

Abstract
Summary
Hsp90 inhibitors have demonstrated unusual selectivity for tumor cells despite its ubiquitous expression. This phenomenon has remained unexplained but could be influenced by ectopically expressed Hsp90 in tumors. We have synthesized novel Hsp90 inhibitors that can carry optical or radioiodinated probes via a PEG tether. We show that these tethered inhibitors selectively recognize cells expressing ectopic Hsp90 and become internalized. The internalization process is blocked by Hsp90 antibodies, suggesting that active cycling of the protein is occurring at the plasma membrane. In mice, we show exquisite accumulation of the fluor-tethered versions within breast tumors at very sensitive levels. Cell-based assays with the radiolabeled version showed picomolar detection in cells that express ectopic Hsp90. Our findings show that fluor-tethered or radiolabeled inhibitors targeting ectopic Hsp90 can be used to detect breast cancer malignancies through non-invasive imaging.
Introduction
The current paradigm for detection and treatment of breast cancer is based on clinical evaluation and anatomic imaging, usually with mammography or less commonly breast magnetic resonance imaging (MRI), followed by biopsy and surgery or surgery plus radiotherapy. Other imaging modalities, such as ultrasound or position emission tomography (PET), are not routinely used for screening although they have specific indications and potential (Smith et al., 2010). While both mammography and MRI demonstrate excellent sensitivity for detecting tissue abnormalities, they lack sufficient specificity for unequivocally distinguishing malignant tissue from benign tissue (Esserman et al., 2009). The question remains as to whether pre-malignant molecular markers can be used non-invasively to detect aggressive cancers.
It is clear that anatomic changes are not the earliest cancer-related transformations. Instead, breast cells with malignant and lethal potential are characterized early on by activated oncogenic signaling nodes. These signaling nodes have been classified into a broad set of characteristics termed the “Hallmarks of Cancer” and are candidate molecular markers of malignant behavior(Hanahan and Weinberg, 2011). Unfortunately, these signaling nodes have been difficult to detect in vivo, particularly when confined to small clusters of cells, as in early stage disease. To date, strategies to visualize these signals in vivo, such as using 18FDG-PET to detect increased glucose uptake, have not achieved the sensitivity or specificity required to appreciably improve breast cancer screening and diagnosis(Warning et al., 2011).
Heat shock protein 90 (Hsp90) is a signaling node that could be exploited as a diagnostic molecular marker to distinguish malignant breast cells from normal tissues(Barrott and Haystead, 2013). Hsp90 has an essential role in cellular homeostasis by chaperoning client proteins. Over 400 putative Hsp90 clients have been identified and many of these regulate signaling pathways governing cellular growth and differentiation(Echeverria et al., 2011; Moulick et al., 2011; Neckers et al., 2009; Samant et al., 2012; Vaughan et al., 2010). Hsp90 and its family members, Grp94 and Trap-1, contain an N-terminal ATP-binding domain with ATPase activity that is necessary for cellular function (Tsutsumi et al., 2009). Hsp90 is regulated both translationally and post-translationally, the latter affecting both ATPase activity and intracellular location(Mollapour and Neckers, 2012). Direct evidence for Hsp90’s participation in oncogenic protein folding/stability in vivo comes from studies with Hsp90 inhibitors that bind competitively to its ATP-binding domain resulting in the degradation of its oncogenic clients(Chiosis et al., 2003; Csermely, 1998; Fadden et al., 2010). This phenomenon has also been demonstrated in human tumor biopsies from patients undergoing Hsp90 inhibitor therapy (Kim et al., 2009). To date, there are 17 different Hsp90 inhibitors targeting its ATP-binding site in clinical development for multiple indications in cancer(Kim et al., 2009; Neckers and Workman, 2012; Trepel et al., 2010; Wang et al., 2010).
Recent studies have linked high expression of Hsp90 with poor prognosis in malignant breast tumors (Cheng et al., 2012; Pick et al., 2007). The role of Hsp90 in mediating malignant behavior may be the result of oncogene driven factors that alter its normal cellular behavior(Whitesell and Lindquist, 2005). Hyperactivation is postulated to result in an increased affinity for ATP and Hsp90 inhibitors and the expression of ectopic Hsp90 (Tsutsumi and Neckers, 2007; Tsutsumi et al., 2008). If oncogenically activated Hsp90 precedes malignant behavior in vivo, we reasoned that this could be used diagnostically (Eustace et al., 2004; McCready et al., 2010; Sims et al., 2011). We therefore developed a series of Hsp90 inhibitors tethered to fluorophores or radioiodine to detect Hsp90 in vivo. When injected into mice bearing human breast tumors, the fluorophore versions are exquisitely targeted to tumors. We show that this targeting is achieved through interactions with ectopic Hsp90, which is undergoing active internalization along with the bound probes. This finding suggests new roles for Hsp90 in which the protein is not only trafficked to the plasma membrane but also reinternalized.
RESULTS
Synthesis and Development of Probes Selectively Targeting Ectopic Hsp90
We recently reported the development of a cleavable tethered Hsp90 inhibitor and demonstrated its use as an affinity resin(Hughes et al., 2012). When bound to the tethered ligand, Hsp90 could be recovered along with one of its established oncogenic clients, Her2, in a competitive manner. To extend our tethered ligands utility, we synthesized several versions tethered to a variety of fluorophores and other molecules to facilitate the detection of Hsp90 in vivo (Figure 1A, S1 and Table 1). In binding studies against immobilized ATP, the tethered inhibitors showed reduced affinity for native Hsp90 (Kd HS-27, 288 nM; HS-69, 49 nM; HS-70, 42 nM) in comparison to the parent compound (HS-10, 3 nM) (Table 1 and Figure S2A) (Fadden et al., 2010; Grenert et al., 1997). Despite some reduction in affinity, the addition of the tethered components was found to increase specificity by eliminating binding to Grp94 (Figure S2B). Previous work had also shown that the addition of the tether at the ortho-position of the parent ligand reduced the specificity towards recombinant and native Trap-1 (Hughes et al., 2012). These findings suggest that the added steric bulk due to the presence of the tether and added fluor, for example, interferes with the ATP-binding site of Grp94 and Trap-1, but not Hsp90. Similar specificity was also observed by adding various non-fluorescent molecules to the tethers, such as ferrocene, iodinated benzylamine or an additional Hsp90 inhibitor, to create a bifunctional inhibitor (Figure S2B, Table 1).
Table 1. Hsp90 and Her2 Tethered inhibitors.
