

Abstract
In rat models of drug relapse and craving, cue-induced cocaine seeking progressively increases after drug withdrawal. This ‘incubation of cocaine craving’ is partially mediated by time-dependent adaptations at glutamatergic synapses in nucleus accumbens. However, the circuit-level adaptations mediating this plasticity remain elusive. Here we studied silent synapses—often regarded as immature synapses that express stable NMDA receptors with AMPA receptors either absent or labile—in basolateral amygdala-to-accumbens projection in incubation of cocaine craving. Silent synapses were detected within this projection during early withdrawal from cocaine. As the withdrawal period progressed, these silent synapses became ‘unsilenced’, a process involving synaptic insertion of calcium-permeable AMPA receptors (CP-AMPARs). In vivo optogenetic stimulation-induced downregulation of CP-AMPARs at amygdala-to-NAc synapses, which re-silenced some of the previously silent synapses after prolonged withdrawal, decreased cocaine incubation. Our finding indicates that silent synapse-based reorganization of the amygdala-to-accumbens projection is critical for persistent cocaine craving and relapse after withdrawal.
Keywords: cocaine, incubation, silent synapse, accumbens, amygdala, NMDA receptor
Introduction
Relapse to drug addiction can occur after prolonged abstinence1 and is often precipitated by exposure to drug-associated cues that provoke drug craving2. In rat models of drug relapse, cue-induced drug craving progressively increases after withdrawal from cocaine and other abused drugs3,4, a phenomenon termed ‘incubation of drug craving’5. Incubation of cocaine craving is partially mediated by delayed time-dependent drug-induced accumulation of GluA2-lacking, calcium permeable AMPA receptors (CP-AMPARs) in nucleus accumbens (NAc)6,7, a brain region critical for relapse to cocaine seeking rat models8,9. The molecular triggering events and the specific glutamatergic projections to NAc that are involved in this form of long-lasting cocaine-induced synaptic plasticity are unknown3,7.
We and others previously reported that, in the NAc, non-contingent exposure to cocaine generates silent excitatory synapses, potentially immature synapses that express stable NMDA receptors (NMDARs) with AMPARs that are either absent or highly labile10–12. Silent synapses are abundant during early developmental stages and subsequently “mature” into fully functional synapses by recruiting AMPARs13,14. The functional significance of cocaine-induced silent synapses in animal models of drug reward and relapse is unknown. We recently hypothesized that the generation and subsequent unsilencing/maturation of silent synapses are a critical component of cocaine-induced brain adaptations mediating cocaine reward and relapse15. Here, we studied the role of cocaine-induced silent synapses in the basolateral amygdala (BLA)-to-NAc glutamatergic projection in the incubation of cocaine craving. We focused on this projection, because of its critical role in cue-induced drug seeking8,16 and appetitive conditioned responses17,18.
Results
Recording of BLA-to-NAc shell excitatory synapses
We examined excitatory synapses within the BLA-to-NAc shell projection19. To determine the BLA subregion that projects to NAc shell, we anesthetized a group of rats (n=5) and sterotaxically injected them with the retrograde tracer fluorogold (FG; 0.1 ng/µl/side) into NAc shell, the region we intended to record from (Fig. 1A). A week later, we obtained BLA slices showing FG-positive neurons enriched throughout this region (Fig. 1B). We then injected ChR2::YFP-expressing AAV2 into the BLA of anesthetized rats (n=5). Three weeks later, we observed ChR2-expressing neurons and neural processes in BLA (Fig. 1C–G). Whole-cell current-clamp recordings from these ChR2-expressing BLA neurons demonstrated that optical stimulations (λ=473 nm × 0.5 ms) induced action potentials (Fig. 1H). In NAc shell slices from these rats, we observed extensive neural fibers expressing ChR2, which presumably originated from BLA ChR2-expressing neurons (detected by their YFP signals; Fig. 1I). Whole-cell voltage-clamp (−70 mV) recording from NAc shell neurons demonstrated that optical stimulation (λ=473 nm×0.5 ms) of these ChR2-expressing fibers induced postsynaptic currents that were inhibited by the AMPAR antagonist NBQX (5 µM; paired t-test, t(3)=23.3, p=0.0002; Fig. 1J), suggesting that synaptic currents generated from BLA-to-NAc projection are predominantly glutamatergic.
Figure 1. Recording of BLA-to-NAc excitatory synapses.

(A) Diagram showing the position within NAc shell where retrograde tracer FG was stereotaxically injected. (B) Diagram and image showing that NAc shell injection of FG resulted in labeling of BLA neurons. (C) Diagram showing the BLA (circled in yellow), where the ChR2-expressing AAV2 was injected. (D) DIC image showing a brain slice with intra-BLA viral injection. Yellow lines sketch the BLA. (E) Magnified DIC image of a portion of D (red square). (F) YFP image showing fluorescence (viral expression) in the BLA. (G) YPF image showing that intra-BLA injection of ChR2/YFP-expressing virus resulted in expression of ChR2/YFP in neurons and neuronal fibers in the BLA. (H) Example traces showing that optical stimulation elicited action potentials in ChR2-expressing BLA neurons. (I) Example image showing extensive ChR2-expressing fibers in NAc shell in a rat given BLA injection of ChR2/YFP AAV2. (J) Example traces showing elimination of optically-elicited synaptic currents in NAc neurons after perfusion of NBQX. (K) Example EPSCs elicited at BLA-to-NAc synapses by optical stimulations in the minimal stimulation assay showing that failed and successful synaptic responses were readily discernible. (L) Optically evoked individual (gray) and averaged EPSCs (black) from an example NAc neuron showing a short latency of BLA-to-NAc synaptic transmission. Vertical green dashed lines indicate that the latency was measured between the stimulation artifact and initiation of EPSCs. Examples in K and L are taken from saline-experienced control rats (1 day after the end of the training phase).
We next verified that that we could perform the minimal stimulation assay10,11,13,20 to estimate the level of silent synapses within the BLA-to-NAc projection. We adjusted the parameters of optical stimulations (duration, intensity of stimulation, or position of the laser path) such that successes and failures of EPSCs could be recorded and clearly separated at −70 and +50 mV (Fig. 1K). Optically-evoked EPSCs exhibited short delays between the light pulse and the onset of synaptic response (0.85±0.16 ms, n=156/85 neurons/rats), indicating monosynaptic transmission (Fig. 1L). Secondary synaptic responses with >2 ms delays were not observed. Thus, we could estimate the level of silent synapses selectively within the BLA-to-NAc excitatory monosynaptic projection. Although BLA-targeted viral injections may potentially infect the neighboring central nucleus (CeA), it is unlikely that CeA contributed to our recorded synaptic responses. CeA neurons are largely GABAergic neurons, and GABA receptor-mediated currents were negligible in light-evoked responses in NAc slices (~7% inhibition by 100-µM picrotoxin, n=22/4 cells/rats). Additionally, CeA neurons do not directly project to NAc19.
Cocaine self-administration generates silent synapses
To examine incubation of cue-induced cocaine craving, we trained rats to nose-poke for intravenous infusions of cocaine for 6 days (one overnight session followed by 2 h/d for 5 days); each infusion was paired with a light cue. As previously demonstrated21–23, this training procedure led to reliable cocaine self-administration over the 5-day training period (Fig. 2A). We then used a within-subjects repeated-measure procedure, previously used in studies on incubation of craving for cocaine and other drugs24,25, to test rats for cue-induced cocaine seeking in extinction sessions performed after 1 withdrawal day or 42–47 days (referred to herein as day 45). During testing, rats were re-exposed to the drug self-administration chambers (the drug environment), and nosepokes (the operational measure of drug seeking and craving) led to contingent presentations of the light cue but not cocaine26. We found that cue-induced cocaine seeking after 45 withdrawal days was higher than after 1 day (incubation of cocaine craving) [Withdrawal day (1, 45) × Nose-poke operandum (active, inactive) interaction, F(1,24)=25.3, p<0.0001, Fig. 2B]. In contrast, no significant time-dependent changes in responding to the light cue during testing were observed in drug-naïve rats that previously nose-poked for saline injections during the training phase (Fig. 2C,D). These dare are in agreement with several reports on incubation of cocaine craving in which a limited access (2-h daily sessions) cocaine self-administration training procedure was used26–28. [Note: in most studies on mechanisms of incubation of cocaine craving3,7,29, investigators have used an extended access (6-h daily sessions) training procedure, which leads to “stronger” incubation after withdrawal than the limited access training procedure26.]
Figure 2. Cocaine self-administration generates silent synapses in the BLA-to-NAc shell projection.

