery tool: application to the Hsp90 molecular chaperone machine. PLoS ONE. 2011;6:e26044. doi: 10.1371/journal.pone.0026044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sidera K, Patsavoudi E. Extracellular HSP90: conquering the cell surface. Cell Cycle. 2008;7:1564–1568. doi: 10.4161/cc.7.11.6054. [DOI] [PubMed] [Google Scholar]
- 70.Eustace BK, Sakurai T, Stewart JK, Yimlamai D, Unger C, Zehetmeier C, Lain B, Torella C, Henning SW, Beste G, et al. Functional proteomic screens reveal an essential extracellular role for hsp90 alpha in cancer cell invasiveness. Nat Cell Biol. 2004;6:507–514. doi: 10.1038/ncb1131. [DOI] [PubMed] [Google Scholar]
- 71.Tsutsumi S, Neckers L. Extracellular heat shock protein 90: a role for a molecular chaperone in cell motility and cancer metastasis. Cancer Sci. 2007;98:1536–1539. doi: 10.1111/j.1349-7006.2007.00561.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cheng CF, Fan J, Fedesco M, Guan S, Li Y, Bandyopadhyay B, Bright AM, Yerushalmi D, Liang M, Chen M, et al. Transforming growth factor alpha (TGFalpha)-stimulated secretion of HSP90alpha: using the receptor LRP-1/CD91 to promote human skin cell migration against a TGFbeta-rich environment during wound healing. Mol Cell Biol. 2008;28:3344–3358. doi: 10.1128/MCB.01287-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li W, Li Y, Guan S, Fan J, Cheng CF, Bright AM, Chinn C, Chen M, Woodley DT. Extracellular heat shock protein-90alpha: linking hypoxia to skin cell motility and wound healing. EMBO J. 2007;26:1221–1233. doi: 10.1038/sj.emboj.7601579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang Y, Rao R, Shen J, Tang Y, Fiskus W, Nechtman J, Atadja P, Bhalla K. Role of acetylation and extracellular location of heat shock protein 90alpha in tumor cell invasion. Cancer Res. 2008;68:4833–4842. doi: 10.1158/0008-5472.CAN-08-0644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Quanz M, Herbette A, Sayarath M, de Koning L, Dubois T, Sun JS, Dutreix M. Heat shock protein 90alpha (Hsp90alpha) is phosphorylated in response to DNA damage and accumulates in repair foci. J Biol Chem. 2012;287:8803–8815. doi: 10.1074/jbc.M111.320887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ullrich SJ, Robinson EA, Law LW, Willingham M, Appella E. A mouse tumor-specific transplantation antigen is a heat shock-related protein. Proc Nat Acad Sci USA. 1986;83:3121–3125. doi: 10.1073/pnas.83.10.3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sidera K, Samiotaki M, Yfanti E, Panayotou G, Patsavoudi E. Involvement of cell surface HSP90 in cell migration reveals a novel role in the developing nervous system. J Biol Chem. 2004;279:45379–45388. doi: 10.1074/jbc.M405486200. [DOI] [PubMed] [Google Scholar]
- 78.Saito K, Dai Y, Ohtsuka K. Enhanced expression of heat shock proteins in gradually dying cells and their release from necrotically dead cells. Exp Cell Res. 2005;310:229–236. doi: 10.1016/j.yexcr.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 79.Yerbury JJ, Stewart EM, Wyatt AR, Wilson MR. Quality control of protein folding in extracellular space. EMBO Rep. 2005;6:1131–1136. doi: 10.1038/sj.embor.7400586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sims JD, McCready J, Jay DG. Extracellular heat shock protein (Hsp)70 and Hsp90alpha assist in matrix metalloproteinase-2 activation and breast cancer cell migration and invasion. PLoS ONE. 2011;6:e18848. doi: 10.1371/journal.pone.0018848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nickel W. The mystery of nonclassical protein secretion. A current view on cargo proteins and potential export routes. Eur J Biochem/FEBS. 2003;270:2109–2119. doi: 10.1046/j.1432-1033.2003.03577.x. [DOI] [PubMed] [Google Scholar]
