

Summary
Therapeutic vaccines are currently developed for chronic viral infections, such as human papillomavirus (HPV), human immunodeficiency virus (HIV), herpesvirus and hepatitis B (HBV) and C (HCV) virus infections. As an alternative to antiviral treatment or to support only partially effective therapy a therapeutic vaccine shall activate the patient's immune system to fight and finally control or ideally even eliminate the virus. Whereas the success of prophylactic vaccination is based on rapid neutralization of the invading pathogen by antibodies, virus control and elimination of infected cells require T cells. Therefore, induction of a multi‐specific and multifunctional T‐cell response against key viral antigens is a paradigm of therapeutic vaccination – besides activation of a humoral immune response to limit virus spread. In this review, we describe options to develop a therapeutic vaccine for chronic viral infections using HBV as a promising example.
Introduction
Any acute viral infection may be resolved, kill the host or become chronic. When becoming chronic, viral replication and spread and the host immune response balance each other. To establish chronic viral infection (i) a virus must evade complete elimination and (ii) the immune system has to limit viral replication and antigen production to a level that avoids permanent damage of the infected tissue, because unrestrained immune attack on virus antigen‐bearing cells causes immunopathology (Virgin et al., 2009).
Some viruses, such as herpes and papovaviruses have co‐evolved with humans over millions of years and manage to persist in the host without causing overt disease. They only become pathogenic when the host immune system is suppressed, e.g. by medical treatment. The fact that most of us do not show obvious symptoms of these infections marks these viruses as part of the human metagenome efficiently controlled by the immune system of normal individuals without causing any harm (Virgin et al., 2009).
In other chronic viral infections, such as HIV or HPV infection or viral hepatitis, an immune response, which cannot clear the infection but continuously fights it, is largely responsible for pathogenicity. Therefore, HIV, HPV, HCV and HBV are the main targets for therapeutic vaccination aiming at virus elimination or at least complete immune control of virus replication and antigen production. However, it remains to be proven that a therapeutic vaccine is able to achieve virus elimination or sustained virus control. So one may ask the question: which is the ideal target to optimize the design of therapeutic vaccines and prove their efficacy?
Chronic viral infections as a target for therapeutic vaccines
Persistent viruses, which cause disease or tissue damage, are the primary target for a therapeutic vaccine since there is an obvious medical need. HIV, HBV and HCV cause long‐term damage to the host or the infected organ. HPV does not cause obvious disease but – as HBV and HCV – sets the infected host at risk to develop cancer.
HIV cannot be eliminated by an immune response since it persists by integrating into the genome of several immune cells (Finzi et al., 1999). There may be a chance to control HIV replication by a therapeutic vaccine and to prolong disease progression as it is observed in long term non‐progressors (Gudmundsdotter et al., 2006). So for HIV, sustained virus control but not virus elimination can be goal of therapeutic vaccination.
HBV, HCV and HPV can be eliminated by the immune system after acute infection or sometimes even when the immune balance in chronic infection tips. Since shifting this balance towards immune control is the aim of therapeutic vaccination, those viruses are primary targets for a proof‐of‐concept of therapeutic vaccination.
Current therapy is able to eliminate HCV in about 50–80% of patients and a series of new and more potent antivirals (e.g. protease inhibitors) will be available in the near future. Treatment results should be awaited to decide whether there is still a need for a therapeutic vaccine (Manns et al., 2007; Munir et al., 2010). HBV cannot be cleared by currently available antiviral therapy and therefore requires long‐term antiviral treatment, which is costly, often selects for drug‐resistant viral variants and may have long‐term side effects (Cornberg et al., 2011). So there is a need for alternative treatment approaches. For HBV highly effective and safe prophylactic vaccines are available. These, however, showed no effect in the setting of chronic infection (Dienstag et al., 1982; Heintges et al., 2001; Dahmen et al., 2002; Hildesheim et al., 2007), indicating the need for a specific therapeutic vaccine design. We here focus on the options to design and develop a therapeutic hepatitis B vaccine.