(A) Cocaine self-administration training. (B) Incubation of cue-induced cocaine craving: Data are numbers of active and inactive nose-pokes from the same groups of rats in extinction tests performed on withdrawal days 1 and 45 after self-administration training. (C) Saline self-administration training. (D) Extinction tests on withdrawal days 1 and 45 after saline self-administration. (E–H) Levels of silent synapses in BLA-to-NAc projection on withdrawal days 1 and 45 after cocaine self-administration: (E, G) EPSCs (at −70 and +50 mV) evoked by minimal stimulation in representative NAc neurons of rats after 1- or 45-day withdrawal (failures and successes are shown in gray and black traces, respectively). (F, H) Trials of EPSCs from the example neurons in E and G showing a higher failure rate at −70 mV (gray dots represent failures and black dots show successes) on withdrawal day 1 and a reduced failure rate on withdrawal day 45. (I–L) Levels of silent synapses in BLA-to-NAc projection on withdrawal days 1 and 45 after saline self-administration: (I, K) EPSCs (at −70 and +50 mV) evoked by minimal stimulation in representative NAc neurons of rats after 1- or 45-day withdrawal. (J, L) Trials of EPSCs from the example neurons in I and K. (M) Summarized results showing that silent synapses were increased on withdrawal day 1 and returned to the basal level on day 45. Error bar, s.e.m. ** p<0.01, *** p<0.001.
Using this self-administration procedure, we trained different groups of rats (Fig. S1) and measured silent synapses in the BLA-to-NAc (shell sub-region) glutamatergic projection after 1 or 45 withdrawal days. We performed the minimal stimulation assay of silent synapses10,11,13,20 through optical stimulation in NAc slices from rats receiving BLA injection of ChR2-expressing AAV2 to assess successful and failed EPSCs at BLA-to-NAc shell synapses. We observed that level (%) of silent synapses within the BLA-to-NAc shell projection was increased on withdrawal day 1 but not day 45 [withdrawal day × training condition (cocaine, saline) interaction, F(1,81)=25.6, p<0.0001, Figs. 2E–M].
Together, cocaine self-administration leads to the formation of silent synapses in the BLA-NAc shell projection; these silent synapses were detected during early (day 1) but not late (day 45) withdrawal.
Un-silencing of cocaine-generated silent synapses
During brain development, silent synapses can be either pruned away or mature into fully functional synapses by recruiting AMPARs, resulting in very low levels of silent synapses in adulthood14,30. While both processes may occur simultaneously after withdrawal from cocaine, the second possibility is supported by evidence of elevated cell surface/synaptic levels of CP-AMPARs in the NAc after prolonged withdrawal from cocaine6,31–34. We therefore tested whether cocaine-generated silent synapses were ‘un-silenced’ during the withdrawal period by recruiting these receptors. CP-AMPARs conduct minimal current at depolarized membrane potentials35. Therefore, synaptic insertion of these receptors can be detected by increased rectification of EPSCs at positive membrane potentials35. In rats trained with the same cocaine regimen (Fig. S1), we found increased rectification of AMPAR EPSCs at BLA-to-NAc synapses after 45 withdrawal days [t(27)=3.5, p=0.002, Fig. 3A–D]. Furthermore, AMPAR EPSCs at these synapses became sensitive to Naspm (200 µM), a selective antagonist of CP-AMPARs35 [t(7)=3.8, p=0.007, Figs. 3E–I]. These data, obtained from rats trained to self-administer cocaine under limited access 2-h daily sessions, are different from those of a recent report34 in which accumulation of CP-AMPARs in NAc after 45 withdrawal days was observed in rats trained to self-administer cocaine for 6-h/d but not 2-h/d. These differences might be due to the fact that we initiated the training phase with an overnight extended access cocaine self-administration. Another difference between our study and previous studies is that we assessed accumulation of CP-AMPARs specifically in the BLA-to-NAc projection, increasing our ability to detect small changes in CP-AMPAR levels, assuming that CP-AMPARs do not accumulate in all NAc synapses after our limited access cocaine self-administration training procedure. Furthermore, the rats used in this study were younger than those previously used in studies on the role of CP-AMPARs in incubation of cocaine craving3,7, and thus may be more susceptible to withdrawal-induced CP-AMPAR accumulation.
Figure 3. Insertion of CP-AMPARs within BLA-to-NAc synapses after 45 withdrawal days.

(A) Example EPSCs elicited at −70 to +50 mV (with 10 mV increment) from BLA-to-NAc synapses; data were collected on withdrawal day 45 in rats previously trained to self-administer saline or cocaine. Whole-cell voltage-clamp recordings were made in the presence of the GABAA receptor-selective antagonist picrotoxin (100 µM) and the NMDA receptor-selective antagonist D-APV (50 µM). (B, C) I–V curves of EPSCs from BLA-to-NAc synapses; the influence of reversal potentials of EPSCs was factored in for each recorded neuron (see Methods). (D) Summary showing that on withdrawal day 45 an increased rectification of EPSCs was detected at BLA-to-NAc synapses from cocaine- but not saline-experienced rats. Scattered dots indicate the values of individual cells. (E–F) Example EPSCs (E) and EPSC peak over trials (F) before and during perfusion of Naspm (a selective antagonist of CP-AMPARs) in saline-trained rats; EPSCs were induced in BLA-to-NAc synapses. (G–H) Example EPSCs (G) and EPSC peak over all trials (H) before and during perfusion of Naspm in cocaine-experienced rats. (I) Summary showing that EPSCs from BLA-to-NAc synapses became sensitive to Naspm on withdrawal day 45 after cocaine self-administration, indicating insertion of CP-AMPARs to BLA-to-NAc excitatory synapses. Error bar, s.e.m. ** p<0.01.
The above results support the possibility that disappearance of silent synapses after prolonged withdrawal from cocaine is mediated by “maturation” or unsilencing process that at least partially involves recruitment of CP-AMPARs. We tested this possibility by assessing both silent synapse formation and CP-AMPARs accumulation on withdrawal day 10 using identical cocaine/saline training and extinction test conditions to those described above. At the behavioral level, cue-induced cocaine seeking in the extinction tests was higher after 10 withdrawal days than after 1 day [withdrawal day (1, 10) × nose-poke operandum interaction, F(1,37)=10.9, p<0.0001] (Fig. 4A). At this intermediate withdrawal day, the level of silent synapses was decreased toward basal levels [saline, 3.9±2.9%; cocaine, 10.1±3.3%; t(19)=1.4, p=0.18; Figs. 4B–D]. Additionally, sensitivity to Naspm, indicative of CP-AMPAR accumulation, had begun to emerge at the BLA-NAc projection in cocaine-trained rats on withdrawal day 10. Planned-comparison contrasts demonstrated a significant effect of Naspm in cocaine-trained rats on withdrawal day 10 [F (1, 20)=16.1; p=0.04], but not in saline-trained rats (p>0.99; Fig. 4E–I). This is a more rapid appearance of CP-AMPARs than found previously7, possibly related to our selective assessment of BLA-to-NAc projection or the initial overnight session that is used in our study but not in previous studies (see above).
Figure 4. Time courses of cocaine incubation, disappearance of silent synapses, and emergence of CP-AMPARs after withdrawal from cocaine self-administration.

(A) Summary showing cue-induced cocaine seeking increased after 10 withdrawal days. Data were from four independent groups; rats given extinction test on withdrawal day 10 but were not tested on day 1. (B) Example EPSCs (left) and their trial course (right) from the minimal stimulation assay of BLA-to-NAc synapses from a rat 10 days after saline self-administration. (C) Example EPSCs (left) and their trial course (right) from the minimal stimulation assay of BLA-to-NAc synapses from a rat 10 days after withdrawal from cocaine self-administration. (D) Summary showing that the level of silent synapses within the BLA-to-NAc projection decreased toward the basal (saline control) level after withdrawal from cocaine. (E–F) Example EPSCs from BLA-to-NAc synapses (left) and their trial course (right) before and during perfusion of Naspm from rats 1 day after withdrawal from saline (E) or cocaine (F) self-administration. (G–H) Example EPSCs from BLA-to-NAc synapses (left) and their trial course (right) before and during perfusion of Naspm from rats 10 days after withdrawal from saline (G) or cocaine (H) self-administration. (I) Summary showing that the sensitivity of EPSCs from BLA-to-NAc synapses to Naspm exhibited a small but significant increase after 10 days of withdrawal from cocaine. (J) Time courses of incubation, disappearance of silent synapses, and insertion of CP-AMPARs after cocaine withdrawal. Data are normalized by setting the withdrawal scores to 0% and withdrawal day 45 scores to 100%. Error bar, s.e.m. * p<0.05, *** p<0.001.