Names, descriptive features and structures for compounds used in the study. The synthesis and detailed analysis of the compounds can be found in Figure S1.
| Designation name |
Feature | Structure |
|---|---|---|
| HS-10 | parent ligand |
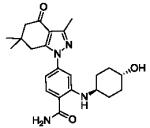
|
| HS-23 | ligand + linker |

|
| HS-27 | ligand + FITC |
|
| HS-32 | bis-ferrocene |

|
| HS-42 | lapatinib + fluorescein |
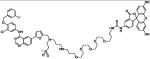
|
| HS-66 | double ligand |
|
| HS-69 | ligand + nIR (645) |
|
| HS-70 | ligand + nIR (800) |
|
| HS-96 | ligand + biotin |
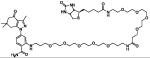
|
| HS-105 | FITC derivative w/o ligand |
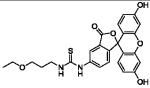
|
| HS-111 | Iodine- containing ligand |
|
Next, we evaluated the specificity of the fluor-probes in several transformed cell lines by fluorescent microscopy. Figure 1b shows that HS-27 is internalized by breast cancer cell lines, but remarkably not by Huh7 cells, a hepatocarcinoma cell line, despite the latter cell line having higher total cellular levels of Hsp90 as determined by immunoblotting (Figure S2C). Time course and titration studies showed HS-27 uptake is variable between breast lines in the following order MCF10<<< MCF7<MDA-MB-468< BT474 (Figure 1C,D). Interestingly, this uptake order correlates with the relative tumorigenicity of the cells to form tumors in SCID mice (Neve et al., 2006). Analysis of Her2 levels in BT474 cells show that, once internalized, HS27 is active as an Hsp90 inhibitor and uptake correlates with Her2 degradation (Figure 1E, S2D). Competition experiments with the untethered ligand, HS-10 (Figure S2E), and comparisons with a control compound FITC-tethered lapatinib, HS-42, demonstrate that HS-27 uptake and selectivity for the breast cells is Hsp90-dependent. In the case of HS-42, despite sharing a common fluorophore with HS-27, HS-42 was rapidly absorbed with identical kinetics in all cells tested, including Huh7 cells (Figure S3A-C). These findings show that the uptake of the two FITC-tethered inhibitors involve different mechanisms that are ligand-dependent. As for HS-42, uptake may reflect binding to EGFR, whereas for HS-27, the apparent mechanism is binding to ectopic Hsp90. This conclusion is consistent with others who have linked extracellular Hsp90 with the metastatic potential of various tumor lines, including breast cancer cells (Koga et al., 2009; Tsutsumi and Neckers, 2007; Tsutsumi et al., 2008).
Figure 1. Fluor-tethered Hsp90 inhibitors label breast cancer cell lines.
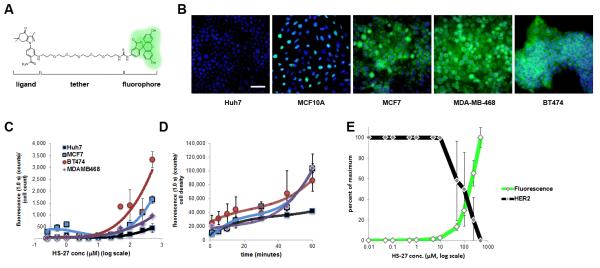
(A) HS-27 structure given to illustrate the generic inhibitor design of the Hsp90 ligand conjugated to a linker series and tethered to a fluorophore. (B) HS-27, at 100 μM, labels native Hsp90 in breast cancer cell lines and correlates to the established malignancy level of the cell lines. HS-27 does not label Huh7 cells under normal cell culture conditions. (C) In a dose titration curve of HS-27, cell lines become stratified at higher doses despite having approximately equivalent levels of total HSP90, (n = 3, ±SEM). (D) A time course study showing the kinetics of HS-27 (100 μM) uptake mirror the dose assay in terms of selectivity against breast cancer cell lines and the Huh7 cell line (n = 2, ±SDM). (E) HS-27 on BT474 cells shows an inverse correlation between fluorescent uptake (green) and Her2 degradation (black). Scale bar, 50 μm. See also Figure S2 and S3.
Additional data supporting that the tethered Hsp90 inhibitors bind to ectopically expressed Hsp90 in breast cell lines came from observations with the permeabilizing agents, β-escin and Triton X-100. In the presence of β-escin, HS-27 enters all cells tested. To test whether HS-27 was non-specifically labeling permeabilized Huh7 cells, we competed HS-27 binding with HS-10, which blocked binding in both cell lines (Figure 2A). These data suggest that Huh7 cells do not express ectopic Hsp90, but they do contain an internal pool of Hsp90 that binds the inhibitor. We also observed other cell lines that were unable to internalize HS-27 without permeabilization, including lymphoma cells purified from patients with CLL, human peripheral blood mononuclear cells or cultured fibroblasts.
Figure 2. HS-27 labels surface Hsp90 in breast cancer cell lines.

(A) HS-27 labels all the cancer lines tested upon permeabilization with 5 μM β-escin. Live cell imaging demonstrates that the fluorescent signal is competed with a pretreatment of 10-fold excess of a non-tethered Hsp90 inhibitor, HS-10. (B) Fixed breast cancer cell lines are treated with or without a detergent (0.3% Triton X-100) and stained by immunofluorescence with an anti-Hsp90 antibody. Cells treated with Triton have intracellular pools that are labeled, while non-permeabilized cells exhibit exclusive labeling of surface Hsp90. (C) Antibody treatment prevents active internalization of Hsp90 and HS-27 in a dose-dependent manner. The first row is the incubation with HS-27 alone and denotes a single field observed with different filters. Rows 2 and 3 are separate fields of cells incubated with the antibody and HS-27. Row 2 demonstrates peripheral staining of Hsp90 (yellow arrow) by both the antibody (red) and HS-27 (green). Row 3 shows cells that are not labeled with the antibody and have internalized HS-27 (white arrows). (D) Diagram of an Hsp90 dimer and the distant labeling of the anti-Hsp90 antibody and HS-27. (E) The ratio of surface HS-27 and internalized HS-27 labeled cells plotted against the Hsp90 antibody titration; R2 = 0.9903. (F) Huh7 cells and Huh7-MCF7 co-cultured cells treated with 100 μM HS-27. White scale bars, 50 μm; yellow scale bar, 20 μm; black scale bar, 10 μm. See also Figure S4.