Chronic hepatitis B as a target for therapeutic vaccines
HBV, liver disease and cancer
HBV infects the liver of humans or humanoid primates. In humans, HBV infection often causes an inflammatory liver disease – hepatitis B. The virus is transmitted by perinatal, percutaneous and sexual exposure, as well as by close person‐to‐person contact among young children presumably by open cuts or sores. Vertical transmission from mothers to their neonates, or infection during the first year of life, results in persistent, often life‐long infection in > 90%. In contrast, infection during adulthood is cleared in > 95% of cases, and results in life‐long protective immunity. The outcome of HBV infection and pathogenesis of liver disease are immune‐mediated and thus determined by the virus‐host interaction (Ganem and Prince, 2004).
Liver disease is characterized by necro‐inflammation, continuous hepatocyte turn‐over and progressing tissue damage. Liver damage results in liver fibrosis or cirrhosis often laying the ground for hepatocellular carcinoma (HCC). The major risk factor for cancer development is the level of virus replication determined by the level of viraemia (Chen et al., 2006). Thus, the primary aim of any therapy for chronic hepatitis B is suppression of viral replication and remission of liver disease. The long‐term goal of treatment is the elimination of the virus in order to prevent disease progression to cirrhosis or HCC.
The clinical course of HBV infection is highly variable (Hoofnagle et al., 2007; McMahon, 2009). Acute infection can be asymptomatic, lead to self‐limiting, acute hepatitis B or even result in fulminant liver failure. Chronic HBV infection is defined by virus persistence for more than 6 months – defined by positive serum testing for hepatitis B surface antigen (HBsAg). A major fraction of the HBV‐specific humoral immune response is mounted against HBsAg, which is composed of three membrane‐bound envelope proteins. These three proteins are called large (L‐), middle (M‐) and small (S‐) envelope protein and share a common C‐terminus, the S‐domain. Compared with the S‐protein, the M‐protein has an N‐terminal extension of 55, the L‐protein of 108 amino acids. Resolution of HBV infection is characterized by loss of HBsAg and seroconversion to anti‐HBs antibodies.
The hepatitis B e antigen (HBeAg) is a non‐particulate, secreted form of the viral capsid protein, which is called hepatitis B core antigen (HBcAg). HBeAg‐specific antibodies (anti‐HBe) appear late during infection and indicate a favourable outcome of infection. In acute hepatitis, clinical recovery is associated with loss of serum HBeAg and seroconversion to anti‐HBe. In chronically evolving hepatitis clearance of serum HBeAg and development of anti‐HBe mark the transition from high replicative to a low replicative, inactive hepatitis B.
If infection is acquired after birth it is usually highly replicative (HBV DNA > 200 000 IU ml−1) and positive for HBeAg, but no or minimal liver inflammation is detected [normal serum alanine aminotransferase (ALT) activity]. This state is termed the high replicative HBsAg carrier state. Years after childhood infection or if HBV infection is acquired during adulthood, virus titres drop (HBeAg positive or negative) and active liver inflammation with elevated serum ALT activity and progressing tissue damage becomes obvious – a stage called active hepatitis B. Active hepatitis B can (spontaneously or treatment induced) convert into an inactive phase characterized by seroconversion from HBeAg to anti‐HBe antibody positivity with usually normal ALT levels and HBV DNA < 104 copies (2000 IU) per ml. Patients with inactive hepatitis B have an improved prognosis but are still at risk for HCC development particularly if liver cirrhosis has developed (Cornberg et al., 2011). Sometimes, HBsAg persists without detectable HBV replication. These patients still can transmit the virus by blood or organ donation.
HBV persistence forms
To prevent late complications of hepatitis B as well as transmission to other individuals, the final goal in hepatitis B therapy must be eradication of HBV‐infected hepatocytes. The virus establishes a stable, circular DNA as a minichromosome in the nucleus of infected cells, the so‐called covalently closed circular (ccc) DNA, which serves as a transcription template and allows the virus to persist without any replication. HBV cccDNA‐positive cells can drive viral rebound and recurrence of disease in the absence of a strong T‐cell response (Levrero et al., 2009). HBV cccDNA may persist at low copy numbers in infected cells for months or years, but is probably lost during cell division. However, RNA‐ and DNA‐containing capsids can readily replenish cccDNA in the nucleus of infected cells (Zhang et al., 2003). Therefore, only a complete block of replication for a time span sufficient to get rid of existing cccDNA molecules by natural turn‐over will allow elimination of HBV cccDNA.
