

Summary
The J‐LEAPS vaccines contain a peptide from β‐2‐microglobulin covalently attached to disease‐related peptides of 8–30 amino acids which contain a T cell epitope. The J‐LEAPS vaccines can initiate a protective Th1 immune response or modulate an ongoing Th17 autoimmune response to the peptide. J‐LEAPS vaccines activate and direct the nature of the subsequent immune response by promoting the maturation of precursor cells into a unique type of dendritic cell that produces interleukin 12, but not IL‐1 or tumour necrosis factor, and presents the antigenic peptide to T cells. Adoptive transfer of JgD‐LEAPS dendritic cells, matured with an anti‐HSV‐1 vaccine, promoted antigen‐specific Th1 protection against lethal challenge with the virus. J‐LEAPS peptide immunogens and J‐LEAPS dendritic cell vaccines have potential applications for antimicrobial prevention and therapy, treatment of autoimmune diseases, and for cancer immunotherapy.
Introduction
The Ligand Epitope Antigen Presentation System (LEAPS) is a vaccine technology platform capable of activating small peptides to become immunogens and simultaneously, directing the nature of the subsequent immune response (Zimmerman and Rosenthal, 2005). Basically, a peptide, consisting of an immune cell‐binding ligand (ICBL), is combined with a T cell antigenic peptide connected through a triglycine linker. The ICBL consists of a small peptide (approximately 13 amino acids) from proteins that interact with immune cells (Table 1). Peptides from the β‐2‐microglobulin component of MHC I [(aa 38–50) (DLLKNGERIEKVE) (J‐ICBL)] (Parham et al., 1983)], β chain of MHC II [(aa 135–149) (NGQEEKAGVVSTGLI) (G‐ICBL)] (Cammarota et al., 1992; Konig et al., 1992; Zhou and Konig, 2008) and IL‐1β[(aa 163–171) (VQGEESNDK) (F‐ICBL)] (Zimmerman et al., 1996a,b)] were chosen for their reported ability to interact with receptors on leucocytes and involvement in immune responses. These ICBLs promote immune responses when attached to antigenic peptides to make the LEAPS vaccine.
Table 1.
Leaps vaccine peptides.
| ICBL – gly–gly–gly – antigenic peptide | |||
|---|---|---|---|
| Source | Protein | Peptide | Sequence |
| Immune cell‐binding ligands | |||
| J | β‐2‐Microglobulin | 38–50 | DLLKNGERIEKVE |
| G | MHC II β chain | 135–149 | NGQEEKAGVVSTGLI |
| F | IL‐1β | 163–171 | VQGEESNDK |
| Infectious diseases | |||
| HIV‐1 B | p17 HGP‐30 | 85–115 | YSVHQRIDVKDTKEALEKIEEEQNKSKKKA |
| Mycobacteria tuberculosis | 38 kDa protein | 350–369 | DQVHFQPLPPAVVKLSDAL |
| MktA.1 | WYPHYAWLL | ||
| Herpes simplex virus 1 | ICP27 | 322–332 | LYRTFAGNPRA |
| Glycoprotein B | 498–505 | SSIEFARL | |
| Glycoprotein D | 8–23 | SLKMADPNRFRGKDLP | |
| Immunotherapies | |||
| Rheumatoid arthritis | Human collagen type 2 | 254–273 | TGGKPGIAGFKGEQGPKGEP |
| Myocarditis | Mouse cardiac myosin | 334–352 | DSAFDVLSFTAEEKAGVYK |
The J‐LEAPS vaccines activate and direct the nature of the subsequent immune response by simultaneously promoting the maturation of precursor cells into dendritic cells (DCs) which produce interleukin 12 (IL‐12) and present antigenic peptide to T cells (Taylor et al., 2010a,b). This both initiates and directs the nature of the T cell response towards the antigenic component of the vaccine. The resulting antigen‐specific Th1 response can provide antimicrobial prophylaxis or an immunomodulating influence on an ongoing immune response (Zimmerman et al., 2010). This review will focus on the activities of the J‐ICBL and vaccines developed from this peptide.
Initiation of an immune response
The goal of a vaccine is to trick the immune system into thinking that it sees an infection and then elicit the desired immune response in a safe manner. A normal immune response is initiated by innate responses that transition to antigen‐specific immune responses. The key actors for the antigen‐specific response are the DC and T cell. DCs sense molecular danger signals related to infection or tissue damage and sample the tissue environment for antigens (Medzhitov, 2008). Microbial cell wall components [e.g. LPS (MPL or lipid A), LTA, peptidoglycan], nucleic acids (CpG, double‐strand RNA, DNA) or cellular components (e.g. heat shock proteins, adenosine) are recognized by Pathogen Pattern Receptors (PPRs) [e.g. Toll‐like receptors (TLRs), RIG‐1a (retinoic acid inducible gene‐1a), NOD‐1 (nucleotide binding oligomerization domain‐1) and NOD‐2] (Koyama et al., 2007; Palm and Medzhitov, 2009) and other receptors to activate and promote the maturation of DCs (Pulendran, 2004). Some adjuvants act in this manner (Coffman et al., 2010).
Maturation of the DC creates a better antigen‐presenting cell with enhanced expression of MHC II molecules, B7 co‐receptors, and a decrease in expression of the PPRs (Czerniecki et al., 1997; Banchereau and Steinman, 1998; Cheong et al., 2010; Castiello et al., 2011). The DC will proteolytically process proteins that it has phagocytized and present them as peptides (11–13 amino acids or longer) to CD4 T cells on MHC II molecules. DCs also present peptides of 8–9 amino acids from phagocytized proteins (cross‐presentation) or from cytoplasmic proteins on MHC I molecules to CD8 T cells. Activation of the T cell requires a combination of interactions between the peptide : MHC complex on the DC with the T cell receptor (TCR) and co‐receptors, adhesion proteins and cytokines.
Cytokines produced by the DC determine whether the immune response will favour a combination of cell‐mediated and antibody responses (Th1), a humoral response (Th2), neutrophil directed inflammatory responses (Th17) or suppressive responses (Treg) (Iwasaki and Medzhitov, 2010). Lipopolysaccharide is an excellent inducer of acute phase cytokines (IL‐1, TNFα and IL‐6) and IL‐12p70, a dimer of p40 and p35 subunits (Snijders et al., 1998; Mosca et al., 2000; Hochrein et al., 2001). Acute phase cytokines plus TGFβ can initiate a Th17 response whereas IL‐12 can initiate and promote a Th1 response. Other microbial stimuli promote the production of acute phase cytokines and IL‐23. IL‐23 utilizes the same p40 subunit as IL‐12 but with a p19 subunit (Kikly et al., 2006). IL‐23 promotes a Th17 response from memory CD4 T cells (Lee et al., 2009a,b). Th2 responses are promoted by DCs and other factors in the absence of IL‐12 or IL‐23.
Th1 immune responses
Th1 immune responses are characterized by the production of interleukin 2 (IL‐2), interferon γ (IFNγ) and lymphotoxin [TNF β (LT)] by CD4 and CD8 T cells (Mosmann and Sad, 1996; Pulendran, 2004). These cytokines promote cell‐mediated and antibody responses which are important for reinforcing local immune responses and controlling intracellular infections and tumour cells. Th2 and Treg responses inhibit the development of Th1 responses. IL‐2 promotes the growth and activation of lymphocytes to expand the immune response. IFNγ, once known as macrophage activation factor, defines the nature of the Th1 immune response by activating DCs, macrophages and other myeloid cells, promoting class switch of B cells to produce IgG and inhibiting the development of Th2 responses. The Th1 response also activates CD8 T killer cells. T cells generated by a Th1 response are also more sensitive to antigen than Th2 T cells and are more likely to detect rare tumour antigen peptide–MHC I complexes on cancer cells (Czerniecki et al., 2007).