Because of its polar nature, HS-27 would not be predicted to enter cells through passive diffusion and the competition studies with HS-10 strongly argue that its internalization requires binding to Hsp90 expressed at the surface. Consistent with this hypothesis, anti-Hsp90 antibodies were found to selectively stain the surface of the more tumorigenic MCF7 cells compared with non-tumorigenic MCF-10A cells (Figure 2B). Additionally, when live MCF7 cells were incubated with anti-Hsp90 antibodies in the presence of HS-27, the probe is retained at the plasma membrane and no longer internalized as shown by co-staining at the surface with the Hsp90 antibody (Figure 2C). Detailed examination of the stained fields, show cells not co-labeled with the antibody at the cell surface continue internalizing HS-27 (Figure 2C). Importantly, the Hsp90 antibody used in this experiment targets the C-terminal domain of the protein and does not interfere with the HS-27 binding at the N-terminal ATP-binding domain (Figure 2D). In antibody titration experiments, the retention of HS-27 at the surface correlates precisely with antibody concentration. At 10 μg/well of antibody, the number of cells that retained HS-27 at the surface to those cells that internalized HS-27 was almost 1:1, whereas the ratio was 1:4 at an antibody treatment of 2.5 μg/well (Figure 2E). Collectively our data highlight an unrecognized pathway in which Hsp90 is not only trafficked to the membrane, but actively internalized. The internalization is likely not attributed to general pinocytosis that results in fusion with the lysosomes. Lysosomes have internal pHs of <6, and HS-27 consists of an Hsp90 inhibitor tethered to FITC which loses its fluorescent properties below pH 6.5 (Figure S4A). Additionally, we failed to detect the co-localization of HS-27 with Rab5, a marker of early endosomes, by fluorescent microscopy or centrifugal enrichment of endosomes. We posit that the internalization mechanism of HS-27 is Rab5-independent.
As aforementioned, Hsp90 is thought to be constitutively secreted only from tumor cells (Cheng et al., 2008; McCready et al., 2010; Wang et al., 2009). To detect the presence of secreted Hsp90, we added HS-27 to media derived from BTB474, MCF7 and Huh7 cells, and removed the free probe by ultrafiltration. BT474 and MCF7 exhibited substantial recovery of fluorescence compared to Huh7, which is consistent with the former cells actively secreting Hsp90 (Figure S4B). To examine whether extracellular Hsp90 was sufficient to facilitate HS-27 uptake into Huh7 cells, MCF7 cells were co-cultured with Huh7 cells and incubated with HS-27. Figure 2F shows that the presence of locally secreted Hsp90 from MCF7 cells is not sufficient to promote subsequent reuptake into the Huh7 cells. This suggests that Huh7 cells lack the machinery necessary for the active internalization of HS-27 bound to Hsp90.
Fluor-tethered Hsp90 Inhibitors are Selective for Active Hsp90 in vitro
Our probes can discriminate between various cell lines, but recent data suggest that there are distinct populations of active and inactive Hsp90 within a given cell (Kamal and Burrows, 2003; Moulick et al., 2011). To test if our probes also discriminate between these cellular pools in vivo, we first isolated these pools using affinity chromatography. Briefly, cell extracts from BT474 cells were repetitively passed over virgin Hsp90 affinity resins. The “active pool” binds and the “non-active pool” flows through the resin (Figure 3A,B) (Hughes et al., 2012). Once separated, Hsp90 from the resin-bound pool demonstrated HS-27 binding that was 15.5-fold higher than inactive Hsp90 in the flow through. To further characterize the HS-27 bound active pool of Hsp90, we separated either purified Hsp90 (from lactating pig mammary gland) or BT474 cell extracts incubated with HS-27 by micro-anion-exchange chromatography. In the extracts from the BT474 cells, we expected to see multiple column fractions with fluorescence in the breast cancer cell extract, consistent with multiple interactions of active Hsp90 with its respective client proteins. However, and in all cases, only a single peak of fluorescence was observed, which correlated precisely with the migration of purified pig mammary Hsp90 bound to HS-27 (Figure 3C). Furthermore, we competed the binding of HS-27 with three structurally distinct Hsp90 inhibitors: HS-10, PUH71 and 17-AAG. MCF7 cells were incubated with the non-fluorescein Hsp90 inhibitors, followed by HS-27. Equal competition was observed with the three Hsp90 inhibitors compared to the DMSO control treatment (Figure 3D). The presence of Hsp90 in the single peak was confirmed after column fractions containing fluorescence were passed over the cleavable Hsp90 affinity resin followed by SDS-PAGE, silver staining and mass spectrometry (Figure 3E). Collectively, these biochemical data strongly argue that the only intracellular target for HS-27 is an active pool of Hsp90 that is largely devoid of client proteins. These data are consistent with previous work by our laboratory using Hsp90 small molecule affinity resins showing that the ligand bound form is not associated in vivo with multiple clients as previously thought (Hughes et al., 2012).
Figure 3. HS-27 binds to the active form of Hsp90 in breast cancer cell lines and normal mouse tissues.
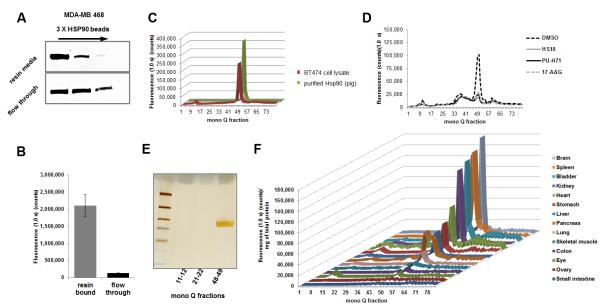
(A) Cell extracts from MDA-MB-468 were passed over immobilized Hsp90 ligand beads three times and the resin media and flow through were analyzed for total Hsp90 by immunoblotting. Hsp90 continued to be detected in the flow through, even after the depletion of the active form on the resin. (B) The resin bound extract and flow through were incubated with HS-27 and unbound probe was filtered away. Fluorescence was measured on a multi label plate reader (n = 3, ±SEM student t-test P value < 0.005). (C) BT474 cell extracts treated with HS-27 (100 μM) was compared to purified Hsp90 from a pig lactating mammary gland with HS-27 and separated on a mono Q anion exchange column. Single fluorescent peaks were consistently observed peaking in the 49th fraction. (D) MCF7 cells were pretreated with 10-fold excess of non-fluorescein Hsp90 inhibitors followed by 10 μM HS-27. Cell lysates were harvested and separated on a mono Q anion exchange column and fluorescence from each fraction was measured. (E) BT474 cell fractions corresponding to the fluorescent peak (48-49) and control fractions (11-12 and 21-22) were passed over the cleavable Hsp90 affinity resin to purify the protein and characterize by gel electrophoresis. The Silver stained band was sequenced by MS which confirmed the presence of Hsp90. (F) Mouse tissues were harvested from BALB/C mice and lysed tissues were incubated with HS-27. After clearing away the unbound probe, samples were analyzed by anion exchange chromatography. Tissues were ordered from lowest expressing tissues to highest. See also Figure S5.
We next explored whether the probes could be used to measure acute activation of Hsp90 in cells in response to heat stress. We show that heat stress produces a consistent 1.2-fold increase in fluorescence eluting in the 49th fraction (Figure S5A,B). We then examined if the probe could be used to quantify the amount of activated Hsp90 distributed in normal tissues by adding HS-27 to homogenized mouse tissue extracts and then fractionating the tissue extracts chromatographically. We show that homogenized tissues contain diverse levels of active Hsp90 which also elute as a single peak (Figure 3F). The significance of these observations is that non-tumorigenic tissues contain an active pool of Hsp90, and in brain, spleen, bladder and kidney the levels were especially high. Irrespective of this finding, only intact cells expressing ectopic Hsp90 are capable of internalizing the fluor-tethered inhibitors. We suggest that malignant tumor cells express ectopic Hsp90 and that this pool of Hsp90 can be used to discriminate malignancies in vivo over normal tissues or more benign tumor cells. We also conclude that although the probe can reflect the tumorigenic state, the drug-bound version must have a low affinity for client proteins in stark contrast to the conclusions reported by others (Moulick et al., 2011).