A hallmark of HBV cccDNA persistence is expression of the small surface (S) protein, which is secreted as HBsAg. When sustained immune control is achieved, HBsAg becomes bound and neutralized by anti‐HBs antibodies, a process described as anti‐HBs seroconversion. However, some cccDNA positive cells may persist despite anti‐HBs seroconversion.
Direct antiviral therapy
Current treatment options for chronic hepatitis B depend on interferon (IFN) α and direct antivirals, i.e. nucleoside or nucleotide analogues. Although there now are seven approved therapies for HBV infection [two IFN formulations and five nucleos(t)ide analogues], elimination of the virus is hardly achieved (Cornberg et al., 2011; Kwon and Lok, 2011). An important risk factor associated with progression of HBV‐related liver disease and development of HCC is high viraemia (Chen et al., 2006). Therefore, current therapeutic goals are sustained suppression of HBV replication and remission of liver disease although HBsAg clearance and anti‐HBs seroconversion are desired (Cornberg et al., 2011; Kwon and Lok, 2011).
Nucleos(t)ide analogues inhibit the viral reverse transcriptase and usually are well tolerated. They control HBV replication but rarely eliminate the virus. HBV cccDNA persists in the host cell nucleus, continues to produce HBsAg, and may drive a viral rebound and recurrent disease if the treatment is stopped. Continuous treatment for a long time is required, which often selects resistant viral variants. Emergence of antiviral‐resistant mutations can lead to negation of the initial response, and in some cases hepatitis flares and hepatic decompensation (Hoofnagle et al., 2007).
HBV elimination will depend on the half‐life of cccDNA and on the long‐term efficacy of antiviral treatment, which may be reduced by the selection of drug‐resistant viral variants (Levrero et al., 2009). The exact half‐life of HBV cccDNA is not known. Data in chimpanzee livers suggested a half‐life between 9 and 14 days during immune clearance (Wieland et al., 2004); in the duck and woodchuck models of HBV infection, cccDNA half‐life is between 35 and 50 days in the absence of an effective immune response (Zhu et al., 2001; Addison et al., 2002). The long cccDNA half‐life in addition to the fact that every possible viral mutant is produced daily in an infected individual, makes viral clearance by direct antivirals very unlikely. Clinical observations support this notion and show that even very effective antivirals such as tenofovir in 3–8% of patients lead to HBsAg seroconversion over 3 years (van Bommel et al., 2010; Heathcote et al., 2011).
Theoretical options for direct targeting of HBV cccDNA are designer molecules like, for example endonucleases fused to a modular zinc finger motive binding to HBV sequences that could specifically cleave HBV cccDNA. This approach – as many other gene therapy approaches – is directly acting in the infected cell. A major hurdle for these approaches, however, is appropriate delivery into all infected hepatocytes, which will hardly be achieved but necessary to clear the virus.
Taken together, true cure of infection (loss of HBsAg and sustained disappearance of viraemia, as measured by stringent PCR assays) is achieved only infrequently with current antivirals (Kwon and Lok, 2011). An increasing number of antiviral drugs targeting reverse transcription but also other steps in the viral life cycle are currently developed, which might improve treatment outcome in the future (Wu et al., 2007). However, without an efficient immune response, long‐time treatment is required. Major concern for long‐term treatment is efficacy and safety, but also high treatment costs (Hoofnagle et al., 2007). Inducing an efficient HBV‐specific immune response – ideally after the amount of infected cells has been reduced to a minimum by direct antivirals – provides an interesting alternative to achieve sustained virus control and be able to stop antiviral treatment.
Immunomodulatory therapies
Interferon‐α was the first therapy approved for chronic hepatitis B and is currently used in pegylated forms with longer serum half‐life (Cornberg et al., 2008). Interferon‐α has direct antiviral effects as well as immunomodulatory properties (Tang et al., 2005). It therefore should stimulate the host immune response to eliminate the virus. However, its side effects (e.g. induction of hepatitis flares, fever, myalgias, thrombocytopenia and depression) make it a difficult treatment for many applications and exclude therapy of advanced or decompensated liver disease. In addition, therapeutic effects are limited: only about 20% of HBeAg‐positive patients profit from IFN‐α treatment, seroconvert to anti‐HBe and lose serum HBV–DNA. Although HBeAg‐negative patients have a better response rate, response is not durable in most cases (Hoofnagle et al., 2007). Loss of HBsAg is achieved in 8–10% of patients with PEG‐IFN‐α (Marcellin et al., 2009) and may further increase during long‐term follow‐up (Moucari et al., 2009).