Under our experimental conditions, the time courses for the incubation of cocaine craving, disappearance of silent synapses, and accumulation of CP-AMPARs exhibit parallel changes during the withdrawal period (Fig. 4J), suggesting that disappearance of silent synapses is mediated in part by an un-silencing process involving CP-AMPAR synaptic insertion. If this speculation is correct, selective inhibition of CP-AMPARs after prolonged withdrawal (day 45) should at least partially re-silence those previously silent synapses. Accordingly, we accessed silent synapses within the BLA-to-NAc projection (Fig. S1) on withdrawal day 45. In cocaine-experienced rats, inhibiting CP-AMPARs with Naspm significantly increased the failure rate of minimal stimulation-induced synaptic responses at −70 mV [t(14)=2.72, p=0.01], but not at +50 mV [t(15)=0.29, p=0.78] within the BLA-to-NAc projection (Fig. 5D). In saline-experienced rats, perfusion of Naspm did not affect the failure rate at either −70 [t(8)=1.0, p=0.33] or +50 mV [t(8)=1.7, p=0.13]. Consequently, inhibition of CP-AMPARs with Naspm caused the reemergence of silent synapses in cocaine-experienced rats [t(16)=2.4, p=0.028; Fig. 5E], but not in saline-experienced rats [t(17)=0.14, p=0.89; Fig. S2]. Note that perfusion of Naspm did not fully recover cocaine-generated silent synapses to the level observed on withdrawal day 1, suggesting that other mechanisms are involved in silent synapse maturation (see Discussion). Nonetheless, these results suggest that a significant portion of cocaine-generated silent synapses within the BLA-to-NAc projection are un-silenced after withdrawal from cocaine by recruiting CP-AMPARs.
Figure 5. Blockade of CP-AMPARs resilences silent synapses in BLA-to-NAc shell projection on withdrawal day 45.

EPSCs elicited by minimal stimulations (recorded at −70 and +50 mV) from BLA-to-NAc synapses 45 days after withdrawal from cocaine self-administration. A–C. Example (A) and trials (B, C) of EPSCs from a cocaine-experienced rat before and during perfusion of Naspm. (D). Summary showing that blockade of CP-AMPARs on withdrawal day 45 caused a re-emergence of silent synapses in the BLA-to-NAc shell projection, suggesting that a large portion of cocaine-generated silent synapses was unsilenced by recruiting CP-AMPARs. Error bar, s.e.m. * p<0.05.
LTD internalizes CP-AMPARs at BLA-to-NAc synapses
The above results depict a silent synapse-based reorganization of the BLA-to-NAc projection during the development of incubation of cocaine craving; silent synapses are generated during cocaine self-administration, and subsequent recruitment of CP-AMPARs un-silences these synapses. To further test this hypothesis, we attempted to optogenetically re-silence cocaine-generated silent synapses within the BLA-to-NAc projection by preferentially internalizing CP-AMPARs using an in vivo LTD-based manipulation on withdrawal day 45 (see below). If our hypothesis is correct, this manipulation would reverse the newly matured/un-silenced silent synapses that contain CP-AMPARs from the “incubated” state (withdrawal day 45) back to their “pre-incubated” silent-synapse state (withdrawal day 1), which would then result in reduced cue-induced cocaine seeking after prolonged withdrawal from the drug.
The LTD protocol was developed based on previous protocols shown to induce internalization of AMPARs from excitatory synapses36, 37. We modified the protocol such that the induction stimulation (1) could be used in our optogenetic setup to selectively target the BLA-to-NAc synapses, (2) did not significantly affect typical AMPARs in saline-experienced rats, and (3) preferentially induced internalization of CP-AMPARs in cocaine-experienced rats. Because inhibiting/removing CP-AMPARs appears to be sufficient to re-silence a significant portion of cocaine-generated silent synapses after prolonged withdrawal from the drug (Figs. 5), an LTD induction protocol that meets the above criteria should selectively re-silence the matured/un-silenced synapses within the BLA-to-NAc projection.
We first verified the efficacy of this LTD protocol in brain slices. We virally expressed ChR2 in the BLA of rats trained to self-administer cocaine or saline (Fig. S1) and recorded EPSCs from BLA-to-NAc synapses on withdrawal day 45. The LTD protocol induced long-lasting depression of EPSCs in cocaine-trained but not saline-trained rats [training condition × LTD protocol interaction, F(57,855)=3.2, p<0.0001, Fig. 6A–B]. Furthermore, although EPSCs from cocaine-trained rats exhibited increased sensitivity to Naspm (Figs. 3E–I,S4), this effect was abolished after LTD induction [t(6)=0.4, p=0.7, Fig. 6C–G]. These results suggest that the AMPARs internalized by LTD induction were primarily CP-AMPARs, which were located at matured silent synapses.
Figure 6. LTD induction at BLA-NAc synapses selectively internalizes CP-AMPARs on withdrawal day 45.

(A–B) Example traces (A) and summarized results (B) showing that the LTD induction protocol induced a persistent reduction of the peak amplitudes of EPSCs at BLA-to-NAc shell synapses from cocaine-experienced, but not saline-experienced, rats. Arrows indicate the time points of application of LTD protocols. (C–F) Example EPSCs and the time course showing that BLA-to-NAc shell excitatory synapses, although highly sensitive to Naspm after withdrawal from cocaine (Fig. 3 E–I), became Naspm insensitive after LTD induction (E–G). In saline-experienced rats, EPSCs within this projection were Naspm-insensitive before (Fig. 3 E–I) or after LTD induction (C, D). (G) Summary showing that, at BLA-to-NAc synapses, LTD induction induced a persistent decrease in the peak amplitude of EPSCs, and that EPSCs lost their sensitivity to Naspm after LTD, suggesting a selectively reduction (internalization) of CP-AMPARs from these synapses. Error bar, s.e.m. ** p<0.01.
Although the mechanisms of the preferential sensitivity of CP-AMPARs to this protocol are unknown, CP-AMPARs inserted during the withdrawal period may be loosely tethered to the postsynaptic density and are therefore more susceptible to regulation31,33. Supporting this possibility, CP-AMPARs that accumulate in the NAc after prolonged withdrawal from cocaine are preferentially removed from synapses by mGluR1 stimulation33. Since optical stimulation of BLA-to-NAc projection predominantly activated excitatory monosynaptic events (Fig. 1J), the LTD protocol could be delivered to the ChR2-expressing BLA-to-NAc projection in vivo via NAc optical fibers with minimal impact on inhibitory synapses or indirect inputs.
Reversal of incubation of cocaine craving by LTD
In the final experiment, we used the above LTD protocol in vivo to reverse the maturation of cocaine-generated silent synapses within the BLA-to-NAc projection on withdrawal day 45 and determined the effect of this manipulation on enhanced (incubated) cue-induced cocaine seeking. Nineteen min before the extinction test for cue-induced cocaine seeking on day 45 we delivered the LTD induction protocol via the NAc optical fibers, which preferentially targeted BLA-to-NAc synapses (Fig. 7A). In a subset of rats, we first verified the in vivo efficacy of this LTD protocol in preferentially internalizing CP-AMPARs within the BLA-to-NAc projection by preparing NAc slices immediately after in vivo LTD induction on day 45. In these slices, EPSCs from BLA-to-NAc synapses had lost the sensitivity to Naspm [one-tailed t-test, t(10)=1.8, p=0.045, Fig. 7B] that would otherwise (without LTD induction) be present. Thus, the optical LTD protocol was effective in vivo, as well as in vitro, in preferentially internalizing CP-AMPARs at BLA-to-NAc synapses.
Figure 7. Reversing the maturation of silent synapses in the BLA-to-NAc projection reverses incubation of cocaine craving.

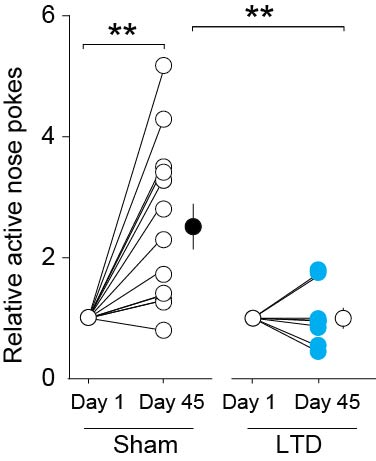
(A) Diagrams showing the timeline of behavioral experiments and the LTD induction protocol in BLA-to-NAc synapses before the test for cue-induced cocaine seeking on withdrawal day 45. (B) Summary showing that after 45 days of withdrawal from cocaine, EPSCs at BLA-to-NAc synapses were significantly inhibited by perfusion of Naspm. After in vivo LTD induction, EPSCs from these synapses became resistant to Naspm. EPSCs were insensitive to Naspm before or after in vivo LTD in saline-experienced rats. (C) In vivo LTD induction prevented incubated cue-induced cocaine seeking in the extinction test. Error bar, s.e.m. ** p<0.01.
We then applied this LTD protocol immediately before the day 45 extinction test. LTD induction reduced cue-induced cocaine seeking to a level similar to that of day 1 [LTD induction × nose-poke operandum interaction, F(1,20)=11.1, p=0.003, Figs. 7C,S3]. Thus, internalization of CP-AMPARs, which likely “re-silenced” the previously silent synapses in the BLA-to-NAc projection, reversed incubation of cue-induced cocaine craving on withdrawal day 45 to the ‘pre-incubated’ day 1 levels.