Peptide vaccines
Development of a peptide vaccine allows selection of T cell epitopes that elicit protective rather than immunopathogenic or inhibitory responses. Immunologically active peptides have sequences that are selected for their ability to pass through the transporter associated with processing (TAP), bind to specific MHC I or II molecules (agretope) and also express antigenic specificities to the TCR (epitope). Helper and cytotoxic T cell epitopes with demonstrated biological activity are described in the literature and were the source of the sequences incorporated into the LEAPS vaccines described herein. In addition, computer analysis of peptide sequences can identify and optimize sequences within target proteins that fulfil the biological properties for them to be potential T cell epitopes (De Groot et al., 2008) and were used to identify sequences from Mycobacterium tuberculosis (Mtb) (Walton et al., 2008) for a J‐LEAPS immunogen.
The requirement for MHC presentation of peptides to T cells limits the population that will be able to respond to that peptide. For mice, most peptides are chosen based on their binding to H2d (BALB/c) or H2b (C57Bl/6) mice. For humans, most peptides are chosen based on their binding to HLA A2, which represents 40% of the population (Buteau et al., 2002). Mixtures of peptides can be used to broaden the population that can respond and the antigenic target of the response.
In order to make the peptide immunogenic, it must be multimerized or attached to a carrier‐like molecule to be presented by DC to T cells (Melief and Van der Burg, 2008). Classically, peptides were attached to protein carriers such as keyhole limpet haemocyanin (KLH). This approach elicits a Th2 response which elicits an antibody response. Other means for activating the immunogenicity of the peptide (Melief and Van der Burg, 2008), including the LEAPS platform (Goel et al., 2005; Rosenthal, 2005; Zimmerman and Rosenthal, 2005), have been described more recently. Steering the nature of the immune response to the peptide towards a Th1 response and eliciting CTLs are additional challenges for designing a peptide vaccine. Th1 immune responses have been elicited to peptides with adjuvants (Coffman et al., 2010) or by attachment of the peptide to the J‐LEAPS ICBL (see below), to TLR agonists, such as flagellin (Coffman et al., 2010), or other carriers (Rosenthal, 2005; Melief and Van der Burg, 2008).
Th1 immune responses induced by J‐LEAPS peptide vaccines
The initial LEAPS vaccines (Zimmerman et al., 1996a,b) attached the immunodominant and Mtb‐specific T cell‐stimulatory epitope from the 38 kDa antigen (amino acids 350–369) (Vordermeier et al., 1992) to various ICBLs. This peptide includes CD4 and CD8 T cell and B cell epitopes (Table 1). The success of the immunization was monitored by production of antibody and as such, the initial results after immunization with the LEAPS vaccines were disappointing. No specific antibody production was detected against the Mtb antigen or the ICBL but there was an increase in the background response suggesting a generic immune activation, a heteroclictic response. The true nature of the LEAPS vaccine stimulation became known when the mice were boosted by immunization with the M protein coupled to KLH. The strength and antibody subtype generated by immunization with the protein booster resembled that of a secondary response with a preponderance of IgG rather than IgM. More interestingly, the IgG subtype ratio differed depending upon the ICBL attached to the antigenic M peptide. For the J‐ICBL, the ratio favoured the IgG2a subtype whereas for the G‐ICBL, it favoured IgG1 and IgG3, while for the F‐ICBL, there was no preference of IgG subtype produced. In the mouse, a preference for production of IgG2a indicates a Th1 response whereas IgG1 and IgG3 indicates a Th2 type of immune response. These results suggested that immunization with the LEAPS vaccines initiated the activation of helper T cells and the J‐, G‐ and F‐ICBLs defined the type of T cell and hence, the cytokine help provided to the B cells to make IgG. The J‐ICBL appeared to promote a Th1 response, the G‐ICBL a Th2 response and the F‐ICBL activates T cells without a clear definition of the nature of the response. Unlike for carrier proteins like KLH, no detectable immune response was elicited against the ICBLs.
JH vaccines to HIV
An important initial target for LEAPS vaccine development was HIV. Major challenges for development of an anti‐HIV vaccine are the large number of antigenic variants of HIV in the population, the genetic instability and antigenic variability of the virus, and the limited efficacy of antibody in protecting and controlling virus infection. A T cell response should be less variable than an antibody response (Allen et al., 2005). A 30‐amino‐acid peptide (‘H’) containing a conserved region of HIV‐1 p17 gag protein with overlapping CD4 T cell, CD8 T cell and B cell epitopes (HGP‐30 peptide) was attached to either the J‐ (JH; LEAPS 102) or the G‐ICBL (GH; LEAPS 101) (Zimmerman et al., 2001; Pisarev et al., 2002). Antibody was obtained after immunization with JH and GH as an oil in water emulsion in incomplete Freund's, MPL‐SE or Seppic ISA51 adjuvants but not when mixed with alum. GH‐immunized mice produced antibody levels similar to those immunized with an ‘H’ peptide conjugate to KLH and much more antibody than from JH‐immunized mice. Unlike the KLH conjugates which produced a mixture of antibody to HGP‐30 and KLH, little or no antibody was elicited towards the J‐ or G‐ICBLs. These results parallel those obtained for LEAPS Mtb and herpes simplex virus (HSV) vaccines in which the G‐ICBL promotes antibody production much better than the J‐ICBL. Better antibody production than for other LEAPS vaccines is likely due to the presence of helper T cell epitopes with the B cell epitopes within the H‐HIV peptide. Study of antibody production to the H peptide provided insight into the nature of the immune response to LEAPS vaccines although the therapeutic benefit of antibody to the H peptide is questionable since it would not neutralize the virus.
Anti‐HSV vaccines
The J‐LEAPS, G‐LEAPS and F‐LEAPS immunogens appeared to activate a response but could they act as a vaccine to elicit a protective response against disease challenge. This was testable using LEAPS vaccines against HSV infection of mice. For HSV, antigen‐specific Th1 immune responses elicit protection from disease whereas Th2 responses may exacerbate disease by inhibiting the development of Th1 responses (Jayaraman et al., 1993). J‐LEAPS and G‐LEAPS vaccines were prepared with T cell epitopes from the ICP27 protein (H1) (Rosenthal et al., 1999), glycoprotein B (gB) (Goel et al., 2005) and glycoprotein D (gD) (Goel et al., 2003) of herpes simplex virus type 1 (HSV‐1). J‐LEAPS vaccines elicited protection from morbidity and mortality whereas the G‐LEAPS vaccines did not.
Immunization with JH1 elicited protection from lethal challenge of BALB/c mice with HSV‐1. The H1 epitope from the ICP27 intracellular protein is the target of a CD8 cytotoxic T cell response in BALB/c mice (Banks et al., 1991; 1993; Rosenthal et al., 1999). Antibody was not detected and antibody to ICP27 would not be protective against HSV disease. DTH responses were present in the immunized, unchallenged mice consistent with the presence of antigen‐specific activated T cells and a Th1 response. Immunization with the GH1 vaccine either had no effect or exacerbated the disease, consistent with eliciting a Th2 response to the infectious challenge. Similar findings were obtained with the JgB peptide vaccine in C57BL/6 mice. The gB peptide is the predominant CTL epitope for this strain of mouse (Goel et al., 2005). The studies with JH1 and JgB proved the principle that a minimal T cell epitope (8–9 amino acids) could be attached to the J‐ICBL and confer protection. In addition, these studies demonstrated that LEAPS peptides activate T cell responses and these responses are sufficient to confer protection from disease and death.