Other cytokines with antiviral activity that non‐specifically enhance host immune responses, such as IFN‐γ, interleukin (IL)‐2, IL‐12 or IL‐18, had little effect on HBV replication or disease activity in humans (Loomba and Liang, 2006).
A potentially promising strategy to clear chronic HBV infection is therapeutic vaccination, which aims to eliminate persisting virus by specifically stimulating endogenous antiviral immune responses. However, it represents a still unresolved challenge to generate effective antiviral T cells during chronic HBV infection due to the tolerogenic environment in the liver, which is the site of HBV replication (Crispe et al., 2006; Thomson and Knolle, 2010) and the high HBV antigenic load present in the infected host (Webster et al., 2004). As the liver is involved in the clearance of foreign antigens from the gastrointestinal tract unnecessary activation of the immune system is avoided by induction of peripheral tolerance towards these antigens (Knolle and Gerken, 2000).
Non‐parenchymal liver cells such as resident dendritic cells (DC), liver sinusoidal endothelial cells, Kupffer cells as well as hepatic stellate cells (Ito cells) contribute to the tolerogenic nature of the liver. These cells are capable of expressing anti‐inflammatory cytokines (e.g. IL‐10 and TGF‐β) and inhibitory cell surface ligands (e.g. PD‐L) negatively regulating T‐cell activation (Thomson and Knolle, 2010).
Novel approaches, which are discussed to activate immune responses, are stimulation of pattern recognition receptors, inhibition of IL‐10 or interfering with PD‐1 or CTLA‐4 inhibitory signals. These approaches will have to undergo pre‐clinical and clinical testing. There is some scepticism that they will be able to induce a strong enough HBV‐specific immune response without causing severe side effects, but they may proof useful in combination with an antigenic stimulus, i.e. a therapeutic vaccine.
Understanding immune responses is the basis for therapeutic vaccine design
While a correlation between the strength of HBV‐specific T‐cell responses and virus clearance has been established, factors determining the strength of a T‐cell response as well as factors shifting the balance from immune tolerance to immune clearance are hardly understood (Rehermann and Nascimbeni, 2005). The innate immune response, early adaptive B‐ and T‐cell responses, regulatory T cells, the liver microenvironment as well as peculiar properties of hepatocytes and non‐parenchymal liver cells to present antigen (Thomson and Knolle, 2010) seem to play a role. Understanding the immunological mechanisms that contribute to HBV clearance or chronicity is of utmost importance for rational design of an effective immunotherapy.
Our understanding of host immunity and viral immune evasion comes largely from studies of resolving acute HBV infection (Webster et al., 2000; Thimme et al., 2003; Dunn et al., 2009). Upon resolution of acute infection, the immune system turns to an uninfected but memory‐armed state including quiescent but easily triggered memory B and T cells, as well as plasma cells that continuously produce antibodies (Virgin et al., 2009). In chronic HBV infection, innate and adaptive arms of the immune system are involved in the control and elimination of an HBV infection.
Innate immunity to HBV infection
Innate immune responses are crucial for early containment of viral infections and favour a timely and efficient induction of virus‐specific adaptive responses. Innate immune responses are essential for elimination of the virus but also contribute to immunopathology (Rouse and Sehrawat, 2010). There is, however, a still ongoing debate about the role of the innate immune system and about how adaptive immunity is triggered thereby. In recent years, there is emerging evidence that the innate immune system plays an important role in influencing both the outcome of acute HBV infection and the pathogenesis of liver disease (Das and Maini, 2010).
Systemic innate immune responses involve macrophages and DC as well as natural killer (NK) cells. A number of cells may be involved in local innate immune responses towards HBV in the liver (Bauer et al., 2011); these include hepatocytes and non‐parenchymal liver cells, such as liver resident macrophages (Kupffer cells), liver sinusoidal endothelial cells, liver resident myofibroblasts and liver resident DC, together with intrahepatic lymphocytes such as NK cells and NK‐T cells. The large number of cells potentially involved in innate immunity against HBV demonstrates the complexity of this response (Thomson and Knolle, 2010).