Discussion
Studies using pharmacological inactivation or site-specific lesions implicate the glutamatergic projection from BLA to NAc in cue-induced cocaine seeking in rat models8,16. Biochemical, electrophysiological and pharmacological evidence indicates that time-dependent delayed accumulation of CP-AMPARs in NAc plays a critical role in incubation of cocaine craving7. Our current results provide a logical link between these two sets of results by demonstrating that during cocaine self-administration, silent synapses are generated within the BLA-to-NAc projection, and that after withdrawal from cocaine, some of these silent synapses are ‘un-silenced’ by recruiting CP-AMPARs, which play a critical role in incubation of cocaine craving.
Cocaine exposure and synaptogenesis
We previously proposed that cocaine-induced generation of silent synapses shares mechanisms with synaptogenesis15. Three features are particularly notable in this regard: (1) synaptogenesis involves formation of new synaptic structures, reflected in postsynaptic morphology as an increase in the number of dendritic spines38, (2) synaptogenesis and circuitry development are often initiated or modulated by postsynaptic activation of CREB39, and (3) one of the pro-synaptogenesis functions of CREB is mediated by up-regulation of GluN2B-containing NMDARs10,40. Similarly, cocaine exposure increases the number of dendritic spines in the NAc41,42 and activates CREB43. Additionally, cocaine-induced CREB activation in NAc plays a role in both silent synapse formation and cocaine-induced increases in dendritic spines10. Finally, cocaine-induced generation of silent synapses is controlled by synaptic insertion of GluN2B-containing NMDARs10,11.
During brain development, generation and subsequent maturation/un-silencing of silent synapses is critical for synaptic stabilization and circuitry formation14. Our results suggest that in the developed brain, generation of silent synapses can be resumed in particular neural circuits after cocaine exposure to reorganize and reshape synapses, neural circuits, and future behaviors15. If the silent synapse-mediated circuitry reorganization is indeed achieved by synaptogenesis as we hypothesize, this process may not only substantially and persistently change the pattern of information flow within this projection, but also create a new path of information flow via newly formed synapses/circuits that did not exist before cocaine exposure15.
Our previous results suggest that cocaine-induced generation of silent synapses occurs in heterogeneous populations of NAc neurons via the same GluN2B-based molecular mechanisms11. However, different mechanisms are likely involved for certain types of NAc neurons. A recent study12 demonstrated that, in mice, prior non-contingent cocaine exposure induces higher levels of silent synapses in ~2 to 3% of Fos-positive NAc neurons that were acutely activated by cocaine. Furthermore, generation of silent synapses in these neurons was not accompanied by increased GluN2-containing NMDARs, and was potentially achieved by internalization of AMPARs from existing synapses, rather than synaptogenesis-related mechanisms12. Thus, although cocaine-induced generation of silent synapses is likely to be a common phenomenon in different NAc neuronal populations, the cellular processes underlying silent synapse formation may differ.
Maturation of silent synapses and cocaine incubation
Our data suggest that, after silent synapses are generated during cocaine self-administration, some of these synapses mature by recruiting CP-AMPARs after prolonged withdrawal from cocaine. The main evidence for this notion is that BLA-to-NAc synapses that were highly sensitive to Naspm after 45 withdrawal days became Naspm-insensitive after LTD induction (Figs. 3,6). Furthermore, at this time-point, pharmacological inhibition of CP-AMPARs caused the re-emergence of silent synapses (Fig. 5). However, Naspm only “recovered” ~60% of the initial level of silent synapses (Fig. 5D), suggesting that some silent synapses may have been pruned away after withdrawal from cocaine, as is the case with some silent synapses during development38. Another possibility is that some silent synapses matured by recruiting Naspm-insensitive, GluA2-containing AMPARs.
We employed a LTD-based in vivo manipulation, rather than NAc application of Naspm, in order to manipulate CP-AMPARs and silent synapses in a projection-specific manner. However, our results cannot rule out two other scenarios: (1) The LTD protocol might also affect synapses that were previously non-silent. This could happen if some previously non-silent synapses recruited CP-AMPARs after prolonged withdrawal from cocaine and thus became susceptible to the LTD protocol; (2) The CP-AMPAR-containing synapses silenced after prolonged withdrawal from cocaine might not be the same silent synapses generated during early withdrawal. This could happen if some previously non-silent synapses lose all their typical AMPARs and recruited new CP-AMPARs after prolonged withdrawal from cocaine. Nevertheless, the observations that the LTD manipulation removed CP-AMPARs from BLA-to-NAc synapses after 45 withdrawal days (Fig. 6) and that inhibiting CP-AMPARs caused a re-emergence of silent synapses at this time point (Fig. 5), suggest that the most parsimonious interpretation of our data is that the LTD manipulation ‘re-silenced’ the ‘un-silenced’ synapses.
Finally, we confirmed the role of NAc CP-AMPARs in incubation of cue-induced cocaine craving and also demonstrated a novel role of silent synapses in this incubation. A question for future research is whether these particular cocaine-induced neuroadaptations also play a role in relapse provoked by re-exposure to the drug itself as assessed by reinstatement of cocaine seeking induced by a drug priming injection44 or resumption (re-acquisition) of drug self-administration under different reinforcement schedules45. We suspect that the mechanisms we have identified in our report are selective to incubation of cue-induced drug craving. This is because there is either little evidence or mixed evidence for time-dependent changes in cocaine-priming-induced reinstatement or resumption of cocaine self-administration after withdrawal from cocaine3,4,26,46.
Concluding remarks
In previous studies we and others demonstrated the generation of silent synapses within the NAc after non-contingent cocaine exposure10–12. Here, we demonstrated that after cocaine self-administration, silent synapses were generated within a particular glutamatergic projection and were necessary for the development of incubation of cocaine craving. These results raise two questions for future research: (1) do generation and subsequent maturation of silent synapses occur in other glutamatergic projections to NAc47, and do they mediate other aspects of cocaine relapse9,44; and (2) does silent synapse-mediated circuitry reorganization occur in other brain regions implicated in incubation of cocaine craving3? Some studies have shown NAc core-shell differences in cocaine-induced plasticity48. However, NAc core and shell undergo similar AMPAR plasticity after prolonged withdrawal from cocaine; both regions exhibit accumulation of CP-AMPARs6,32,49. Furthermore, CP-AMPARs in both NAc subregions play a necessary role in the expression of incubation of cocaine craving6. These results warrant future studies examining withdrawal-period-associated generation and maturation of silent synapses in NAc core. However, complementing the present results, removal of CP-AMPARs from NAc core synapses after prolonged withdrawal from cocaine represents a promising pharmacotherapeutic approach to attenuate incubation of cocaine craving33,50. Future studies will delineate the role of silent synapse-based re-organization of neural circuits involving the NAc in drug craving and the potential for exploiting this understanding to devise novel therapies for cocaine relapse.
On Line Methods Section
Subjects
Male Sprague-Dawley rats (Harlan, MD), postnatal day 28–30 at the beginning of the experiments were used. Rats were single housed under a 12 hr light/dark cycle (on at 700, off at 1900). Temperature (22 ± 1°C) and humidity (60 ± 5%) were controlled. Behavioral experiments were performed during the day time. The rats were used in all experiments in accordance with protocols approved by the Institutional Animal Care and Use Committees at Washington State University or University of Pittsburgh.
Viral vectors
Recombinant adeno-associated vectors (rAAV2) expressing venus tagged ChR2 H134R were pseudotyped with AAV1/2 capsid proteins. HEK293T cells were co-transfected with the plasmids pF6 (adenoviral helper plasmid), pRVI (cap and rep genes for AAV serotype 2), pH21 (cap gene for AAV serotype 1 and rep gene for serotype 2) and the rAAV plasmid, using linear polyethylenimine assisted transfection23. The helper plasmids were kindly provided by Dr. M. Schwarz51. Cultures grown in DMEM (Biochrom) with 10% substituted FBS (Biochrom, #S0115) were harvested from 15 by 15 cm dishes after 48h. rAAV were isolated and purified as described51. Briefly, HEK293T cells were lysed with sodium desoxycholate and repeated freeze-thaw cycles in the presence of Benzonase-Nuclease HC (Novagen). From the supernatant, rAAVs were isolated by iodixanol gradient centrifugation from the 40% and 54% interphase. rAAVs were then desalted by ultrafiltration, filtered through 0.2um Millex-GV filter units (Millipore), and stored at 4°C in 500 µlPBS with 10mM MgCl2 and 25mM KCl. The ChR2 gene was packaged with an YFP gene into a recombinant, replication-defective form of the adenovirus (AAV).