Fluor-Tethered Hsp90 inhibitors Specifically Target Human Breast Tumors in Mice
To test if fluor-tethered Hsp90 inhibitors might be exploited to selectively visualize malignancies non-invasively, we injected the probes into mice bearing breast tumor derived xenografts. With MDA-MB-468 xenografts, we detected the tumor mass within 5 minutes post injection either with HS-27 or the near infrared (nIR) HS-69 and HS-70 versions in a IVIS Kinetic fluorescent imager (Perkin-Elmer, Inc.). In the case of HS-27, the tumor is clearly visible through the fur and discriminated from the natural background fluorescence normally observed at 520 nm (Figure 4A). With HS-27, an 8-fold increase was consistently observed over control tumors in mice that did not receive HS-27. With both nIR probes, we achieved a 150-fold increase at the tumor site due to the low background signal at 660 nm or 800 nm (Figure 4B-D, S6A). With nIR versions, post injection, the probe was observed in the extremities (i.e. ears, nose, and paws) and eyes reflecting the circulating unbound probe in the blood pool. This was not visible with the fluorescein versions because of the light scattering at 520 nm. Pharmacokinetic studies by various methods show dose dependent uptake of either the visible or nIR forms, peaking within the tumor mass by 30 minutes and with a detectable signal remaining for up to 72 hours. To test whether the probes were binding to Hsp90 in vivo, or just accumulating in the tumors because of a blood pooling effect, excised tumor lysates were fractionated chromatographically. As shown in figure 3 a single major peak of fluorescence was observed that contained Hsp90 (Figure 4E).
Figure 4. Fluor-tethered Hsp90 inhibitors target human breast tumors in mice.

(A) SCID mice bearing MDA-MB-468 xenografts in the right flank were injected with HS-27, HS-69 or saline and imaged using an IVIS kinetic imager. (B) HS-27 (green), (C) HS-69 (red), and HS-70 (dark red) were injected with increasing doses into mice and tumors excised (n = 2, ±SDM). (D) Examples of excised tumors from various treatments by IVIS. (E) Tumors from treated mice were excised, lysed and fractionated on an anion exchange column. (F,G) Fluorescence spectra measured from a normal site and the tumor at different times post-injection. Spectra are corrected for the effects of scattering and absorption from the tissue. Tumor shows signature FITC fluorescence at 6 hours and a reduced signal at 24 hours post injection which is also absent in the normal site. (H) Cryosections of biopsies taken from xenografts show the presence of the internalized probe within the tumor cells. (I) Flow cytometry of tumor cells, splenocytes and hepatocytes over the course of 72 hours demonstrates the selectivity of the probe for tumor cells over the spleen and liver (n = 3, ±SEM). (J) Varying tissues were excised from mice bearing tumors and injected with HS-27 and were analyzed for fluorescence by IVIS imaging (n = 2). See also Figure S6.
Pharmacokinetic studies over 96 hours by optical spectroscopy more elegantly confirmed selective uptake of the tethered inhibitors. In this in vivo approach, a spectral pen was placed either at the tumor site or an adjacent skin patch, and the fluorescence spectrum measured from 500-620 nm. Figure 4F and 4G show the signature spectrum of fluorescein at the tumor site and not at the adjacent skin sites over a period of 6-24 hours post injection. To rule out the possibility that the fluorescence measured in the tumor mass by IVIS imaging or the optical method was due to local blood pooling, we analyzed biopsied tissue sections by fluorescent microscopy (Figure 4H). Figure 4h shows discrete uptake of HS-27 within tumor cells. As a test of tumor selectivity in vivo, we simultaneously harvested tumor cells, splenocytes and hepatocytes from SCID mice bearing MDA-MB-468 tumors over the course of 72 hours and analyzed the viable cells by flow cytometry for the presence of HS-27. Figure 4I shows specific uptake of the probe by tumor cells, while splenocytes and hepatocytes did not. Selectivity of HS-27 for tumor cells was further illustrated in pharmacokinetic studies. Mice were injected with HS-27 and its distribution and tissue uptake were analyzed either by IVIS kinetic imaging or from fluorescent reading of tissue lysates. In all cases the probe was retained within the tumor with no evidence of accumulation elsewhere (Figure 4J).
To ensure that the in vivo probe accumulation within the tumor was ligand-dependent, a control compound HS-105 was synthesized. HS-105 consists of the fluorophore and tether, minus the ligand. In affinity chromatography studies against Hsp90 bound to immobilized ATP, HS-105, showed no affinity for the protein, and therefore any tumor retention would be non-specific (Figure S6B). Using IVIS kinetic imaging, HS-27 was detected through the skin in live animals at one hour, whereas HS-105 was below detection (Figure 5A). In more detailed necropsies, by 24 hours we found no trace of HS-105 by fluorescence whereas HS-27 was still present within the tumor (Figure 5B, S6B). Next we sought to quantify the amount of HS-27 accumulation in the tumor by comparing the average radiant efficiency in the tumor to a standard curve of HS-27 concentrations measured by the IVIS kinetic imager (Figure 5C). We calculated that in a cohort of 5 mice, the mean accumulation of HS-27 was 6.5 μM ±2.6 (SEM). To test the utility of fluor-tethered Hsp90 inhibitors as potential means of non-invasive early tumor detection, we designed an assay to test the sensitivity of the nIR version, HS-70, in mice. MDA-MB-468 cells were treated ex vivo with HS-70 or control. A fixed number of cells were injected in an equal volume to the right flank of SCID mice. We found that HS-70 could be detected at as little as 100,000 cells (Figure 5D,E). Current imaging approaches by MRI or PET/CT are estimated to reliably detect tumor masses at 1 cm3 with an estimated cell mass of ~10 million cells (Ide and Suzuki, 2005; Schoder and Gonen, 2007). By these criteria, 100,000 cells would suggest that nIR probes could theoretically detect masses as low as 0.01 cm3.
Figure 5. Tumor detection limits and specificity using optical or radioiodinated tethered Hsp90 inhibitors.