The next LEAPS anti‐HSV vaccine utilized a peptide from the N‐terminus of HSV‐1 gD that is longer than the H1 or gB peptides and includes T and B cell epitopes (Goel et al., 2003). This peptide has sequence homology between HSV‐1 and HSV‐2 and is recognized by mouse and man (Cohen et al., 1984; Eisenberg et al., 1985; Damhof et al., 1993). Immunization with JgD, but not GgD, emulsified in Seppic ISA 51 elicited protection from disease and death in inbred BALB/c, C57BL/6, C3H, FVB mice and outbred Swiss Webster, CD1 and SKH1 mice. The GgD vaccine did not elicit protection. As for JH1 and JgB, antibody was not necessary for protection. Antibody was only observed if there was sufficient virus breakthrough disease to generate protein to elicit a B cell response.
JgD activation of anti‐HSV response
How does the J‐ICBL within JgD enhance the immunogenicity and direct the nature of the immune response to the antigenic peptide? Does it activate a Th1 immune response by binding to antigen‐specific T cells to promote a direct stimulation of T cells or through an antigen‐presenting cell? What is the role of the adjuvant in activating the response? These questions will be addressed in the following sections.
Antibody ablation studies demonstrated that CD8‐bearing cells were important to initiate the response to JgD (and likely other J vaccines) and CD4‐bearing cells, IFNγ and CD8‐bearing cells were important for delivering protection from lethal challenge with HSV‐1 (Goel et al., 2003). In mice, CD8 is expressed on T cells and also on a DC subset (Kronin et al., 1997; 2001; Kessler et al., 1998) and either cell type could be involved in the initiation of the response.
Cytokine production in mice immunized with either JgD or JH provided a hint towards the nature of the response to these vaccines (Taylor et al., 2010a). Protein array analysis was used as a sensitive means to assay for the presence of 22 cytokines and chemokines in the blood of mice at 4, 10 and 20 days after immunization. JgD or JH was administered as an oil in water emulsion in Seppic ISA51 adjuvant. Immunization with the unconjugated J‐ICBL or the gD peptide did not elicit a significant response. The response at 4 days included IL‐12p70, IL‐12p40, and chemokines RANTES, MCP1 and MCP5 but with no increase in IL‐1 or TNFα. On day 10, the predominant responses were IL‐12p70, IL‐17, IL‐2 and IFNγ and on day 24, IL‐12p70 and IFNγ were the predominant responses.
The initial IL‐12p40, IL‐12p70, and chemokine response to JgD or JH are likely from an activated monocyte, macrophage or DC. The cytokine response indicates that a transient Th17 response subsequently matures into a Th1 response. Unlike other IL‐12p70‐associated responses, the pro‐inflammatory cytokines, IL‐1 and TNFα, were not elicited by JgD or JH treatment.
In an attempt to establish an ex vivo system to study the vaccine, spleen cells were treated with JgD, but with no response. The lack of response of spleen cells may be due to the presence of more mature forms of these cells and indicated that mature DCs, B and T cells are not targets for the J‐LEAPS vaccines. In contrast, treatment of mouse bone marrow cells resulted in a profound response. JgD or JH treatment promoted a large portion of the cells to develop dendrites and to cluster, increase expression of MHC II and CD86, and produce IL‐12p70, but not IL‐1 or TNFα. Similar results were obtained after JgD treatment of a population of bone marrow cells treated with IL‐6, Flt3 ligand and IL‐4 for 6 days and then GM‐CSF for 24 h, a process known to develop and select immature DCs (Cohen et al., 2008). The ex vivo results with JgD and JH recapitulated the serum cytokine repertoire of immunized mice.
The JgD or JH treated bone marrow cells that were producing IL‐12 also expressed CD8. This finding is consistent with the role for CD8‐expressing cells demonstrated earlier in the antibody ablation of protection studies (Goel et al., 2003). Interestingly, in the mouse, CD8‐expressing DCs are also more active at cross‐presentation of antigen to CD8 T cells (Kronin et al., 1997; 2001; Kessler et al., 1998) which would elicit a cytotoxic T cell response.
Treatment of bone marrow cells with JH (HIV vaccine) generated DCs with the same properties as for JgD. The cytokines produced in response to JH treatment were dominated by IL‐12p70 just like for JgD. These results indicate that the J‐LEAPS vaccines interact with precursors of DCs and promote their maturation into a unique type of DC that makes IL‐12 without the pro‐inflammatory cytokines.
JgD‐generated mouse DCs
The cells generated by treatment of bone marrow with JgD or JH had properties consistent with that of DCs and are termed JgD‐DC or JH‐DC depending upon the J‐LEAPS vaccine used to induce the response (Taylor et al., 2010a). If JgD‐DCs are DCs and responsible for eliciting a Th1 response, then they must be able to present antigen and boost an antigen‐specific IFNγ and IL‐2 response from T cells. JgD‐DC, washed free of unbound vaccine or cytokine, were able to induce IL‐2 and IFNγ production by spleen cells from JgD‐immunized mice (IL‐2 and IFNγ as detected by protein array and by ELISA for IFNγ). Surprisingly, JH‐DC treatment of the JgD‐immunized spleen cells also induced IL‐2 and IFNγ but the levels were below detection by ELISA. There was no response when JgD was added directly to spleen cells without bone marrow cells.
The ability of both JgD and JH‐DC to induce Th1 cytokine production from splenic T cells indicates that the J‐LEAPS‐DC can activate Th1 responses from naïve and memory T cells. The lower level of cytokine induction by JH‐DC may be the result of activation of a new T cell response or a generic activation of T cells by IL‐12‐producing DCs. The antigen‐specific booster response in IFNγ and IL‐2 production demonstrates that the JgD‐DC are antigen‐presenting cells and can steer immune responses towards Th1.
Ultimately, a DC must be able to initiate an immune response in naïve T cells and this was demonstrated by the ability of JgD‐DC to confer protection to unimmunized mice from lethal HSV‐1 infection. Mice receiving two intradermal and intraperitoneal injections (separated by 2 weeks) of JgD‐DC but not JH‐DC, J‐treated bone marrow cells, naive bone marrow cells or buffer were protected from a lethal dose of HSV‐1 administered to cause a zosteriform disease presentation. All the JgD‐DCs survived and only minor disease was observed in two out of seven mice. In contrast, mice receiving the other treatments had significant disease and most died. Only the antigen‐specific response induced by JgD‐DC was capable of inducing protection in the immunologically naïve mice. The protection did not require an adjuvant or other treatment. JgD‐DC are therefore capable of initiating an antigen‐specific immune response in naïve mice defining them as a DC and elicitation of protection is consistent with their induction of a Th1 immune response.
In summary, JgD is sufficient to promote the development of a DC population that makes IL‐12p70 but not acute phase cytokines. JgD does not directly induce cytokine responses from splenic T cells. The J‐ICBL and peptide must be conjugated but additional cytokines or an adjuvant is not required to elicit these responses. The J‐LEAPS‐DCs can activate new Th1 responses in splenic T cells which may (JgD‐DC) or may not (JH‐DC) be antigen‐specific and can initiate a new immune response that is sufficient to elicit protection as a vaccine.