Fluorogold ChR2-YFP imaging procedures
For retrograde tracing, rats were anesthetized with a ketamine/xylzazine mixture (50/5 mg/kg, i.p.) and placed in a stereotaxic apparatus (Stoelting). Fluorogold solution (FG; 1% in saline) was injected iontophoretically (+5 µA, 7-s on/7-s off, 10 min) into the nucleus accumbens (NAc) shell with a glass micropipette (tip diameter: 20 µm). The coordinates used to target the NAc shell were: +1.8 (AP), 0.9 (ML) and 6.5 (DV, from brain surface). After a 2-wk survival period, rats were perfused transcardially with 0.1 M phosphate-buffered saline (PBS) followed by 4% formaldehyde in PBS. Brains were removed carefully and given an additional 48-hr postfix in 4% formaldehyde, and then transferred to 30% sucrose in PBS for 48 h before sectioning. Coronal sections (50 µm) were cut with a Leica RM 2000R freezing microtome. Sections were washed in PBS and mounted on gelatin-coated slides and then coverslipped with DPX mounting medium. Expression of fluorogold in BLA was examined using a Zeiss Axioplane2 imaging microscope and images were taken at 10× original magnification, using infinity digital camera. For imaging of ChR2-YFP, rats were injected with AAV-ChR2-YFP into the BLA (see below). After approximately 3 weeks, rats were transcardially perfused with 0.1 M PBS followed by 4% formaldehyde in PBS. Brains were removed carefully and given an additional 48-hr postfix in 4% formaldehyde, and then transferred to 30% sucrose in PBS for 48 hrs before sectioning. Coronal sections (50 µm) were cut with a Leica RM 2000R freezing microtome. Sections were washed in PBS and mounted on gelatin-coated slides and then coverslipped with DPX and Prolong Antifade (Invitrogen) mounting medium. Expression of ChR2-YFP in the BLA and NAc Shell was examined using a Zeiss LSM5210 Meta confocal microscope and images were taken at 10x and 40x original magnification.
Behavioral studies
Drugs
Cocaine HCl (Provided by NIDA Drug Supply Program) was dissolved in 0.9% NaCl saline. Ketamine and xylazine were mixed for anesthesia (purchased from WSU College of Veterinary Medicine or DEA-designated vendor at University of Pittsburgh).
Viral delivery
A 26 gauge injection needle was used to bilaterally inject 1 µl (0.2 µl/min) of the AAV2-ChR2-YFP solution via Hamilton syringe into the BLA (AP: −2.50, ML: ±4.80, DV: −8.50), using a Thermo Orion M365 pump. Injection needles were left in place for 5 min following injection. ChR2-YFP was given 1–2 weeks before any experimental manipulation to ensure maximal infection; therefore, electrophysiological analyses were conducted ~3 weeks (1 d withdrawal) or ~8 weeks (45 d withdrawal) post infection.
Catheter Implantation
Self-administration surgery was as described in ref.10, 21. Briefly, a silastic catheter was inserted into the right auricle through the external jugular vein, and the distal end was led subcutaneously to the back between the scapulas. Catheters are constructed from silastic tubing (~5 cm; inner diameter 0.020 in, outer diameter 0.037 in) connected to a Quick Connect Harness (SAI Infusion). Rats were allowed to recover for 5–14 days. During recovery, the catheter was flushed daily with 0.1 ml of heparin (10 U/ml) and gentamicin antibiotic (5 mg/ml) in sterile saline to help protect against infection and catheter occlusion.
Self-administration apparatus
Experiments were conducted in operant-conditioning chambers enclosed within sound-attenuating cabinets (Med Associates). Each chamber contains an active and inactive nose poke, a food dispenser, the conditioned stimulus (CS) light in each nose poke and a house light. No food or water was provided in the chambers during the training sessions.
Intravenous self-administration training
Cocaine self-administration training began 5–14 days after surgery. On day 1, rats were placed in the self-administration chamber for an overnight training session on a fixed ratio (FR) 1 reinforcement schedule. Nose poking in the active hole resulted in a cocaine infusion (0.75 mg/kg in 0.10 ml over 6 sec) and illumination of a CS light inside the nose poke hole. The CS light remained on for 6 sec, whereas the house-light was illuminated for 20 sec during which active nose-pokes were counted but resulted in no cocaine infusions. After the 20 sec, the house-light was turned off, and the next nose-poke in the active hole resulted in a cocaine infusion. Nose-pokes in the inactive hole had no reinforcement consequences but were recorded. Rats that received at least 40 cocaine infusions in the overnight session were allowed to self-administer cocaine for 2 h for 5 consecutive days ~24 h after the overnight training on a fixed-ratio-1 (FR1) reinforcement schedule. Same or similar cocaine self-administration procedures/standards were used in our previous studies21–23. Rats that did not meet the overnight number of infusions criterion (n=5) were removed from the study.
Measurement of cue-induced cocaine seeking after withdrawal
We assessed incubation of cue-induced cocaine craving in extinction tests (1 hr) conducted after 1, 10, or 45 days of withdrawal from cocaine self-administration. During the test sessions, active nose-pokes resulted in contingent delivery of the CS light cue but not cocaine. For behavioral assays without electrophysiology, we used within-subject assessment of incubation24, 26; the same rats were tested for cocaine seeking on withdrawal day 1 and 45, or day 1 and 10. For electrophysiology experiments, we used between-subject assessments; different groups of rats were killed on either withdrawal day 1, 10, or 45 without the cocaine seeking extinction tests.
Optogenetic procedures
Construction of Optical Neural Interface (ONI)
For in vivo optical control of BLA projections, two 105-µm core optic fibers were modified for attachment to an internal cannula creating the Optical Neural Interface (ONI). When the ONI was secured in vivo to the guide cannula, the stripped fiber extended 1.0 mm past the tip of the cannula. This experimental set-up was based on a previously verified setup52 with slight modifications. The ONI was secured in vivo to the cannula head-mount only during stimulation.
In vivo stimulation
The ONI was attached with an FC/PC adaptor to a 473 nm blue laser diode (IkeCool), and light pulses were generated through a stimulator (A-M Systems). Optic fiber light intensity was measured using a light sensor (Thor Labs - S130A) and light intensity was adjusted to ~10 mW. Prior to attaching the ONI to head-mount guide cannula, the rat was briefly sedated with isoflurane to allow smooth insertion and to prevent damage to the optic fibers. Once the rat was awake, an LTD protocol was administered (5 Hz blue light pulses were administered for 3 min and repeated 3 times with 5 min intervals; pulse duration, 1.0 ms) in their home cage. Once this 19-min LTD protocol was concluded, the rat was placed in a testing chamber. Control rats were also briefly sedated with isoflurane and a sham optic fiber was attached to the head-mount guide cannula. Upon awakening, they remained in their home cage for 19 min and subsequently were placed into a testing chamber.
In vitro stimulation
All evoked responses were delivered using an IkeCool laser at 473 nM, ~10 mW, through the microscope’s 40x objective. The duration (0.01–1 ms) of the light pulse was decreased until an optimal evoked response was achieved.
Electrophysiological studies
Slice preparation
Before decapitation, the rats were anesthetized with isoflurane and subsequently transcardially perfused with 4°C cutting solution (in mM: 135 N-methyl-D-glucamine, 1 KCl, 1.2 KH2PO4, 0.5 CaCl2, 1.5 MgCl2, 20 choline-HCO3, 11 glucose, pH adjusted to 7.4 with HCl, and saturated with 95% O2 /5% CO2). The rat was decapitated, and then the brain was removed and glued to a block before being sliced using a Leica VT1200s vibratome in 4°C cutting solution. Coronal slices of 300 µm thickness were cut such that the preparation contained the signature anatomical landmarks (e.g., the anterior commissure) that clearly delineate the NAc subregions. After allowing 1–2 h for recovery, one slice was transferred from a holding chamber to a submerged recording chamber where it was continuously perfused with oxygenated aCSF maintained at 30 ± 1°C.
Drugs
D-Aminophosphonovaleric acid (D-APV), referred to herein as APV, was used at a concentration of 50 µM to inhibit NMDAR-mediated responses. Picrotoxin (100 µM) was used to inhibit GABAAR-mediated responses. Spermine (100 µM) was added freshly to the internal solution to restore the endogenous polyamine that blocks GluA2-lacking AMPARs. 2, 3-Dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX) (5 µM) was used to inhibit AMPAR-mediated responses. 1-Naplhthylacetyl spermine trihydrochloride (Naspm) (200 µM) was used to selectively inhibit GluA2-lacking AMPARs. APV, NBQX, and Naspm were purchased from R & D Systems, and all other chemicals were purchased from Sigma-Aldrich.