(A) One hour post injection IVIS kinetic images of mice bearing MDA-MB-468 tumors and injected with HS-27, HS-105 or control. (B) 24 hours post injection IVIS Kinetic images of excised tissues from SCID mice bearing MDA-MB-468 tumors and injected with HS-27, HS-105 or control. Liver (L) and lung (R) are in the top rows and the tumors are in the bottom row. (C) The IVIS average radiant efficiency plotted against the concentration of HS-27, mean ± SEM. (D,E) The IVIS average radiant efficiency of live mice injected with MDA-MB-468 cells treated ex vivo with HS-70 or control, mean ± SDM. (F-I) MCF7 and Huh7 cells treated with [125I]HS-111 and cell lysates fractioned on a mono Q anion exchange column. (F) MCF7 and Huh7 cells under normal conditions, (G) MCF7 cells treated with and without HS-10 (H) MCF7 and Huh7 cells with β-escin, (I) MCF7 and Huh7 cells with β-escin and HS-10. *P value = 0.0424. See also Figure S6.
Based upon our findings with the nIR probes, an obvious application for early malignancy detection would be surface tumors in which up regulation of Hsp90 has been indicated, such as head and neck, colorectal, bladder and melanoma(Wang et al., 2010; Yin et al., 2010). Because nIR probes are limited to 3-4 cm in tissue depth, to enable whole body imaging we investigated an alternative approach using tethered Hsp90 inhibitors capable of carrying the radioisotope 125I ([125I]HS-111). Picomolar amounts of [125I]HS-111were added to either MCF7, BT474 or Huh7 cells and as observed with the fluor versions, the breast cancer cells exhibited uptake above Huh7 cells (Figure S6C). Uptake of [125I]HS-111into MCF7 and Huh7 cells was then characterized after chromatographic fractionation and the probe was detected by its radioactivity. As observed with the fluor-tethered versions, in MCF7 cells, the majority of the radioactivity migrated as a single peak, in stark contrast to Huh7 cells which showed no peak recovery (Figure 5F). Importantly, the signal in MCF7 cells was effectively competed by the free ligand, HS-10 (Figure 5G). As with the fluor versions, β-escin permeabilization of Huh7 cells permitted the labeling of Hsp90 with [125I]HS-111in a competitive manner (Figure 5H,I).
Importantly, HS-111 behaves similarly to the fluor-tethered versions in terms of its selectivity towards the breast cancer cell lines and entry to these cells also clearly requires active internalization through binding to ectopic Hsp90. Interestingly, HS-111 is more potent than the fluor versions in Her2 knock down assays suggesting faster kinetics of internalization and performed most closely to HS-10, the parent untethered compound. This is likely due to the differences in added steric bulk of the attached imaging moieties. HS-111 consists of a benzalamide moiety of 121.1 Da, whereas HS-27 carries a bulky fluorescein moiety of 389.0 Da. Similarly, a lower rate of entry was observed with HS-96, a biotin-tethered Hsp90 inhibitor, which is considered to not passively diffuse across cell membranes (Figure S2D). These data strongly suggest that one can readily manipulate the entry rate of tethered inhibitors into cells expressing ectopic Hsp90 by changing the properties of the tethered imaging moiety. The finding that tethered inhibitors with structurally diverse imaging prosthetic groups only enter cells expressing ectopic Hsp90, and at variable rates, discredits the possibility that the entry of the tested probes occurs through simple diffusion.
Discussion
Our data have shown that an important frontline cancer target, Hsp90, can be exploited through its role in the oncogenic process as a diagnostic marker for real-time imaging of metastatic status. This observation represents somewhat of a paradigm shift in the way we currently view cutting edge therapeutic targets such as Hsp90, and the concept could be extended to other therapeutic targets. Tethered Hsp90 inhibitors as imaging agents potentially add an element of diagnostic detail that cannot be garnered from expression analysis or deep sequencing techniques. Specifically for Hsp90, these techniques do not measure the activation state of the protein or its localization, two important parameters linked to disease progression in breast cancer (Moulick et al., 2011; Tsutsumi and Neckers, 2007). The urgency to exploit this approach is made more relevant by the recent landmark studies of Gerlinger et al. who demonstrated the degree to which tumors exhibit phenotypic heterogeneity, even within the same tumor (Gerlinger et al., 2012). Because expression analysis and deep sequencing only represent a fraction of a tumor’s global heterogeneous expression pattern at a single time point, these findings signal the limited utility of these approaches to stratify and diagnose tumors. However, because tumor growth requires the continued activation of signal pathways, then perhaps the development of fluor-tethered and other imaging inhibitors targeting proteins like Hsp90, or constitutively activated oncogenes like Her2, can offer a new alternative strategy to more accurately stratify disease progression through real-time, non-invasive imaging.
If expression of ectopic Hsp90 signifies metastatic behavior in vivo, the fluor-or 125I-carrying-tethered Hsp90 inhibitors could be used in conjunction with current biopsy practice to diagnose aggressive tumors. This would simply involve histological examination to determine probe uptake following microdose administration prior to the biopsy procedure. Moreover, non-invasive whole body PET imaging could be employed using 124I-containing tethered versions. The PET based approach is attractive because one could simultaneously determine the fate and distribution of the tethered inhibitor in all tissues and in real-time. If PET analysis of a 124I-carrying-version showed highly selective targeting to the tumor in any one individual, one could proceed to a 131I version (which emits cytotoxic β-particles) with the goal of achieving complete body-wide tumor ablation without unwanted damage in normal tissues. The known μM accumulations of Hsp90 inhibitors (5-20 μM) within tumors and established protocols for the treatment of thyroid cancers with 131I should help accelerate the development of such treatment strategies(Fadden et al., 2010).
The importance of ectopically expressed Hsp90 to the metastatic process has been established in a variety of ways including using a functional proteomic screen (Eustace et al., 2004) and a non-cell permeable form of geldanamycin (DMAG-N-oxide) and anti-Hsp90 antibodies, both of which block cellular migration in metastatic tumor lines (Koga et al., 2009; Sidera et al., 2011; Stellas et al., 2010; Tsutsumi et al., 2008; Xu et al., 2005). The observation that fluor-tethered versions are selectively internalized by binding to ectopically expressed Hsp90 has revealed new roles for the protein in vivo. Although the expression of surface Hsp90 appears to be connected to cell migration and metastasis, the molecular mechanism by which ectopic Hsp90 signals to the tumor cell to promote these events is not known. Both migration to the surface and reinternalization of Hsp90 could involve low copy clients, which may provide a means of signaling that promotes cellular migration and metastasis. This conclusion is supported by anti-Hsp90 antibodies that blocked reinternalization of Hsp90 as reflected by inhibition of HS-27 uptake into MCF7 cells. Irrespective of ectopic Hsp90 roles in metastatic progression, the knowledge that the protein is reinternalized could be exploited to improve the safety margins of existing Hsp90 therapeutics or selectively deliver chemotoxic payloads to tumor cells. Compared to other cancer therapeutics, Hsp90 inhibitors are generally well tolerated by humans although some dose limiting side effects have been observed such as night blindness (Zhou D, 2012).