- 82.McCready J, Sims JD, Chan D, Jay DG. Secretion of extracellular hsp90alpha via exosomes increases cancer cell motility: a role for plasminogen activation. BMC Cancer. 2010;10:294. doi: 10.1186/1471-2407-10-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stellas D, El Hamidieh A, Patsavoudi E. Monoclonal antibody 4C5 prevents activation of MMP2 and MMP9 by disrupting their interaction with extracellular HSP90 and inhibits formation of metastatic breast cancer cell deposits. BMC Cell Biol. 2010;11:51. doi: 10.1186/1471-2121-11-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang X, Song X, Zhuo W, Fu Y, Shi H, Liang Y, Tong M, Chang G, Luo Y. The regulatory mechanism of Hsp90alpha secretion and its function in tumor malignancy. Proc Natl Acad Sci USA. 2009;106:21288–21293. doi: 10.1073/pnas.0908151106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dowling P, Walsh N, Clynes M. Membrane and membrane-associated proteins involved in the aggressive phenotype displayed by highly invasive cancer cells. Proteomics. 2008;8:4054–4065. doi: 10.1002/pmic.200800098. [DOI] [PubMed] [Google Scholar]
- 86.Taiyab A, Rao ChM. HSP90 modulates actin dynamics: inhibition of HSP90 leads to decreased cell motility and impairs invasion. Biochim Biophys Acta. 2011;1813:213–221. doi: 10.1016/j.bbamcr.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 87.Wong K, Koczywas M, Goldman JW, Paschold EH, Horn L, Lufkin JM, Blackman RK, Teofilovici F, Shapiro G, Socinski MA. An open-label phase II study of the Hsp90 inhibitor ganetespib (STA-9090) as monotherapy in patients with advanced non-small cell lung cancer (NSCLC) J Clin Oncol. 2011;29(suppl 7500) [Google Scholar]
- 88.Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 89.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Garraway LA, Janne PA. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2012;2:214–226. doi: 10.1158/2159-8290.CD-12-0012. [DOI] [PubMed] [Google Scholar]
- 92.Shimamura T, Shapiro GI. Heat shock protein 90 inhibition in lung cancer. J Thoracic Oncol. 2008;3:S152–S159. doi: 10.1097/JTO.0b013e318174ea3a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Holmes JL, Sharp SY, Hobbs S, Workman P. Silencing of HSP90 cochaperone AHA1 expression decreases client protein activation and increases cellular sensitivity to the HSP90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Cancer Res. 2008;68:1188–1197. doi: 10.1158/0008-5472.CAN-07-3268. [DOI] [PubMed] [Google Scholar]
- 94.Gray PJ, Jr, Stevenson MA, Calderwood SK. Targeting Cdc37 inhibits multiple signaling pathways and induces growth arrest in prostate cancer cells. Cancer Res. 2007;67:11942–11950. doi: 10.1158/0008-5472.CAN-07-3162. [DOI] [PubMed] [Google Scholar]
- 95.McDowell CL, Bryan Sutton R, Obermann WM. Expression of Hsp90 chaperone [corrected] proteins in human tumor tissue. Int J Biol Macromol. 2009;45:310–314. doi: 10.1016/j.ijbiomac.2009.06.012. [DOI] [PubMed] [Google Scholar]
- 96.Au Q, Zhang Y, Barber JR, Ng SC, Zhang B. Identification of inhibitors of HSF1 functional activity by high-content target-based screening. J Biomol Screening. 2009;14:1165–1175. doi: 10.1177/1087057109347472. [DOI] [PubMed] [Google Scholar]