Human DCs
Having shown that JgD and JH are capable of converting mouse precursors into DCs, the ability of these J‐LEAPS vaccines to convert human precursors into DCs was examined (Taylor et al., 2010b). IL‐12‐producing DCs can be generated by treatment of purified monocytes with IL‐4 and then GM‐CSF and Flt‐3 ligand or other combinations (Koski et al., 2001; 2004; Shuwen et al., 2003; Napolitani et al., 2005; Czerniecki et al., 2007; Roses et al., 2008; Cheong et al., 2010; Castiello et al., 2011; Paustian et al., 2011). JgD or JH treatment of human monocytes generated DCs within 72 h (Taylor et al., 2010b). Treatment of monocytes produced DCs (JgD‐hmDC; JH‐hmDC) that changed from individual and round cells to clumped cells with dendritic processes with increased expression of CD86 and HLA‐DR. As for the mouse, the LEAPS‐hmDC produce IL‐12p70 but not IL‐1 or TNFα. The conversion occurred whether the monocytes were pre‐treated or co‐treated with GM‐CSF or pre‐selected to become immature DCs by treatment with GM‐CSF and IL‐4. In addition, similar results were obtained for cells from three donors of different HLA type. Interestingly, the cytokine profile produced by the LEAPS‐hmDCs generated from GM‐CSF and IL‐4 pre‐selected monocyte differed from LEAPS‐hmDCs generated from monocytes or GM‐CSF‐treated monocytes (Table 2). This implies that treatment of monocytes with GM‐CSF plus IL‐4 generates a different precursor than those treated with only GM‐CSF such that the J‐LEAPS‐DC that are generated have different functions and potential roles in promoting immune responses.
Table 2.
Cytokine production by JgD‐treated human monocytes and immature DCs.
| Cytokine | Monocyte | GMCSFa | GMCSF + IL‐4a |
|---|---|---|---|
| IL‐12p70 | +++ | +++ | +++ |
| MCP‐1b | +++ | +++ | + |
| MCP‐2 | +++ | +++ | ++ |
| RANTES | ++ | ++ | ++ |
| PDGF‐BB | ++ | ++ | ++ |
| ENA‐78 | + | + | + |
| MIG | + | + | + |
| MIP‐1 delta | + | + | ++ |
| MCSF | + | + | ++ |
| MDC | + | + | ++ |
| Angiogenin | + | + | ++ |
| Oncostatin | + | + | ++ |
| TARC | ∼c | ∼ | + |
| VEGF | ∼ | ∼ | ++ |
| GCSF | 0.00 | 0.00 | ++ |
| IL‐1α | 0.00 | 0.00 | + |
| IL‐10 | 0.00 | 0.00 | + |
| TNFα | 0.00 | 0.00 | ∼ |
| IL‐1β | 0.00 | 0.00 | ∼ |
Monocytes either received no treatment or received treatment with either GM‐CSF or GM‐CSF and IL‐4 for 24 h prior to addition of JgD.
MCP‐1 and ‐2, monocyte chemoattractant proteins; RANTES, regulated upon activation normal T cell express sequence; PDGF‐BB, platelet‐derived growth factor; MIP‐1 delta, macrophage inflammatory protein‐1 delta; ENA‐78, epithelial neutrophil‐activating peptide 78; MCSF, macrophage colony‐stimulating factor; MDC, macrophage‐derived chemokine; MIG, monokine induced by interferon γ; TARC, thymus and activation‐regulated chemokine; VEGF, vascular endothelial growth factor; GCSF, granulocyte colony‐stimulating factor; EGF, epidermal growth factor, GMCSF, granulocyte macrophage colony‐stimulating factor.
The symbol ‘∼’ signifies low but detectable levels.
The JgD‐hmDC were also able to present antigen to T cells and initiate a Th1‐biased immune response. Upon mixing of JgD‐hmDC from one donor with T cells from another donor, an allogenic response was elicited from the T cells that generated IL‐2 and IFNγ over the course of 6 days. Since DCs, not macrophages, are required to initiate such an allogenic response (Roses et al., 2008), the JgD‐hmDC not only look like DCs, produce cytokines like DCs, but they also behave like DCs.
Immunotherapy with LEAPS vaccines
The pathogenesis of autoimmune diseases, like many infectious diseases, is mediated by inflammatory responses that are maintained or enhanced by T cells and cytokines. The ability of J‐LEAPS vaccines to generate IL‐12‐producing DCs could potentially modulate an ongoing deleterious immune response. Many autoimmune diseases appear to be mediated by Th17 responses which generate TNFα, activate neutrophils and promote inflammation and tissue remodelling (Shahrara et al., 2008; Nistala and Wedderburn, 2009).
Rheumatoid arthritis (RA) is an example of an autoimmune disease that is driven by Th17 responses (Shahrara et al., 2008). TNFα antagonists, such as etanercept, are used to treat this disease (Van Vollenhoven, 2009). In a mouse model of RA, immunization with a J‐LEAPS vaccine, which incorporates a peptide from human collagen (CEL‐2000), was able to stop the progression of disease as well as, or better than, etanercept with much fewer treatments (Zimmerman et al., 2010).
CEL‐2000 consists of the J‐ICBL attached to a peptide from human collagen type II (amino acids 254–273). RA‐like disease was induced in these mice by injection of bovine collagen in complete Freund's adjuvant followed by bovine collagen in incomplete Freund's adjuvant. Treatment was initiated when disease signs were evident. By day 28, untreated mice were incapacitated with all joints involved in the disease process. Treatment with CEL‐2000 as an oil in water emulsion with either the MAS‐1 or the Seppic ISA‐51 adjuvant limited disease progression as demonstrated by the low number of affected joints and less footpad swelling. Efficacy was confirmed by histopathological microscopic examination of tissues. Disease progression ceased in mice receiving etanercept every other day or CEL‐2000 on days 0, 14, 29, 42 and 56. Analysis of blood from these mice indicated that both etanercept treatment and CEL‐2000 treatment caused a reduction in IL‐17, IL‐6 and TNFα as well as MCP1 and IL‐12p40. IL‐12 production was increased for CEL‐2000‐treated mice and to a much lesser extent for etanercept‐treated mice. A decrease in IL‐12p40 with an increase in IL‐12p70 (p40 + p35) suggests a large decrease in IL‐23 (p40 + p19) which would reduce the activation of Th17 cells and decrease the inflammation.
Immunomodulation of an ongoing Th17 response is likely to be the means by which CEL‐2000 treatment curtails the disease progression in the mouse model of RA. The rapid cessation of disease progression is likely due to the production of IL‐12 upon treatment with CEL‐2000. Antigen‐specific interactions between autoimmune T cells and CEL‐2000‐DCs would also influence cytokine production and inflammation. The Th17 response is plastic and upon exposure to IL‐12p70, the T cells will downregulate production of IL‐17 which would stop the promotion of the disease‐defining inflammation (Lexberg et al., 2008; Lee et al., 2009a,b).
Unlike ablative (Šenolt et al., 2009) or antagonistic anti‐cytokine treatments such as ustekinumab (neutralizing IL‐12/23 antibody) (Segal et al., 2008) or etanercept (Van Vollenhoven, 2009), the CEL‐2000 approach converts the pathogenic immune response into a different response. Although the mice tolerated multiple treatments with CEL‐2000 extremely well, the nature of this alternative response is not known, as yet.
Immunization with a J‐LEAPS vaccine (J‐My‐1) that incorporates the myocarditogenic peptide of murine cardiac myosin (MuCMα334‐352) (My‐1) also blocks the initiation and progression of autoimmune cardiomyositis in a mouse model of the disease (Cihakova et al., 2008). Prophylactic immunization with J‐My‐1 [on days −14 and −7 before experimental autoimmune myocarditis (EAM) induction] or therapeutic immunization with J‐My‐1 (J‐My‐1 injected on days 7, 14 and 21 or 10, 17 and 24) significantly decreased the incidence and severity of EAM as indicated by histopathology or heart weight. This is another example of J‐LEAPS vaccines immunomodulation of autoimmunity.
How do J‐LEAPS vaccines induce immunity?