Whole-cell recordings
Standard whole-cell current- or voltage-clamp recording were used with a MultiClamp 700B amplifier (Molecular Device). During recordings, slices were superfused with aCSF that was heated to 30 ± 1°C by passing the solution through a feedback-controlled in-line heater (Warner Instruments) before entering the chamber. Recordings were made under visual guidance (40x, differential interference contrast optics) with electrodes (3–5 MΩ). Expression of ChR2-YFP in neurons or processes was verified using an Olympus BX51WI fluorescent/DIC microscope before recordings. The intracellular and extracellular solutions can be found in our published papers10, 11. For all recordings, series resistance was 8 to 14 MΩ and was left uncompensated. Series resistance was monitored continuously during all recordings, and a change beyond 15% resulted in exclusion of the cell from data analyses. Synaptic currents were recorded with a MultiClamp 700B amplifier (Molecular Devices), filtered at 3 kHz, amplified 5 times, and then digitized at 20 kHz with a Digidata 1440A analog-to-digital converter (Molecular Devices).
Silent synapse recordings
Neurons in the NAc shell were randomly selected for recording. Minimal stimulation experiments were performed as previously reported11, 13, 20. After obtaining a small (<50 pA) EPSC at −70 mV, the duration of the light pulse was reduced in small increments to the point that failures vs. successes of synaptically evoked events (EPSCs) could be clearly distinguished visually. Pulse duration and frequency were then kept constant for the rest of the experiment. The amplitude of both AMPAR and NMDAR ESPCs resulting from single vesicle release is relatively large (e.g., ~15 pA for AMPAR mEPSCs)11, which facilitates the judgment of success vs. failures of EPSCs; therefore, they were defined visually. For each cell, 50–100 traces were recorded at −70 mV, and 50–100 traces were recorded at +50 mV. Recordings were then repeated at −70 mV and +50 mV for another round or two. Each cell was recorded >2 rounds. Only cells with relatively constant failure rates (changes <10%) between rounds were included for calculation of % silent synapses. Percent silent synapses were calculated using the equation: 1–Ln(F−70)/Ln(F+50), in which F−70 was the failure rate at −70 mV and F+50 was the failure rate at +50 mV. The major difference between minimal stimulations delivered by optical fibers and electrical electrode is that electrical stimulation can be confined to one or a few afferents around the well-defined stimulation site (i.e., the tip of the stimulation electrode), whereas minimal optical stimulation will preferentially influence the afferents with the highest expression of ChR2 despite not necessarily being located together. This difference may not be a major concern because: i) the amplitudes of successful synaptic responses elicited by minimal optical stimulations are similar or very close to that of mEPSCs (see example traces throughout the manuscript), suggesting that one or very few afferents are stimulated each time. Thus, the premise that only a very small number of synapses are activated each time is met, even without knowing the actual activation spot; ii) Given that only fibers with the highest expression of ChR2 are activated with short-duration stimulation, we assume that this same set of fibers is repeatedly activated throughout the trial, similar to minimal electrical stimulation in which the same set of fibers that is most sensitive to electrical stimulation is assumed to be activated repeatedly; iii) When performing the minimal optical stimulation assay, the stimulation intensity/duration was adjusted such that failure rate was around 0.5 (ranging from 0.2 – 0.8 with median of 0.5). Successes and failures were readily separable, with the pattern similar to that observed in previous studies detecting silent synapses10, 11, 13, 20, 53 using minimal electrical stimulation. Thus, even with potentially different properties of optical stimulation, the minimal optical stimulation can still detect silent synapses, at least semi-quantitatively.
Rectification recording
To examine AMPAR subunit composition, an IV curve was plotted. Evoked BLA-to-NAc AMPAR-mediated EPSCs were measured at the membrane potentials of −70, −50, −30, −10, 0, 10, 30, and 50 mV. The rectification index was calculated by comparing the peak amplitude at +50 mV to −70 mV after offsetting the reversal potential54.
LTD recording
Light-evoked LTD was induced at BLA-to-NAc synapses in cocaine- or saline-treated rats. After 10 min of stable baseline recording, an induction protocol was used that consisted of three trains of stimuli at 5 Hz (pulse duration, 0.5 ms; train duration, 3 min; 5 minutes apart), while holding the cells at −70 mV.
Specific experiments
Experiment 1: Incubation of cocaine craving and measurement of silent synapses in the BLA-NAc projection on withdrawal day 1, 10, and day 45 (Figs. 2, 4)
We first verified a “short” incubation procedure. Briefly, we trained the rats with one overnight session on an FR1 reinforcement schedule with either saline (0.1 ml/infusion) or cocaine HCl (0.75 mg/kg/0.1 ml per infusion). If the rats had at least 40 cocaine infusions during the overnight session, they began 5 consecutive 2-h training days under the same reinforcement schedule on the second following day. Once completing the 5-day training with at least 10 infusions per day, the rats were placed back in the home cages for withdrawal. On withdrawal day 1, rats were placed in the operant chambers for a 1-h extinction test, during which nose pokes in the active holes resulted in presentation of light cues but not cocaine infusion. The number of nose pokes in the active holes was used as a measure of cocaine seeking. After this test, the rats were placed back in the home cages for prolonged withdrawal. On withdrawal day1, 10, or 45, the rats were returned to the operant chambers for another 1-h extinction test. For these experiments, saline self-administering rats were used as control. In a different group of rats, we assessed extinction responding after 10 withdrawal days (Fig. 4). Animal use: withdrawal day 1: saline 17, cocaine 20; withdrawal day 10: saline 8, cocaine 9; withdrawal day 45: saline 9, cocaine 11.
We next examined cocaine-induced generation of silent synapses in rats that were treated with the same incubation-inducing cocaine regimen as described above. For this study, rats were bilaterally injected with AAV2-ChR2-YFP into the BLA and then implanted with a silastic catheter into their right jugular vein. After approximately two weeks of recovery, self-administration training was conducted as described in the previous paragraph. After completing the 5-day training, the rats were used for electrophysiological analysis of silent synapses after 1, 10, or 45 days of withdrawal in their home cages (saline self-administration, day 1 n/m 26/5; day 10 n/m = 11/8, day 45 n/m = 21/6; cocaine self-administration, day 1 n/m = 12/4, day 10 n/m = 13/5, day 45 n/m = 26/11). To estimate the number of silent synapses, minimal stimulation experiments were performed. Optogenetic stimulation was used to obtain a small EPSC at BLA-to-NAc synapses while holding the cells at −70mV. Then, the duration of the light pulse was reduced in small increments to a point at which failures vs. successes could be clearly defined. The recordings at the two membrane potentials were then repeated 1–2 rounds to ensure the stability. Responses immediately after (within 2.5 min) the change of holding potentials were not included for analysis. Percent silent synapses were calculated using the equation: 1−Ln(F−70)/Ln(F+50), in which F−70 was the failure rate at −70 mV and F+50 was the failure rate at +50 mV11.
Experiment 2: AMPAR subunit composition in the BLA-to-NAc projection on withdrawal day 1, 10 and 45 (Figs. 3, 4)
The self-administration procedures were identical to the ones used in Experiment 1. Rats were euthanized for electrophysiological analyses after 1, 10, or 45 day withdrawal (saline self-administration, day 1, n/m = 9/4, day 10, n/m = 12/6, day 45, n/m = 8/8; cocaine self-administration, day 1, n/m = 10/4, day 10, n/m = 11/6, day 45, n/m = 21/10). Picrotoxin and APV were added to the bath to isolated AMPAR-mediated responses and spermine was added to the internal solution to restore the endogenous polyamines that contribute to the rectification at depolarized potentials, which are indicative of GluA2-lacking AMPARs. Light pulses, <1 ms in duration, were used to evoke BLA-to-NAc EPSCs for generation of the I–V curve. The rectification index was calculated by comparing the peak amplitude at +50 mV to −70 mV after correcting for the reversal potential. Additional verification of the CP-AMPAR component was achieved by locally applying Naspm. Light-evoked AMPAR-mediated EPSCs were obtained from saline self- (n/m = 8/7) or cocaine self-administration (n/m = 8/4) rats while locally perfusing aCSF through a large (ID, 100 – 150 µm) bore pipette. Once a stable baseline response was achieved, the solution was switched to one that contained Naspm. Upon stabilization and diffusion of solutions, data were collected for at least 5 min.
Experiment 3: Reemergence of silent synapse after blockade of CP-AMPARs on withdrawal day 45 (Fig. 5)
The self-administration procedures were identical to the ones used in Experiment 1. Rats were euthanized for electrophysiological analysis after long-term withdrawal (42–47 days) (saline self-administration, n/m = 18/5; cocaine self-administration, n/m = 17/6). Minimum stimulation assay (described in Experiment 1) was performed first in the absence and then in the presence of Naspm. A large bore pipette, as described in Experiment 2, was used to locally supply either bath solution (control) or bath solution with the addition of Naspm.