Significance
To many, the fact that inhibition of Hsp90 can be used therapeutically at all may seem paradoxical. After all, Hsp90 is thought to represent 1-3% of the expressed protein in most cells. This paradox is made even more extraordinary in the light of biochemical studies shown herein, with the finding that extracts from normal tissues contain considerable levels of active Hsp90. Various explanations have been offered in the past concerning the extraordinary selectivity of various Hsp90 inhibitors for tumor cells, including the idea that tumor cells express an oncogenically activated form of Hsp90 with a higher affinity for Hsp90 inhibitors (Kamal and Burrows, 2003). Our results suggest that the expression of ectopic Hsp90 may play a substantial role in the entry of Hsp90 inhibitors in general. This finding also suggests that in addition to tumor imaging, it may be possible to extend the therapeutic window of Hsp90 inhibitors by developing a range of molecules that do not passively diffuse across the plasma membrane, but can only enter cells expressing ectopic Hsp90.
Experimental Procedures
Studies with HSP90 antibodies
MCF7 and MCF10A cells were cultured and fixed in wells using 4% PFA/PBS. Successively, cells were incubated in blocking solution (5% goat serum, 0.2% Na Azide, PBS) with or without 0.3% Triton X-100 for 1 hour. After blocking with or without the detergent, cells were incubated with a polyclonal antibody for HSP90 at 1:100 (sc-7947, Santa Cruz Biotechnology, Santa Cruz, California). Cells were sequentially incubated with a goat-anti-rabbit Alexa Fluor 488-conjugated antibody at 1:1000 (A-11008, Life Technologies, Grand Island, New York). Cells were imaged using the Olympus IX 71 epifluorescence microscope. To evaluate ectopic expression of HSP90, HSP90 antibody was incubated with live MCF7cells (2.5 hours), then 100 nM of HS-27 was added. Concurrently, cells were stained with DAPI and goat-anti-rabbit Alexa Fluor 568 secondary antibody (A11011, Life Technologies, NY) at 1:1000 and incubated the cells for one hour at 37°C. Cells were washed in PBS and imaged using the Olympus IX 71 epifluorescence microscope. Ratios of cells that retained HS-27 at the cell periphery to cells that internalized HS-27 was calculated and plotted against the antibody mass.
Anion exchange chromatography
Samples from either cell or tissue lysates underwent buffer exchange using 30K filter devices. Lysis buffers were exchanged for 25 mM Tris-HCl and 1 mM DTT. Samples were centrifuged (13000 rpm, 10 minutes), then passed through a 0.2 μm syringe filter. Next the supernatants were loaded onto a Pharmacia mono Q anion-exchange SMART column (0.1 × 1.0 cm) that has been previously equilibrated in 25 mM Tris-HCl, 1 mM dithiothreitol buffer, pH 7.4 as described(Ray and Haystead, 2003). The column was eluted using a linear salt gradient over 80 minutes (100 μL/minute) to 1 M NaCl in the same buffer. Samples were fractionated into an opaque plate that was read for fluorescence on the multi label plate reader. Peak fractions of fluorescence were passed over a cleavable affinity resin to purify Hsp90 as described (Hughes et al., 2012).
IVIS Kinetic mouse imaging
All protocols using mice were approved beforehand by the IACUC at Duke University and strictly adhered to throughout the studies. SCID mice bearing MDA-MB-468 tumors were anesthetized with ketamine. Mice received tail vein injections or abdominal injections and imaged post injection at the indicated time using an IVIS Kinetic imager (Caliper Life Science, MA) as part of the Optical Molecular Imaging and Analysis shared resource in the Duke Cancer Institute. The following filters were used for the corresponding small molecule inhibitors (emission/excitation): HS-27 (468/GFP), HS-105 (468/GFP), HS-69 (640/Cy5.5), HS-70 (745/ICG).
Tissue Harvest and Analysis
The following mouse tissues were taken immediately post mortem: blood, brain, eyes, heart, kidney, liver, lung, spleen, and tumor. With the exception of the blood, the tissues were rinsed in PBS and blotted followed by image analysis using the IVIS kinetic imager. Tissues were stored on dry ice or at −80°C. For analysis of tissue lysates using SMART mono Q fractionation and multi label fluorescent reader, tissues were dounced in mono Q buffer (25 mM Tris-HCl, 1 mM DTT). We centrifuged the samples and transferred supernatants to be analyzed for fluorescence and protein concentration.
Fluorescence spectroscopy of drug uptake
SCID mice bearing MDA-MB-468 tumors were anesthetized with ketamine and received tail vein injections of drug. Using an optical spectroscopy instrument and a fiber optic probe the fluorescence spectrum of the FITC-conjugated drug was measured in vivo at 6, 24, 48, 72 and 96 hours post injection (Brown et al., 2009). With optical spectroscopy, tissue is illuminated using light of interest and the reflected light is analyzed to study the morphology and biochemical composition of the underlying tissue. The pen-shaped fiber optic probe has a diameter of approximately 2 mm and was placed in gentle contact with the tumor or adjacent normal site. Incident light at 490 nm corresponding to FITC excitation was delivered through the probe into tissue and resulting longer wavelength fluorescent light was collected. Although 490 nm does not correspond to maximal absorption by FITC, this wavelength was used to minimize fluorescent contributions from flavins (FAD) which fluoresces in the same wavelength range. Measured fluorescence spectra were corrected for tissue absorption and scattering using an intrinsic fluorescence recovery model described in the literature (Palmer and Ramanujam, 2008). Briefly, the model calculates the tissue optical properties that are a function of light scattering and absorption and uses these properties to correct distortions in the measured fluorescence. The intrinsic fluorescence model has been shown to accurately recover fluorophore concentrations in tissue-mimicking phantoms (Liu et al., 2012). In addition, the intrinsic fluorescence calculated using this model has been used to monitor intra-tumor drug concentrations in vivo and shown to be strongly correlated with concentrations measured using HPLC (Palmer et al., 2010). Because optical spectroscopy measures wavelength-dependent fluorescence, the background fluorescence can be accurately measured prior to injection. This allows monitoring of the fluorophore concentration in vivo and determination of when the fluorescent tether has cleared from the tumor.
Active Hsp90 depletion using affinity resin chromatography
MDA-MB-468 cell lysate was diluted to 1 mg/mL in low stringency wash buffer and 1 mL was added to 1 mL of the Hsp90 affinity resin at 50% slurry. For three consecutive washes fresh resin was used and 25 μL of flow through (< 3%) was set aside for flow through analysis. The resin was washed thoroughly with low stringency wash buffer and then Hsp90 was eluted off with 10% SDS. Both flow through and resin samples were characterized by 1D SDS-PAGE and silver staining. In a separate experiment, flow through and resin samples were incubated with 100 nM HS-27 and then washed using a 10K kDa filter. Samples were concentrated and analyzed on a multilabel plate reader for fluorescence.