The J‐ICBL promotes Th1 responses for peptides that contain a T cell epitope and are sufficient, without additional cytokines or adjuvants, to activate precursor cells to mature into a unique type of DC. The LEAPS‐DC makes IL‐12p70, but not acute phase cytokines, presents the antigenic peptide to T cells, and can initiate a new antigen‐specific response and boost an established response. Attachment of the J‐ICBL to antigen directs a different response than the G‐ICBL or the F‐ICBL suggesting its interaction with a specific receptor. The G‐ICBL is known to interact with CD4 molecules (Konig et al., 1992) but the receptor for the J‐ICBL is not known. The (DLLKNGERIEKVE) peptide from β‐2‐microglobulin in the J‐ICBL is near the site of binding to CD8 (Meijers et al., 2005) but may interact with another cell surface β‐2‐microglobulin receptor on the DC precursor such as CD85 or another LIR molecule (Borges et al., 1997). CD4 and CD8 molecules are expressed on murine DC subsets but a CD8 analogue has not been detected on human DCs. A search is on for other receptors for the J‐peptide and the activation pathways induced by the binding of the J‐LEAPS vaccines.
The ICBL must be covalently linked to the antigenic peptide since neither the individual components, nor combinations of the individual components elicit the response. Binding to and cross‐linking of cell surface receptors for the ICBL and the T cell epitope (MHC molecule) is likely to activate the maturation process.
The maturation of mouse bone marrow cells or human monocytes to antigen‐presenting DCs may require multiple steps generated by receptor interactions and require continuous exposure to vaccine for at least 24 h. This would be consistent with early findings that slow release of vaccine from an adjuvant oil‐in‐water emulsion, but not phagocytosis of an alum complex of vaccine, is required to immunize mice.
Once initiated, the IL‐12p70‐producing DC can steer cytokine production by T cells towards Th1‐related IFNγ and IL‐2 and also initiate antigen‐specific T cell responses (Fig. 1). As demonstrated for the JH‐DC interaction with JgD‐immunized splenic T cells, antigen‐specific interactions may not be required to induce production of IFNγ and IL‐2 but as demonstrated in the adoptive transfer studies, antigen‐specific interactions are required to elicit an immune response sufficient for protective immunity. The ability of CEL‐2000 to prevent the onset of RA symptoms in the mice is likely to result from a combination of an antigen non‐specific IL‐12p70 modulation and an antigen‐specific alteration in the ongoing autoimmune Th17 responses.
Figure 1.
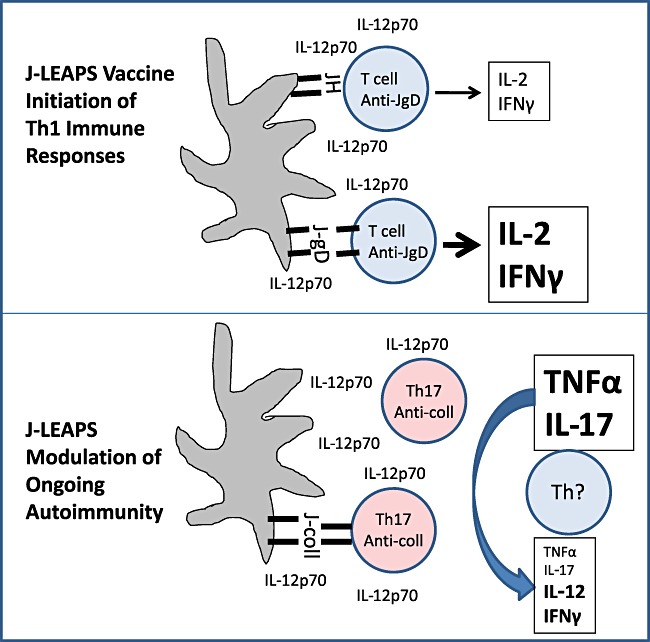
Models for J‐LEAPS vaccine initiation of Th1 immune responses and modulation of ongoing autoimmunity. J‐LEAPS vaccines induce the differentiation of precursors into IL‐12‐producing DCs that are capable of promoting Th1‐related cytokines (IL‐2, IFNγ) production from naïve T cells and as an antigen‐specific boost in response. DCs generated by CEL‐2000 (J‐coll) modulate the ongoing Th17 autoimmune response in rheumatoid arthritic mice to block the progression of autoimmune disease.
The immune response elicited by the J‐LEAPS vaccines is restricted to T cell responses. As demonstrated for HSV, this is sufficient to elicit protection from disease. As for DNA vaccines and defective adenovirus hybrid vaccines which prime the immune system, antibody production following J‐LEAPS vaccination requires administration of an antigenic protein boost (Woodland, 2004; Lu, 2009).
Other applications of LEAPS vaccines and LEAPS‐DCs
Most approaches to enhance peptide immunogenicity promote only antibody production. J‐LEAPS peptide vaccines have the unique capability to activate small peptides into immunogens that elicit IL‐12 production, without acute phase cytokines, to induce protective Th1 responses and immunomodulate ongoing responses to prevent disease. The J‐LEAPS peptides have defined sequences, are synthetically produced, rather than purified from biological mixtures, and can be administered as a predetermined mixture to include several epitopes and to accommodate different MHC types (Rosenthal, 2005; Zimmerman and Rosenthal, 2005). These properties can be utilized for many potential applications, some of which are described below. Among the directions that have already been pursued are vaccines for infectious disease targets other than HSV, a cytokine‐based blood‐screening test for tuberculosis (Walton et al., 2010), and therapies for autoimmune diseases.
The criteria for incorporation of a peptide into a LEAPS vaccine are: length of 8–30 amino acids containing potential T cell epitopes that are involved in CTL or neutralizing antibody formation. The peptides must also behave well in solution and not dimerize, oxidize or decompose under physiological conditions.
Cytokine blood test for Mtb
A J‐LEAPS immunogen containing peptides from Mtb was used to develop an assay that can discriminate between infection with Mtb and immunization with Bacillus Calmette Guerin (BCG) (Walton et al., 2010). The tuberculin skin test cannot make this distinction. Commercially available assays capable of identifying and distinguishing Mtb‐infected people from BCG‐immunized people detect the production of IFNγ upon incubation of human blood with a mixture of ESAT‐6 and CFP‐10 proteins (Brock et al., 2004). These proteins are produced by Mtb and not BCG. Walton et al. used computer programs to identify peptides unique to Mtb that have the potential to be processed and presented on murine H2 or human HLA molecules to CD8 T cells. Attachment of one of these peptides, the Mtb.K1 epitope, to the J‐ICBL created an antigen (J‐Mtb.K1) that selectively elicits IFNγ production upon treatment of blood from Mtb‐immunized mice. Little or no response was obtained upon treatment of blood cells from BCG‐immunized mice. This assay also utilizes a unique detection system for a positive reaction.
DC vaccines
Although it can be dangerous to translate immunological results obtained in mice to humans, the generation of J‐LEAPS‐hmDCs that can drive a Th1 allotypic immune response support the prophylactic and therapeutic potential of the LEAPS technology in humans. Use of J‐LEAPS‐hmDCs would provide personalized and potent immunotherapy for cancer and other diseases. Adoptive transfer of autologous DCs generated from monocytes and loaded with antigen ex vivo is being used for cancer immunotherapy (Palucka et al., 2011). Vaccines that elicit Th1 T cell immune responses are very important for anti‐cancer therapy since Th1‐driven T cells are more sensitive to antigen than Th2 T cells and are better for anti‐cancer therapy (Czerniecki et al., 2007). Treatment by adoptive transfer of J‐LEAPS‐hmDCs would minimize the exposure of the individual to the vaccine peptide and abrogate the need for use of an adjuvant or incipient. Preparation of DCs with a mixture of J‐LEAPS vaccines would ensure coverage for different MHC types and also different antigenic targets. For anti‐cancer immunotherapy, the procedure for producing DCs would be simpler, more chemically defined than the current cytokine and TLR agonist approaches (Castiello et al., 2011) and could reduce the time between obtaining monocytes and treatment of the patient to 24 h, potentially making this an overnight procedure.