Experiment 4: LTD at BLA-to-NAc synapses (Fig. 6A,B)
The self-administration procedures were identical to the ones used in Experiment 1. Rats were euthanized for electrophysiological analysis after long-term withdrawal (42–47 days) (saline self-administration, n/m = 8/5; cocaine self-administration, n/m = 9/6). Picrotoxin was added to the bath in some conditions to verify the synaptic properties. Light pulses, <1 ms in duration, were used to evoke BLA-to-NAc EPSCs while holding the cell at −70mV. Once a stable baseline was obtained for ~10 min, a light–evoked LTD-induction protocol was administered: 0.5–1-ms flashes, 5 Hz for 3 min, three times with 5 min intervals. Following this induction, recordings were continued for at least 30 min.
Experiment 5: Removal of the Naspm-sensitive component of BLA-to-NAc EPSCs by LTD on withdrawal day 45 (Fig. 6C–G)
This set of recordings examined the CP-AMPAR component in rats with a previous history of saline self-administration (n/m = 10/7) or cocaine self-administration (n/m=8/5) before and after LTD induction on withdrawal day 45. We measured light-evoked AMPAR-mediated EPSCs while locally perfusing aCSF containing picrotoxin through a large bore pipette. Once a stable response was achieved, we applied a light-evoked LTD protocol (5Hz for 3 times, with 5-min intervals). After completion of the LTD induction, the local perfusion solution was switched to one that contained Naspm. Upon stabilization, data were collected for at least 10 min.
Experiment 6: In vivo LTD attenuates cue-induced cocaine seeking on withdrawal day 45 (Fig. 7)
The self-administration procedures were identical to those used in Experiment 1. Two groups of rats (n = 8–14 per group) were tested for extinction responding (1-h session) on withdrawal days 1 and 45. Prior to the final test, on withdrawal day 35, rats were anesthetized with sodium pentobarbital (40 mg/kg) and placed in a stereotaxic apparatus. A bilateral 26 gauge guide cannula was inserted 1.0 mm above the NAc shell and secured with cranial screws and dental cement. On withdrawal day 45, the rats were divided into two groups: one group had a sham optic fiber inserted into the guide cannula; the other group received a true optic fiber-ONI. The ONI was adjusted to a power level ~10 mW at the optic fiber tip and the rat was briefly anesthetized to allow smooth insertion of optic fiber. Sham controls received the same brief anesthesia and installation of optical fiber. Once the rats recovered and were freely moving, we initiated the LTD protocol with an IkeCool laser connected to a pulse generator (5 Hz for 3 min, 3 times with 5 min intervals) in the rats’ home cage. The sham control group received an identical treatment except that the optic fiber was not functional. Once the LTD stimulation train was completed, the rats were placed in the operant chamber for a 1-h extinction test.
Data acquisition and analysis
In all electrophysiology experiments, the data were coded before analysis. Data were then de-coded for the final results. All results are shown as mean ± SEM. Each experiment was replicated in at least 4 rats (1–3 cells were recorded from each rat) for electrophysiological analysis and 8 rats for behavioral tests. No data points were excluded unless specified in the experimental procedure. For Experiments 1–6 described above, a total of 257 rats were used, among which 5 rats were excluded before data collection because of the catheter leakage or clogging, 12 rats were excluded before data collection because of surgery-associated infection or significant (>15%) loss of body weight, 9 rats were excluded during data collection because misplacement of cannula (found during preparation of brain slices), and 5 rats were excluded because they did not meet the cocaine self-administration criterion (see above). Data from the repeated runs for the same experiment were pooled together for statistical analysis. Technical replicates were utilized for some of the key experiments, such as insertion of CP-AMPARs, in which biophysical (rectification of AMPAR EPSCs) and pharmacological (sensitivity to Naspm) approaches were both employed. Sample size for each experiment was determined either based on our previous experience with similar experiments or those that have been routinely used in similar studies published in this journal. Sample size was presented as n/m, where "n" refers to the number of cells examined and "m" refers to the number of rats. Normal distribution was assumed for all statistics. This is based on our previous work related to silent synapses, in which a typical normal distribution was observed10. Variance was estimated for most major results and no significant difference was found between control and manipulation groups. Statistical significance was assessed using t-tests (when two groups are compared) or one/two-way ANOVAs (when multiple groups or repeated measured were involved), followed by Bonferroni posttests.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgement
We thank Drs. Barbara Sorg, David Dietz, R. Lane Brown, and Heiko Jansen for technical consultations. This research was supported [in part] by the Intramural Research Program of the NIDA-NIH (YS), extramural funds DA028020 (BRL), DA029565, DA030379 (MEW, YD), DA009621 (MEW), DA029099 (MEW), DA015835 (MEW), DA007359 (EJN), DA014133 (EJN), DA023206 (YD), DA031551(YD), DA034856 (YD) from NIDA NIH, grants from NSFC 81328011, and DFG (OMS). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Author contribution: BRL, YYM, YHH, SRS, MEW, EJN, YS, OMS and YD contributed to the design of the experiments, design of the analyses, and the writing of the manuscript. BRL, YYM, XW, MO, MI, PAN, NMG, TEB, AS, CG, and MKL conducted the experiments and formed the analyses.
The authors declare that they do not have any conflicts of interest related to the data presented in this manuscript.
References
- 1.Hunt WA, Barnett LW, Branch LG. Relapse rates in addiction programs. J Clin Psychol. 1971;27:455–456. doi: 10.1002/1097-4679(197110)27:4<455::aid-jclp2270270412>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 2.Gawin FH, Kleber HD. Abstinence symptomatology and psychiatric diagnosis in cocaine abusers. Clinical observations. Arch Gen Psychiatry. 1986;43:107–113. doi: 10.1001/archpsyc.1986.01800020013003. [DOI] [PubMed] [Google Scholar]
- 3.Pickens CL, et al. Neurobiology of the incubation of drug craving. Trends Neurosci. 2011;34:411–420. doi: 10.1016/j.tins.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu L, Grimm JW, Dempsey J, Shaham Y. Cocaine seeking over extended withdrawal periods in rats: different time courses of responding induced by cocaine cues versus cocaine priming over the first 6 months. Psychopharmacology. 2004;176:101–108. doi: 10.1007/s00213-004-1860-4. [DOI] [PubMed] [Google Scholar]
- 5.Grimm JW, Hope BT, Wise RA, Shaham Y. Incubation of cocaine craving after withdrawal. Nature. 2001;412:141–142. doi: 10.1038/35084134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conrad KL, et al. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature. 2008;454:118–121. doi: 10.1038/nature06995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wolf ME, Tseng KY. Calcium-permeable AMPA receptors in the VTA and nucleus accumbens after cocaine exposure: when, how, and why? Frontiers in molecular neuroscience. 2012;5:72. doi: 10.3389/fnmol.2012.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bossert JM, Marchant NJ, Calu DJ, Shaham Y. The reinstatement model of drug relapse: recent neurobiological findings, emerging research topics, and translational research. Psychopharmacology. 2013 doi: 10.1007/s00213-013-3120-y. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kalivas PW. The glutamate homeostasis hypothesis of addiction. Nature reviews. Neuroscience. 2009;10:561–572. doi: 10.1038/nrn2515. [DOI] [PubMed] [Google Scholar]
- 10.Brown TE, et al. A Silent Synapse-Based Mechanism for Cocaine-Induced Locomotor Sensitization. The. J. Neurosci. 2011;31:8163–8174. doi: 10.1523/JNEUROSCI.0016-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang YH, et al. In vivo cocaine experience generates silent synapses. Neuron. 2009;63:40–47. doi: 10.1016/j.neuron.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koya E, et al. Silent synapses in selectively activated nucleus accumbens neurons following cocaine sensitization. Nat Neurosci. 2012 doi: 10.1038/nn.3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Isaac JT, Crair MC, Nicoll RA, Malenka RC. Silent synapses during development of thalamocortical inputs. Neuron. 1997;18:269–280. doi: 10.1016/s0896-6273(00)80267-6. [DOI] [PubMed] [Google Scholar]