Ex vivo cell treatment and injection into mice
MDA-MB-468 cells were treated with 5 μM β-escin for 5 minutes. Cells were harvested and treated with either 10 μM HS-70 or PBS. Cells were counted and aliquots of 10 million to 10 thousand cells were made in 200 μL of saline. SCID mice were anesthetized with ketamine and received two flank injections of the treated cells and the control cells. Mice were imaged using the IVIS kinetic imager and average radiant efficiency was measured.
125I-labeled Hsp90 inhibitor treatment of cells
Cells were permeabilized by treatment with 5 μM β-escin for 5 minutes. Competition experiments were performed by treatment of cells with 1 μM of HS-10 for 5 minutes prior to exposure to the [125I]HS-111. β-escin and HS-10 were washed away and then cells were incubated in 5 mL of medium with the [125I]HS-111 that had about 10 μCi of activity per 10 cm dish of cells for 45 minutes. Cells were washed extensively with PBS and then lysed on an ethanol/dry ice bath and harvested in 25 mM Tris buffer and centrifuged. Supernatants were fractionated on a Pharmacia mono Q anion-exchange SMART column and fractions were counted using a PerkinElmer 1480 Wizard 3 gamma counter (Turku, Finland) for 30 seconds per fraction sample.
Supplementary Material
Highlights.
Tethered Hsp90 inhibitors are selectively internalized in cancer cells
Ectopic Hsp90 facilitates probe internalization and is unique to tumors
Non-malignant tissue contain cytoplasmic Hsp90 that bind inhibitors after lysis
125I and nIR tethered inhibitors can be detected at pM amounts and 100,000 cells
Acknowledgments
This work was funded by 1R01-AI089526-01 and 1R01AI090644 - 01 to TAJH and the Department of Defense Transformative Vision Award to TAJH, NLS and HKL. There is a patent pending application by PFH and TAH for several of the small molecule inhibitors listed in this manuscript. Reagents are available upon request.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barrott JJ, Haystead TA. Hsp90, an Unlikely Ally in the War on Cancer. The FEBS journal. 2013 doi: 10.1111/febs.12147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JQ, Wilke LG, Geradts J, Kennedy SA, Palmer GM, Ramanujam N. Quantitative optical spectroscopy: a robust tool for direct measurement of breast cancer vascular oxygenation and total hemoglobin content in vivo. Cancer research. 2009;69:2919–2926. doi: 10.1158/0008-5472.CAN-08-3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CF, Fan J, Fedesco M, Guan S, Li Y, Bandyopadhyay B, Bright AM, Yerushalmi D, Liang M, Chen M, et al. Transforming growth factor alpha (TGFalpha)-stimulated secretion of HSP90alpha: using the receptor LRP-1/CD91 to promote human skin cell migration against a TGFbeta-rich environment during wound healing. Molecular and cellular biology. 2008;28:3344–3358. doi: 10.1128/MCB.01287-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q, Chang JT, Geradts J, Neckers LM, Haystead T, Spector NL, Lyerly HK. Amplification and high-level expression of heat shock protein 90 marks aggressive phenotypes of human epidermal growth factor receptor 2 negative breast cancer. Breast cancer research: BCR. 2012;14:R62. doi: 10.1186/bcr3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiosis G, Lucas B, Huezo H, Solit D, Basso A, Rosen N. Development of purine-scaffold small molecule inhibitors of Hsp90. Curr Cancer Drug Targets. 2003;3:371–376. doi: 10.2174/1568009033481778. [DOI] [PubMed] [Google Scholar]
- Csermely P, Tamás Schnaider, Csaba S ti, Zoltán Prohászka, Gborá Nardai. The 90-kDa Molecular Chaperone Family: Structure, Function, and Clinical Applications. A Comprehensive Review. Pharmacol Ther. 1998;79:129–168. doi: 10.1016/s0163-7258(98)00013-8. [DOI] [PubMed] [Google Scholar]
- Echeverria PC, Bernthaler A, Dupuis P, Mayer B, Picard D. An interaction network predicted from public data as a discovery tool: application to the Hsp90 molecular chaperone machine. PloS one. 2011;6:e26044. doi: 10.1371/journal.pone.0026044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esserman L, Shieh Y, Thompson I. Rethinking screening for breast cancer and prostate cancer. JAMA: the journal of the American Medical Association. 2009;302:1685–1692. doi: 10.1001/jama.2009.1498. [DOI] [PubMed] [Google Scholar]
- Eustace BK, Sakurai T, Stewart JK, Yimlamai D, Unger C, Zehetmeier C, Lain B, Torella C, Henning SW, Beste G, et al. Functional proteomic screens reveal an essential extracellular role for hsp90 alpha in cancer cell invasiveness. Nature cell biology. 2004;6:507–514. doi: 10.1038/ncb1131. [DOI] [PubMed] [Google Scholar]
- Fadden P, Huang KH, Veal JM, Steed PM, Barabasz AF, Foley B, Hu M, Partridge JM, Rice J, Scott A, et al. Application of chemoproteomics to drug discovery: identification of a clinical candidate targeting hsp90. Chem Biol. 2010;17:686–694. doi: 10.1016/j.chembiol.2010.04.015. [DOI] [PubMed] [Google Scholar]
- Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenert JP, Sullivan WP, Fadden P, Haystead TA, Clark J, Mimnaugh E, Krutzsch H, Ochel HJ, Schulte TW, Sausville E, et al. The amino-terminal domain of heat shock protein 90 (hsp90) that binds geldanamycin is an ATP/ADP switch domain that regulates hsp90 conformation. J Biol Chem. 1997;272:23843–23850. doi: 10.1074/jbc.272.38.23843. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hughes PF, Barrott JJ, Carlson DA, Loiselle DR, Speer BL, Bodoor K, Rund LA, Haystead TA. A highly selective Hsp90 affinity chromatography resin with a cleavable linker. Bioorganic & medicinal chemistry. 2012;20:3298–3305. doi: 10.1016/j.bmc.2012.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ide M, Suzuki Y. Is whole-body FDG-PET valuable for health screening? For. European journal of nuclear medicine and molecular imaging. 2005;32:339–341. doi: 10.1007/s00259-005-1774-3. [DOI] [PubMed] [Google Scholar]
- Kamal A, Lia Thao, John Sensintaffar, Lin Zhang, Boehm Marcus F., Burrows LCFFJ. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–410. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- Kim YS, Alarcon SV, Lee S, Lee MJ, Giaccone G, Neckers L, Trepel JB. Update on Hsp90 Inhibitors in Clinical Trial. Current topics in medicinal chemistry. 2009;9:1479–1492. doi: 10.2174/156802609789895728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga F, Kihara K, Neckers L. Inhibition of cancer invasion and metastasis by targeting the molecular chaperone heat-shock protein 90. Anticancer research. 2009;29:797–807. [PubMed] [Google Scholar]