Conclusion
The ability of J‐LEAPS‐DCs to activate Th1 T cell immune responses has potential for prevention of infections by other viruses, intracellular bacteria or parasites (e.g. leishmania) and as therapy for chronic infections, such as with Mtb, HIV or CMV. Similarly, this approach could be used for immunomodulatory treatment of ongoing autoimmune disease.
In developed countries, the hygiene hypothesis (Okada et al., 2010) suggests that immunity in individuals favours Th2 responses and associated allergic and asthmatic disease rather than Th1 immune responses. J‐LEAPS vaccines and J‐LEAPS‐DC elicit Th1 immune responses that may be able to provide prophylaxis or therapy to rebalance or emphasize the required immune responses to combat certain diseases. As with all means of immune manipulation, the effects of J‐LEAPS vaccines on the balance of the immune system will have to be watched.
Acknowledgments
The authors thank Sarah Stone for assistance with the manuscript. Portions of this work were supported by an unrestricted grant from CEL‐Sci.
References
- Allen T.M., Altfeld M., Geer S.C., Kalife E.T., Moore C., O'Sullivan K.M. Selective escape from CD8− T‐cell responses represents a major driving force of human immunodeficiency virus type 1 (HIV‐1) sequence diversity and reveals constraints on HIV‐1 evolution. J Virol. 2005;79:13239–13249. doi: 10.1128/JVI.79.21.13239-13249.2005. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banchereau J., Steinman R. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- Banks T.A., Allen E.M., Dasgupta S., Sandri‐Golden R., Rouse B.T. HSV‐1 specific cytotoxic T lymphocytes recognize immediate early protein ICP27. J Virol. 1991;65:3185–3191. doi: 10.1128/jvi.65.6.3185-3191.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks T.A., Nair S., Rouse B.T. Recognition by and in vitro induction of cytotoxic T lymphocytes against predicted epitopes of the immediate‐early protein ICP27 of herpes simplex virus. J Virol. 1993;37:613–616. doi: 10.1128/jvi.67.1.613-616.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges L., Hsu M., Fanger N., Kubin M., Cosman D. A family of human lymphoid and myeloid Ig‐like receptors, some of which bind to MHC class I molecules. J Immunol. 1997;159:5192–5196. [PubMed] [Google Scholar]
- Brock I., Weldingh K., Lillebaek T., Follmann F., Andersen P. Comparison of tuberculin skin test and new specific blood test in tuberculosis contacts. Am J Respir Crit Care Med. 2004;170:65–69. doi: 10.1164/rccm.200402-232OC. [DOI] [PubMed] [Google Scholar]
- Buteau C., Markovic S.N., Celis E. Challenges in the development of effective peptide vaccines for cancer. Mayo Clin Proc. 2002;77:339–349. doi: 10.4065/77.4.339. [DOI] [PubMed] [Google Scholar]
- Cammarota G., Scheirle A., Takacs B., Doran D., Knorr R., Bannwarth W. Identification of a CD4 binding site on the beta 2 domain of HLA‐DR molecules. Nature. 1992;356:799–801. doi: 10.1038/356799a0. et al. [DOI] [PubMed] [Google Scholar]
- Castiello L., Sabatino M., Jin P., Clayberger C., Marincola F., Krensky A., Stroncek D.F. Monocyte‐derived DC maturation strategies and related pathways: a transcriptional view. Cancer Immunol Immunother. 2011;60:457–466. doi: 10.1007/s00262-010-0954-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong C., Matos I., Choi J.H., Dandamudi D.B., Shrestha E., Longhi M.P. Microbial stimulation fully differentiates monocytes to DC‐SIGN/CD209 dendritic cells for immune T cell areas. Cell. 2010;143:416–429. doi: 10.1016/j.cell.2010.09.039. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cihakova D., Barin J.G., Baldeviano G.C., Kimura M., Talor M.V., Zimmerman D.H. L.E.A.P.S. heteroconjugate is able to prevent and treat experimental autoimmune myocarditis by altering trafficking of autoaggressive cells to the heart. Int J Immunopharmacol. 2008;8:624–633. doi: 10.1016/j.intimp.2008.01.004. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffman R.L., Sher A., Seder R.A. Vaccine adjuvants: putting innate immunity to work. Immunity. 2010;33:492–503. doi: 10.1016/j.immuni.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen G.H., Dietzschold B., Ponce de Leon M., Long D., Golub E., Varrichio A. Localization and synthesis of an antigenic determinant of herpes simplex virus glycoprotein D that stimulates the production of neutralizing antibody. J Virol. 1984;49:102–108. doi: 10.1128/jvi.49.1.102-108.1984. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P., Koski G., Czerniecki B., Bunting K., Fu X‐Y., Wang Z. Stat 3 and Stat 5 dependent pathways competitively regulate the pan differentiation of CD 34 pos cells into tumor competent dendritic cells. Blood. 2008;112:1832–1843. doi: 10.1182/blood-2007-12-130138. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czerniecki B., Carter C., Rivoltini L., Koski G., Kim H., Weng D. Calcium ionophore‐treated peripheral blood monocytes and dendritic cells rapidly display characteristics of activated dendritic cells. J Immunol. 1997;159:3823–3837. et al. [PubMed] [Google Scholar]
- Czerniecki B., Koski G., Koldovsky U., Xu S., Cohen P., Mick R. Targeting HER‐2/neu in early breast cancer development using dendritic cells with staged interleukin‐12 burst secretion. Cancer Res. 2007;67:1842–1852. doi: 10.1158/0008-5472.CAN-06-4038. et al. [DOI] [PubMed] [Google Scholar]
- Damhof R., Drijfhout J.W., Sheffer A.J., Wilterdink J.B., Welling G.W., Welling‐Wester S. T cell responses to synthetic peptides of herpes simplex virus type 1 glycoprotein D in naturally infected individuals. Arch Virol. 1993;130:187–193. doi: 10.1007/BF01319007. [DOI] [PubMed] [Google Scholar]
- De Groot A.S., McMurry J., Moise L. Prediction of immunogenicity: in silico paradigms, ex vivo and in vivo correlates. Curr Opin Pharmacol. 2008;8:620–626. doi: 10.1016/j.coph.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Eisenberg R.J., Cerini C.P., Heilman C.J., Joseph A.D., Dietzschold B., Golub E. Synthetic glycoprotein D‐related peptides protect mice against herpes simplex virus challenge. J Virol. 1985;56:1014–1017. doi: 10.1128/jvi.56.3.1014-1017.1985. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel N., Rong Q., Zimmerman D., Rosenthal K.S. A L.E.A.P.S. heteroconjugate vaccine containing a T cell epitope from HSV‐1 glycoprotein D elicits Th1 responses and protection. Vaccine. 2003;21:4410–4420. doi: 10.1016/s0264-410x(03)00429-8. [DOI] [PubMed] [Google Scholar]