- 14.Kerchner GA, Nicoll RA. Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nat Rev Neurosci. 2008;9:813–825. doi: 10.1038/nrn2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee BR, Dong Y. Cocaine-induced metaplasticity in the nucleus accumbens: silent synapse and beyond. Neuropharmacology. 2011;61:1060–1069. doi: 10.1016/j.neuropharm.2010.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Belin D, Jonkman S, Dickinson A, Robbins TW, Everitt BJ. Parallel and interactive learning processes within the basal ganglia: Relevance for the understanding of addiction. Behav. Brain Res. 2009;199:89–102. doi: 10.1016/j.bbr.2008.09.027. [DOI] [PubMed] [Google Scholar]
- 17.Setlow B, Holland PC, Gallagher M. Disconnection of the basolateral amygdala complex and nucleus accumbens impairs appetitive pavlovian second-order conditioned responses. Behav Neurosci. 2002;116:267–275. doi: 10.1037//0735-7044.116.2.267. [DOI] [PubMed] [Google Scholar]
- 18.Stuber GD, et al. Excitatory transmission from the amygdala to nucleus accumbens facilitates reward seeking. Nature. 2011;475:377–380. doi: 10.1038/nature10194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pitkanen A. Connectivity of the rat amygaloid complex. In: Aggleton JP, editor. The amygdala: a functional analysis. Oxford: Oxford University Press; 2000. pp. 31–115. [Google Scholar]
- 20.Liao D, Hessler NA, Malinow R. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- 21.Mu P, et al. Exposure to cocaine dynamically regulates the intrinsic membrane excitability of nucleus accumbens neurons. J. Neurosci. 2010;30:3689–3699. doi: 10.1523/JNEUROSCI.4063-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Otaka M, et al. Exposure to cocaine regulates inhibitory synaptic transmission in the nucleus accumbens. J. Neurosci. 2013;33:6753–6758. doi: 10.1523/JNEUROSCI.4577-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suska A, Lee BR, Huang YH, Dong Y, Schluter OM. Selective presynaptic enhancement of the prefrontal cortex to nucleus accumbens pathway by cocaine. Proc Natl Acad Sci U S A. 2013;110:713–718. doi: 10.1073/pnas.1206287110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu L, et al. Role of ventral tegmental area glial cell line-derived neurotrophic factor in incubation of cocaine craving. Biological Psychiatry. 2009;66:137–145. doi: 10.1016/j.biopsych.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Theberge FR, et al. Effect of chronic delivery of the Toll-like receptor 4 antagonist (+)-naltrexone on incubation of heroin craving. Biological psychiatry. 2013;73:729–737. doi: 10.1016/j.biopsych.2012.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu L, Grimm JW, Hope BT, Shaham Y. Incubation of cocaine craving after withdrawal: a review of preclinical data. Neuropharmacology. 2004;47(Suppl 1):214–226. doi: 10.1016/j.neuropharm.2004.06.027. [DOI] [PubMed] [Google Scholar]
- 27.Sorge RE, Stewart J. The contribution of drug history and time since termination of drug taking to footshock stress-induced cocaine seeking in rats. Psychopharmacology. 2005;183:210–217. doi: 10.1007/s00213-005-0160-y. [DOI] [PubMed] [Google Scholar]
- 28.Hollander JA, Carelli RM. Cocaine-associated stimuli increase cocaine seeking and activate accumbens core neurons after abstinence. J. Neurosci. 2007;27:3535–3539. doi: 10.1523/JNEUROSCI.3667-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolf ME, Ferrario CR. AMPA receptor plasticity in the nucleus accumbens after repeated exposure to cocaine. Neurosci Biobehav Rev. 2010;35:185–211. doi: 10.1016/j.neubiorev.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Durand GM, Kovalchuk Y, Konnerth A. Long-term potentiation and functional synapse induction in developing hippocampus. Nature. 1996;381:71–75. doi: 10.1038/381071a0. [DOI] [PubMed] [Google Scholar]
- 31.Ferrario CR, et al. Alterations in AMPA receptor subunits and TARPs in the rat nucleus accumbens related to the formation of Ca(2)(+)-permeable AMPA receptors during the incubation of cocaine craving. Neuropharmacology. 2011;61:1141–1151. doi: 10.1016/j.neuropharm.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCutcheon JE, Wang X, Tseng KY, Wolf ME, Marinelli M. Calcium-permeable AMPA receptors are present in nucleus accumbens synapses after prolonged withdrawal from cocaine self-administration but not experimenter-administered cocaine. J. Neurosci. 2011;31:5737–5743. doi: 10.1523/JNEUROSCI.0350-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCutcheon JE, et al. Group I mGluR activation reverses cocaine-induced accumulation of calcium-permeable AMPA receptors in nucleus accumbens synapses via a protein kinase C-dependent mechanism. J. Neurosci. 2011;31:14536–14541. doi: 10.1523/JNEUROSCI.3625-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Purgianto A, et al. Different Adaptations in AMPA Receptor Transmission in the Nucleus Accumbens after Short vs Long Access Cocaine Self-Administration Regimens. Neuropsychopharmacology. 2013;38:1789–1797. doi: 10.1038/npp.2013.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cull-Candy S, Kelly L, Farrant M. Regulation of Ca2+-permeable AMPA receptors: synaptic plasticity and beyond. Curr. Opin. Neurobiol. 2006;16:288–297. doi: 10.1016/j.conb.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 36.Thomas MJ, Beurrier C, Bonci A, Malenka RC. Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat Neurosci. 2001;4:1217–1223. doi: 10.1038/nn757. [DOI] [PubMed] [Google Scholar]
- 37.Carroll RC, Beattie EC, von Zastrow M, Malenka RC. Role of AMPA receptor endocytosis in synaptic plasticity. Nat Rev Neurosci. 2001;2:315–324. doi: 10.1038/35072500. [DOI] [PubMed] [Google Scholar]
- 38.Waites CL, Craig AM, Garner CC. Mechanisms of vertebrate synaptogenesis. Annu Rev Neurosci. 2005;28:251–274. doi: 10.1146/annurev.neuro.27.070203.144336. [DOI] [PubMed] [Google Scholar]
- 39.Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- 40.Barria A, Malinow R. Subunit-specific NMDA receptor trafficking to synapses. Neuron. 2002;35:345–353. doi: 10.1016/s0896-6273(02)00776-6. [DOI] [PubMed] [Google Scholar]
- 41.Robinson T. Structural plasticity associated with exposure to drugs of abuse. Neuropharmacology. 2004;47:33–46. doi: 10.1016/j.neuropharm.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 42.Russo SJ, et al. The addicted synapse: mechanisms of synaptic and structural plasticity in nucleus accumbens. Trends Neurosci. 2010;33:267–276. doi: 10.1016/j.tins.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carlezon WA, Jr, Duman RS, Nestler EJ. The many faces of CREB. Trends Neurosci. 2005;28:436–445. doi: 10.1016/j.tins.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 44.Shaham Y, Shalev U, Lu L, De Wit H, Stewart J. The reinstatement model of drug relapse: history, methodology and major findings. Psychopharmacology. 2003;168:3–20. doi: 10.1007/s00213-002-1224-x. [DOI] [PubMed] [Google Scholar]
- 45.Marchant NJ, Li X, Shaham Y. Recent developments in animal models of drug relapse. Curr Opin Neurobiol. 2013 doi: 10.1016/j.conb.2013.01.003. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hollander JA, Carelli RM. Abstinence from cocaine self-administration heightens neural encoding of goal-directed behaviors in the accumbens. Neuropsychopharmacology. 2005;30:1464–1474. doi: 10.1038/sj.npp.1300748. [DOI] [PubMed] [Google Scholar]
- 47.Voorn P, Vanderschuren LJ, Groenewegen HJ, Robbins TW, Pennartz CM. Putting a spin on the dorsal-ventral divide of the striatum. Trends Neurosci. 2004;27:468–474. doi: 10.1016/j.tins.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 48.Martin M, Chen BT, Hopf FW, Bowers MS, Bonci A. Cocaine self-administration selectively abolishes LTD in the core of the nucleus accumbens. Nat Neurosci. 2006;9:868–869. doi: 10.1038/nn1713. [DOI] [PubMed] [Google Scholar]
- 49.Mameli M, et al. Cocaine-evoked synaptic plasticity: persistence in the VTA triggers adaptations in the NAc. Nat Neurosci. 2009;12:1036–1041. doi: 10.1038/nn.2367. [DOI] [PubMed] [Google Scholar]
- 50.Loweth JA, Tseng KY, Wolf ME. Using metabotropic glutamate receptors to modulate cocaine's synaptic and behavioral effects: mGluR1 finds a niche. Current opinion in neurobiology. 2013 doi: 10.1016/j.conb.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pilpel N, Landeck N, Klugmann M, Seeburg PH, Schwarz MK. Rapid, reproducible transduction of select forebrain regions by targeted recombinant virus injection into the neonatal mouse brain. Journal of neuroscience methods. 2009;182:55–63. doi: 10.1016/j.jneumeth.2009.05.020. [DOI] [PubMed] [Google Scholar]
- 52.Lobo MK, et al. Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science. 2010;330:385–390. doi: 10.1126/science.1188472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marie H, Morishita W, Yu X, Calakos N, Malenka RC. Generation of silent synapses by acute in vivo expression of CaMKIV and CREB. Neuron. 2005;45:741–752. doi: 10.1016/j.neuron.2005.01.039. [DOI] [PubMed] [Google Scholar]
- 54.Kamboj SK, Swanson GT, Cull-Candy SG. Intracellular spermine confers rectification on rat calcium-permeable AMPA and kainate receptors. The Journal of physiology. 1995;486(Pt 2):297–303. doi: 10.1113/jphysiol.1995.sp020812. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.