- Liu C, Rajaram N, Vishwanath K, Jiang T, Palmer GM, Ramanujam N. Experimental validation of an inverse fluorescence Monte Carlo model to extract concentrations of metabolically relevant fluorophores from turbid phantoms and a murine tumor model. Journal of biomedical optics. 2012;17:077012. doi: 10.1117/1.JBO.17.7.077012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCready J, Sims JD, Chan D, Jay DG. Secretion of extracellular hsp90alpha via exosomes increases cancer cell motility: a role for plasminogen activation. BMC cancer. 2010;10:294. doi: 10.1186/1471-2407-10-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollapour M, Neckers L. Post-translational modifications of Hsp90 and their contributions to chaperone regulation. Biochim Biophys Acta. 2012;1823:648–655. doi: 10.1016/j.bbamcr.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulick K, Ahn JH, Zong H, Rodina A, Cerchietti L, Gomes DaGama EM, Caldas-Lopes E, Beebe K, Perna F, Hatzi K, et al. Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nature chemical biology. 2011;7:818–826. doi: 10.1038/nchembio.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neckers L, Mollapour M, Tsutsumi S. The complex dance of the molecular chaperone Hsp90. Trends Biochem Sci. 2009;34:223–226. doi: 10.1016/j.tibs.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neckers L, Workman P. Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res. 2012;18:64–76. doi: 10.1158/1078-0432.CCR-11-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer GM, Boruta RJ, Viglianti BL, Lan L, Spasojevic I, Dewhirst MW. Non-invasive monitoring of intra-tumor drug concentration and therapeutic response using optical spectroscopy. Journal of Controlled Release. 2010;142:457–464. doi: 10.1016/j.jconrel.2009.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer GM, Ramanujam N. Monte-Carlo-based model for the extraction of intrinsic fluorescence from turbid media. Journal of biomedical optics. 2008;13:024017. doi: 10.1117/1.2907161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pick E, Kluger Y, Giltnane JM, Moeder C, Camp RL, Rimm DL, Kluger HM. High HSP90 expression is associated with decreased survival in breast cancer. Cancer research. 2007;67:2932–2937. doi: 10.1158/0008-5472.CAN-06-4511. [DOI] [PubMed] [Google Scholar]
- Ray R, Haystead TA. Phosphoproteome analysis in yeast. Methods Enzymol. 2003;366:95–103. doi: 10.1016/s0076-6879(03)66008-8. [DOI] [PubMed] [Google Scholar]
- Samant RS, Clarke PA, Workman P. The expanding proteome of the molecular chaperone HSP90. Cell cycle. 2012;11:1301–1308. doi: 10.4161/cc.19722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoder H, Gonen M. Screening for cancer with PET and PET/CT: potential and limitations. Journal of nuclear medicine: official publication, Society of Nuclear Medicine. 2007;48(Suppl 1):4S–18S. [PubMed] [Google Scholar]
- Sidera K, El Hamidieh A, Mamalaki A, Patsavoudi E. The 4C5 cell-impermeable anti-HSP90 antibody with anti-cancer activity, is composed of a single light chain dimer. PloS one. 2011;6:e23906. doi: 10.1371/journal.pone.0023906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims JD, McCready J, Jay DG. Extracellular heat shock protein (Hsp)70 and Hsp90alpha assist in matrix metalloproteinase-2 activation and breast cancer cell migration and invasion. PloS one. 2011;6:e18848. doi: 10.1371/journal.pone.0018848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RA, Cokkinides V, Brooks D, Saslow D, Brawley OW. Cancer screening in the United States, 2010: a review of current American Cancer Society guidelines and issues in cancer screening. CA: a cancer journal for clinicians. 2010;60:99–119. doi: 10.3322/caac.20063. [DOI] [PubMed] [Google Scholar]
- Stellas D, El Hamidieh A, Patsavoudi E. Monoclonal antibody 4C5 prevents activation of MMP2 and MMP9 by disrupting their interaction with extracellular HSP90 and inhibits formation of metastatic breast cancer cell deposits. BMC cell biology. 2010;11:51. doi: 10.1186/1471-2121-11-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nature reviews Cancer. 2010;10:537–549. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsumi S, Mollapour M, Graf C, Lee CT, Scroggins BT, Xu W, Haslerova L, Hessling M, Konstantinova AA, Trepel JB, et al. Hsp90 charged-linker truncation reverses the functional consequences of weakened hydrophobic contacts in the N domain. Nat Struct Mol Biol. 2009;16:1141–1147. doi: 10.1038/nsmb.1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsumi S, Neckers L. Extracellular heat shock protein 90: a role for a molecular chaperone in cell motility and cancer metastasis. Cancer science. 2007;98:1536–1539. doi: 10.1111/j.1349-7006.2007.00561.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsumi S, Scroggins B, Koga F, Lee MJ, Trepel J, Felts S, Carreras C, Neckers L. A small molecule cell-impermeant Hsp90 antagonist inhibits tumor cell motility and invasion. Oncogene. 2008;27:2478–2487. doi: 10.1038/sj.onc.1210897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan CK, Neckers L, Piper PW. Understanding of the Hsp90 molecular chaperone reaches new heights. Nat Struct Mol Biol. 2010;17:1400–1404. doi: 10.1038/nsmb1210-1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Song X, Zhuo W, Fu Y, Shi H, Liang Y, Tong M, Chang G, Luo Y. The regulatory mechanism of Hsp90alpha secretion and its function in tumor malignancy. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:21288–21293. doi: 10.1073/pnas.0908151106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Trepel JB, Neckers LM, Giaccone G. STA-9090, a small-molecule Hsp90 inhibitor for the potential treatment of cancer. Current opinion in investigational drugs. 2010;11:1466–1476. [PubMed] [Google Scholar]
- Warning K, Hildebrandt MG, Kristensen B, Ewertz M. Utility of 18FDG-PET/CT in breast cancer diagnostics--a systematic review. Danish medical bulletin. 2011;58:A4289. [PubMed] [Google Scholar]
- Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nature reviews Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- Xu W, Yuan X, Xiang Z, Mimnaugh E, Marcu M, Neckers L. Surface charge and hydrophobicity determine ErbB2 binding to the Hsp90 chaperone complex. Nat Struct Mol Biol. 2005;12:120–126. doi: 10.1038/nsmb885. [DOI] [PubMed] [Google Scholar]
- Yin X, Zhang H, Lundgren K, Wilson L, Burrows F, Shores CG. BIIB021, a novel Hsp90 inhibitor, sensitizes head and neck squamous cell carcinoma to radiotherapy. International journal of cancer Journal international du cancer. 2010;126:1216–1225. doi: 10.1002/ijc.24815. [DOI] [PubMed] [Google Scholar]
- Zhou D TF, Liu Y, Ye J, Ying W, Shin Ogawa L, Inoue T, Lee W, Adjiri-Awere A, Kolodzieyski L, Tatsuta N, Wada Y, Sonderfan AJ. Associating retinal drug exposure and retention with the ocular toxicity profiles of Hsp90 inhibitors. Journal Clinical Oncology. 2012;30 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.