- Goel N., Zimmerman D., Rosenthal K.S. Ligand epitope presentation system vaccines against herpes simplex virus. Front Biosci. 2005;10:966–674. doi: 10.2741/1591. [DOI] [PubMed] [Google Scholar]
- Hochrein H., Shortman K., Vremec D., Scott B., Hertzog P., O'Keefe M. Differential production of IL‐12, IFN‐alpha, and IFN‐gamma by mouse dendritic cell subsets. J Immunol. 2001;166:5448–5455. doi: 10.4049/jimmunol.166.9.5448. [DOI] [PubMed] [Google Scholar]
- Iwasaki A., Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science. 2010;327:291–295. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman S., Heiligenhaus A., Rodriguez A., Soukiasian S., Dorf M.E., Foster C.S. Exacerbation of murine herpes simplex virus‐mediated stromal keratitis by Th2 type T cells. J Immunol. 1993;151:5777–5789. [PubMed] [Google Scholar]
- Kessler B., Hudrisier D., Schroeter M., Tschopp J., Cerottini J.C., Luescher I.F. Peptide modification of blocking of CD8, resulting in weak TCR signaling, can activate CTL for FAS‐but not perforin‐dependent cytotoxicity or cytokine production. J Immunol. 1998;161:6939–3946. [PubMed] [Google Scholar]
- Kikly K., Liu L., Na S., Sedgwick J.D. The IL‐23/Th17 axis: therapeutic targets of autoimmune inflammation. Curr Opin Immunol. 2006;18:670–675. doi: 10.1016/j.coi.2006.09.008. [DOI] [PubMed] [Google Scholar]
- Konig R., Huang L.Y., Germain N. MHC class II interaction with CD4 mediated by a region analogous to the MHC class I binding site for CD8. Nature. 1992;356:796–798. doi: 10.1038/356796a0. [DOI] [PubMed] [Google Scholar]
- Koski G.K., Lyakh L.A., Rice N.R. Rapid lipopolysaccharide‐induced differentiation of CD14(+) monocytes into CD83(+) dendritic cells is modulated under serum‐free conditions by exogenously added IFN‐gamma and endogenously produced IL‐10. Eur J Immunol. 2001;31:3773–3781. doi: 10.1002/1521-4141(200112)31:12<3773::aid-immu3773>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Koski G.K., Kariko K., Xu S., Weissman D., Cohen P.A., Czerniecki B.J. Cutting edge: innate immune system discriminates between RNA containing bacterial versus eukaryotic structural features that prime for high‐level IL‐12 secretion by dendritic cells. J Immunol. 2004;172:3989–3993. doi: 10.4049/jimmunol.172.7.3989. [DOI] [PubMed] [Google Scholar]
- Koyama S., Ishii K.J., Kumar H., Tanimoto T., Coban C., Uematsu S. Differential role of TLR‐ and RLR‐signaling in the immune responses to influenza A virus infection and vaccination. J Immunol. 2007;179:4711. doi: 10.4049/jimmunol.179.7.4711. et al. [DOI] [PubMed] [Google Scholar]
- Kronin V., Vremec D., Winkel K., Classon B.J., Miller R.G., Mak T.W. Are CD8+ dendritic cells (DC) veto cells? The role of CD8 in DC development and in the regulation of CD4 and CD8 T cell responses. Int Immunol. 1997;9:1061–1064. doi: 10.1093/intimm/9.7.1061. et al. [DOI] [PubMed] [Google Scholar]
- Kronin V., Fitzmaurice C., Caminschi I., Shortman K., Jackson D., Brown L. Differential effect of CD8+ and CD8− dendritic cells in the stimulation of secondary CD4+ T cells. Int Immunol. 2001;13:465–473. doi: 10.1093/intimm/13.4.465. [DOI] [PubMed] [Google Scholar]
- Lee Y.K., Mukasa R., Hatton R.D., Weaver C.T. Weaver developmental plasticity of Th17 and Treg cells. Curr Opin Immunol. 2009a;21:274–280. doi: 10.1016/j.coi.2009.05.021. [DOI] [PubMed] [Google Scholar]
- Lee Y.K., Turner H., Maynard C.L., Oliver J.F., Chen D., Elson C.O., Weaver C.T. Late developmental plasticity in the T helper 17 lineage. Immunity. 2009b;30:92–107. doi: 10.1016/j.immuni.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lexberg M.H., Taubner A., Förster A., Albrecht I., Richter A., Kamradt T. Th memory for interleukin‐17 expression is stable in vivo. Eur J Immunol. 2008;38:2654–2664. doi: 10.1002/eji.200838541. et al. [DOI] [PubMed] [Google Scholar]
- Lu S. Heterologous prime‐boost vaccination. Curr Opin Immunol. 2009;21:346–351. doi: 10.1016/j.coi.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- Meijers R., Lai C.C., Yang Y., Liu J.H., Zhong W., Wang J.H., Reinherz E.L. Crystal structures of murine MHC Class I H‐2 D(b) and K(b) molecules in complex with CTL epitopes from influenza A virus: implications for TCR repertoire selection and immunodominance. J Mol Biol. 2005;345:1099–1110. doi: 10.1016/j.jmb.2004.11.023. [DOI] [PubMed] [Google Scholar]
- Melief C.J.M., Van der Burg S.H. Immunotherapy of established (pre)malignant disease by synthetic long peptide vaccines. Nat Rev Cancer. 2008;8:351–360. doi: 10.1038/nrc2373. [DOI] [PubMed] [Google Scholar]
- Mosca P., Hobeika A., Clay T., Nair S., Thomas E., Morse M., Lyerly H. A subset of human monocyte‐derived dendritic cells express high levels of interleukin‐12 in response to combined CD40 ligand and interferon‐gamma treatment. Blood. 2000;96:3499–3504. [PubMed] [Google Scholar]
- Mosmann T.R., Sad S. The expanding universe of T‐cell subsets: Th1, Th2 and more. Immunol Today. 1996;17:138–146. doi: 10.1016/0167-5699(96)80606-2. [DOI] [PubMed] [Google Scholar]
- Napolitani G., Rinaldi A., Bertoni F., Sallusto F., Lazavecchia A. Selected toll‐like receptor agonist combinations synergistically trigger a T helper type 1‐polarizing program in dendritic cells. Nat Immunol. 2005;6:769–776. doi: 10.1038/ni1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nistala K., Wedderburn L.A. Th17 and regulatory T cells: rebalancing pro‐ and anti‐inflammatory forces in autoimmune arthritis. Rheumatology. 2009;48:602–606. doi: 10.1093/rheumatology/kep028. [DOI] [PubMed] [Google Scholar]
- Okada H., Kuhn C., Feillet H., Bach J.F. The ‘hygiene hypothesis’ for autoimmune and allergic diseases: an update. Clin Exp Immunol. 2010;160:1–9. doi: 10.1111/j.1365-2249.2010.04139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm N.W., Medzhitov R. Pattern recognition receptors and control of adaptive immunity. Immunology. 2009;227:221. doi: 10.1111/j.1600-065X.2008.00731.x. [DOI] [PubMed] [Google Scholar]
- Palucka K., Ueno H., Fay J., Banchereau J. Dendritic cells and immunity against cancer. J Intern Med. 2011;269:64–73. doi: 10.1111/j.1365-2796.2010.02317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parham P., Androlewicz M.J., Holmes N.J., Rothenberg B.E. Arginine 45 is a major part of the antigenic determinant of human b2‐microglobulin recognized by mouse monoclonal antibody BBM1. J Biol Chem. 1983;258:6179–6186. [PubMed] [Google Scholar]