- 97.Dai C, Santagata S, Tang Z, Shi J, Cao J, Kwon H, Bronson RT, Whitesell L, Lindquist S. Loss of tumor suppressor NF1 activates HSF1 to promote carcinogenesis. J Clin Invest. 2012;122:3742–3754. doi: 10.1172/JCI62727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schmitt E, Maingret L, Puig PE, Rerole AL, Ghiringhelli F, Hammann A, Solary E, Kroemer G, Garrido C. Heat shock protein 70 neutralization exerts potent antitumor effects in animal models of colon cancer and melanoma. Cancer Res. 2006;66:4191–4197. doi: 10.1158/0008-5472.CAN-05-3778. [DOI] [PubMed] [Google Scholar]
- 99.Carbone DL, Doorn JA, Kiebler Z, Ickes BR, Petersen DR. Modification of heat shock protein 90 by 4-hydroxynonenal in a rat model of chronic alcoholic liver disease. J Pharmacol Experiment Therapeutics. 2005;315:8–15. doi: 10.1124/jpet.105.088088. [DOI] [PubMed] [Google Scholar]
- 100.Scroggins BT, Robzyk K, Wang D, Marcu MG, Tsutsumi S, Beebe K, Cotter RJ, Felts S, Toft D, Karnitz L, et al. An acetylation site in the middle domain of Hsp90 regulates chaperone function. Mol Cell. 2007;25:151–159. doi: 10.1016/j.molcel.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB, Yao TP. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18:601–607. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 102.Murphy PJ, Morishima Y, Kovacs JJ, Yao TP, Pratt WB. Regulation of the dynamics of hsp90 action on the glucocorticoid receptor by acetylation/deacetylation of the chaperone. J Biol Chem. 2005;280:33792–33799. doi: 10.1074/jbc.M506997200. [DOI] [PubMed] [Google Scholar]
- 103.Bali P, Pranpat M, Bradner J, Balasis M, Fiskus W, Guo F, Rocha K, Kumaraswamy S, Boyapalle S, Atadja P, et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem. 2005;280:26729–26734. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- 104.Kekatpure VD, Dannenberg AJ, Subbaramaiah K. HDAC6 modulates Hsp90 chaperone activity and regulates activation of aryl hydrocarbon receptor signaling. J Biol Chem. 2009;284:7436–7445. doi: 10.1074/jbc.M808999200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 105.Wandinger SK, Suhre MH, Wegele H, Buchner J. The phosphatase Ppt1 is a dedicated regulator of the molecular chaperone Hsp90. EMBO J. 2006;25:367–376. doi: 10.1038/sj.emboj.7600930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Duval M, Le Boeuf F, Huot J, Gratton JP. Src-mediated phosphorylation of Hsp90 in response to vascular endothelial growth factor (VEGF) is required for VEGF receptor-2 signaling to endothelial NO synthase. Mol Biol Cell. 2007;18:4659–4668. doi: 10.1091/mbc.E07-05-0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kurokawa M, Zhao C, Reya T, Kornbluth S. Inhibition of apoptosome formation by suppression of Hsp90beta phosphorylation in tyrosine kinase-induced leukemias. Mol Cell Biol. 2008;28:5494–5506. doi: 10.1128/MCB.00265-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lees-Miller SP, Anderson CW. Two human 90-kDa heat shock proteins are phosphorylated in vivo at conserved serines that are phosphorylated in vitro by casein kinase II. J Biol Chem. 1989;264:2431–2437. [PubMed] [Google Scholar]