- Paustian C., Caspell R., Johnson T., Cohen P.A., Shu S., Xu S. Effect of multiple activation stimuli on the generation of Th1‐polarizing dendritic cells. Hum Immunol. 2011;72:24–31. doi: 10.1016/j.humimm.2010.10.004. et al. [DOI] [PubMed] [Google Scholar]
- Pisarev P., Parajuli P., Mosely R.L., Sublet J., Kelsey L., Zimmerman D.H. Flt3 ligand and an IL‐1 beta bioactive heteroconjugate as adjuvants for a type 1 cell mediated immune response to HIV gag p17 peptide. Vaccine. 2002;20:2358–2368. doi: 10.1016/s0264-410x(02)00096-8. et al. [DOI] [PubMed] [Google Scholar]
- Pulendran B. Modulating Th1/Th2 responses with microbes, dendritic cells, and pathogen recognition receptors. Immunology. 2004;29:187–196. doi: 10.1385/IR:29:1-3:187. [DOI] [PubMed] [Google Scholar]
- Rosenthal K.S. Immune peptide enhancement of peptide‐based vaccines. Front Biosci. 2005;10:478–482. doi: 10.2741/1543. [DOI] [PubMed] [Google Scholar]
- Rosenthal K., Mao H., Horne W.T., Zimmerman D.H. Immunization with a L.E.A.P.S. heteroconjugate vaccine containing a CTL epitope and a peptide from beta‐2‐microglobulin elicits a protective and DTH response to herpes simplex virus type 1. Vaccine. 1999;17:535–542. doi: 10.1016/s0264-410x(98)00231-x. [DOI] [PubMed] [Google Scholar]
- Roses R., Xu S., Xu M., Koldovsky U., Koski G., Czerniecki B. Differential production of IL‐23 and IL‐12 by myeloid‐derived dendritic cells in response to TLR agonists. J Immunol. 2008;181:5120–2127. doi: 10.4049/jimmunol.181.7.5120. [DOI] [PubMed] [Google Scholar]
- Segal B.M., Constantinescu C.S., Raychaudhuri A. Repeated subcutaneous injections of IL12/23 p40 neutralising antibody, ustekinumab, in patients with relapsing‐remitting multiple sclerosis: a Phase II, double‐blind, placebo‐controlled, randomized, dose‐ranging study. Lancet Neurol. 2008;7:796–804. doi: 10.1016/S1474-4422(08)70173-X. et al. [DOI] [PubMed] [Google Scholar]
- Šenolt L., Vencovský J., Pavelka K., Ospelt C., Gay S. Prospective new biological therapies for rheumatoid arthritis. Autoimmun Rev. 2009;9:102–107. doi: 10.1016/j.autrev.2009.03.010. [DOI] [PubMed] [Google Scholar]
- Shahrara S., Huang Q., Mandelin A.M., 2nd, Pope R.M. TH‐17 cells in rheumatoid arthritis. Arthritis Res Ther. 2008;10:R93. doi: 10.1186/ar2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuwen X., Koski G., Faries M., Bedrosian I., Mick R., Maeurer M. Rapid high efficiency sensitization of CD8+ T cells to tumor antigens by dendritic cells leads to enhanced functional avidity and direct tumor recognition through an IL‐12‐dependent mechanism. J Immunol. 2003;171:2251–2261. doi: 10.4049/jimmunol.171.5.2251. et al. [DOI] [PubMed] [Google Scholar]
- Snijders A., Kalinski P., Hilkens C.M., Kapsenberg M.L. High‐level IL‐12 production by human dendritic cells requires two signals. Int Immunol. 1998;10:1593–1598. doi: 10.1093/intimm/10.11.1593. [DOI] [PubMed] [Google Scholar]
- Taylor P.R., Koski G.K., Paustian C.C., Bailey E., Cohen P.A., Moore F.B.G. J‐LEAPS vaccines initiate murine Th1 responses by activating dendritic cells. Vaccine. 2010a;28:5533–5542. doi: 10.1016/j.vaccine.2010.06.043. et al. [DOI] [PubMed] [Google Scholar]
- Taylor P.R., Paustian C.C., Koski G.K., Zimmerman D.H., Rosenthal K.S. Maturation of dendritic cell precursors into IL12 producing DCs by J‐LEAPS immunogens. Cell Immunol. 2010b;262:1–5. doi: 10.1016/j.cellimm.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Vollenhoven R.F. Treatment of rheumatoid arthritis: state of the art 2009. Nat Rev Rheumatol. 2009;5:531–541. doi: 10.1038/nrrheum.2009.182. [DOI] [PubMed] [Google Scholar]
- Vordermeier H.M., Harris D.P., Mehrotra P.K., Roman E., Elsaghier A., Moreno C., Ivanyi J. M. tuberculosis‐complex specific T‐cell stimulation and DTH reactions induced with a peptide from the 38‐kDa protein. Scand J Immunol. 1992;35:711–718. doi: 10.1111/j.1365-3083.1992.tb02979.x. [DOI] [PubMed] [Google Scholar]
- Walton C.B., Inos A.B., Andres O.A., Jube S., deCouet H.G., Douglas J.T. Immunization with hybrid recombinant Mycobacterium tuberculosis H37Rv proteins increases the TH1 cytokine response in mice following a pulmonary instillation of irradiated mycobacteria. Vaccine. 2008;34:4396–4402. doi: 10.1016/j.vaccine.2008.05.093. et al. [DOI] [PubMed] [Google Scholar]
- Walton C.B., Jube S., Schorlemmer A., Patek P., Zimmerman D., Rosenthal K., Borthakur D. Ex vivo stimulation assay for T‐cell responses for tuberculosis using LEAPS‐peptide heteroconjugates. Curr Trends Microbiol. 2010;6:1–12. [Google Scholar]
- Woodland D.L. Jump‐starting the immune system: prime‐boosting comes of age. Trends Immunol. 2004;25:98–104. doi: 10.1016/j.it.2003.11.009. [DOI] [PubMed] [Google Scholar]
- Zhou W., Konig R. T cell receptor‐independent CD4 signalling: CD4–MHCII interactions regulate intracellular calcium and cyclic AMP. Cell Signal. 2008;15:751–762. doi: 10.1016/s0898-6568(03)00037-8. [DOI] [PubMed] [Google Scholar]
- Zimmerman D.H., Rosenthal K.S. The LEAPS approach to vaccine development. Front Biosci. 2005;10:790–798. doi: 10.2741/1572. [DOI] [PubMed] [Google Scholar]
- Zimmerman D., Bergmann K.F., Rosenthal K.S., Elliot D.A. A new approach to T cell activation: natural and synthetic conjugates capable of activating T cells. Vaccine Res. 1996a;5:91–102. [Google Scholar]
- Zimmerman D., Morris S., Rouse D., Worthington K.F., Elliot D.A., Rosenthal K.S. Immunization with peptide heteroconjugates primes a TH1 associated antibody (IgG2a) response which recognizes the native epitope on the 38‐kDa protein of Mycobacterium tuberculosis. Vaccine Res. 1996b;5:103–118. [Google Scholar]
- Zimmerman D., Lloyd J.P., Heisey D., Winship M.D., Siwek M., Talor E., Sarin P.S. Induction of cross clade reactive specific antibodies in mice by conjugates of HGP‐30 (peptide analog of HIV‐1SF p17) and peptide segments of human beta‐2‐Microglobulin or MHC II beta chain. Vaccine. 2001;19:4750–4759. doi: 10.1016/s0264-410x(01)00247-x. [DOI] [PubMed] [Google Scholar]
- Zimmerman D.H., Taylor P., Bendele A., Carambula R., Duzant Y., Lowe V. CEL‐2000: a therapeutic vaccine for rheumatoid arthritis arrests disease development and alters serum cytokine/chemokine patterns in the bovine collagen type II induced arthritis in the DBA mouse model. Int Immunopharmacol. 2010;10:412–421. doi: 10.1016/j.intimp.2009.12.016. et al. [DOI] [PubMed] [Google Scholar]