- 109.Mollapour M, Tsutsumi S, Neckers L. Hsp90 phosphorylation, Wee1 and the cell cycle. Cell Cycle. 2010;9:2310–2316. doi: 10.4161/cc.9.12.12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mollapour M, Tsutsumi S, Donnelly AC, Beebe K, Tokita MJ, Lee MJ, Lee S, Morra G, Bourboulia D, Scroggins BT, et al. Swe1Wee1-dependent tyrosine phosphorylation of Hsp90 regulates distinct facets of chaperone function. Mol Cell. 2010;37:333–343. doi: 10.1016/j.molcel.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mollapour M, Tsutsumi S, Truman AW, Xu W, Vaughan CK, Beebe K, Konstantinova A, Vourganti S, Panaretou B, Piper PW, et al. Threonine 22 phosphorylation attenuates Hsp90 interaction with cochaperones and affects its chaperone activity. Mol Cell. 2011;41:672–681. doi: 10.1016/j.molcel.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mollapour M, Tsutsumi S, Kim YS, Trepel J, Neckers L. Casein kinase 2 phosphorylation of Hsp90 threonine 22 modulates chaperone function and drug sensitivity. Oncotarget. 2011;2:407–417. doi: 10.18632/oncotarget.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Xu W, Mollapour M, Prodromou C, Wang S, Scroggins BT, Palchick Z, Beebe K, Siderius M, Lee MJ, Couvillon A, et al. Dynamic tyrosine phosphorylation modulates cycling of the HSP90-P50 (CDC37)-AHA1 chaperone machine. Mol Cell. 2012;47:434–443. doi: 10.1016/j.molcel.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lees-Miller SP, Anderson CW. The human double-stranded DNA-activated protein kinase phosphorylates the 90-kDa heat-shock protein, hsp90 alpha at two NH2-terminal threonine residues. J Biol Chem. 1989;264:17275–17280. [PubMed] [Google Scholar]
- 115.Adinolfi E, Kim M, Young MT, Di Virgilio F, Surprenant A. Tyrosine phosphorylation of HSP90 within the P2X7 receptor complex negatively regulates P2X7 receptors. J Biol Chem. 2003;278:37344–37351. doi: 10.1074/jbc.M301508200. [DOI] [PubMed] [Google Scholar]
- 116.Old WM, Shabb JB, Houel S, Wang H, Couts KL, Yen CY, Litman ES, Croy CH, Meyer-Arendt K, Miranda JG, et al. Functional proteomics identifies targets of phosphorylation by B-Raf signaling in melanoma. Mol Cell. 2009;34:115–131. doi: 10.1016/j.molcel.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Huang SY, Tsai ML, Chen GY, Wu CJ, Chen SH. A systematic MS-based approach for identifying in vitro substrates of PKA and PKG in rat uteri. J Proteome Res. 2007;6:2674–2684. doi: 10.1021/pr070134c. [DOI] [PubMed] [Google Scholar]
- 118.Lei H, Venkatakrishnan A, Yu S, Kazlauskas A. Protein kinase A-dependent translocation of Hsp90 alpha impairs endothelial nitric-oxide synthase activity in high glucose and diabetes. J Biol Chem. 2007;282:9364–9371. doi: 10.1074/jbc.M608985200. [DOI] [PubMed] [Google Scholar]
- 119.Martinez-Ruiz A, Villanueva L, Gonzalez de Orduna C, Lopez-Ferrer D, Higueras MA, Tarin C, Rodriguez-Crespo I, Vazquez J, Lamas S. S-nitrosylation of Hsp90 promotes the inhibition of its ATPase and endothelial nitric oxide synthase regulatory activities. Proc Nat Acad Sci USA. 2005;102:8525–8530. doi: 10.1073/pnas.0407294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Retzlaff M, Stahl M, Eberl HC, Lagleder S, Beck J, Kessler H, Buchner J. Hsp90 is regulated by a switch point in the C-terminal domain. EMBO Rep. 2009;10:1147–1153. doi: 10.1038/embor.2009.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Blank M, Mandel M, Keisari Y, Meruelo D, Lavie G. Enhanced ubiquitinylation of heat shock protein 90 as a potential mechanism for mitotic cell death in cancer cells induced with hypericin. Cancer Res. 2003;63:8241–8247. [PubMed] [Google Scholar]