Adaptive immune responses
Although there is increasing evidence that an impairment of the innate immune response contributes to chronicity, the hallmark of chronic HBV infection is the lack of a robust HBV‐specific CD8+ and CD4+ T‐cell response (Wang and Zhang, 2009; Rouse and Sehrawat, 2010). HBV‐specific adaptive immune responses include CD4+ T helper cells, cytotoxic CD4+ and CD8+ T cells and B cells responsible for a neutralizing antibody response. CD4+ T cells are robust cytokine producers, required for cross‐priming of CD8+ T cells and T cell‐dependent B cell responses. Cytotoxic T lymphocytes (CTL) clear HBV‐infected hepatocytes, while the humoral immune response neutralizes free virus thereby preventing virus spread. There is a clear correlation between timing, strength and specificity of the adaptive cellular immune response and the outcome of infection (summarized in Rehermann and Nascimbeni, 2005).
Several mechanisms are proposed to contribute to immunological failure during development of chronicity. Hyporesponsiveness of specific T cells can be caused by deletion via apoptosis and functional tolerance. Functional tolerance of HBV‐specific T cells is characterized by anergy or exhaustion, dysregulation of cytokine production and expression of inhibitory receptors (Fisicaro et al., 2010). Functional T‐cell tolerance is probably caused by continuous exposure to high levels of viral antigens (Kakimi et al., 2002). Das and co‐workers suggest that functional skewing of lymphocyte subpopulations, which contributes to the failure of viral control additionally, favours the inflammatory liver environment (Das et al., 2008). According to their data intrahepatic CD8+ T cells are impaired in patients with chronic HBV infection regardless of their specificity. Compared with healthy donors a large influx of non‐antigen‐specific CD8+ T cells into the liver was observed. These liver‐associated CD8+ T cells show poor IL‐2 production and proliferative capacity but maintain their capacity to produce pro‐inflammatory cytokines.
In addition, CD4+Foxp3+CD25high regulatory T‐cell (Treg) frequencies are increased in chronic HBV infection. They do not only suppress effectors of the immune response to HBV, thereby contributing to HBV persistence, but also exhibit protective effects by limiting liver immunopathology (Stoop et al., 2005; Manigold and Racanelli, 2007; L. Stross and U. Protzer, manuscript submitted). To terminate persistent viral infection the mechanisms underlying T‐cell tolerance and hypo‐responsiveness need to be overcome by an effective therapeutic vaccine without promoting immune‐mediated liver damage. Counteracting Treg‐mediated suppression by selective depletion of Treg prior to therapeutic vaccination is one possible approach, but it is essential to investigate whether short‐term suppression of Treg is sufficient and to ensure that inhibition of Treg activity does not mediate autoimmunity.
Multifunctional and multi‐specific CD4+ as well as CD8+ T cells, secreting IFN‐γ, TNF‐α and IL‐2 are required for control or elimination of viral infections including HBV (Thimme et al., 2003; Seder et al., 2008). Hence, for induction of efficient CTL any therapeutic vaccine should elicit an additional Th1 type CD4+ helper response. Elimination and protective immunity, however, requires also T cell‐dependent B‐cell responses, which guarantee production of sufficient amounts of neutralizing antibodies (Rehermann and Nascimbeni, 2005). Innate immunity needs to be activated to ensure appropriate presentation of vaccine antigens and provide immune‐stimulatory signals for efficient priming of B and T cells. For this purpose potent vaccine adjuvants are needed, which, by activation of pattern recognition receptors, induce pro‐inflammatory signals required for priming and modulation of the adaptive immune responses (Coffman et al., 2010).
Therapeutic vaccine approaches used for chronic hepatitis B
During past years a series of different therapeutic vaccination strategies (protein‐based vaccines, DNA/peptide vaccines, cell‐based vaccines) have been investigated using different animal models (i.e. HBV‐transgenic mice, woodchuck hepatitis virus model or chronically HBV‐infected chimpanzees) (Kosinska et al., 2010). Several of those strategies have reached clinical development stages with different outcomes (reviewed in Beckebaum et al., 2002; Akbar and Onji, 2005; Michel et al., 2011). Ideally, one would create vaccines that have all the properties of natural pathogens with the exception of causing disease. Key features of pathogens that can be mimicked by vaccine delivery systems are their size, shape and surface molecule organization. In addition, pathogen‐associated molecular patterns can be added to induce innate immune responses that promote adaptive immunity (Bachmann and Jennings, 2010). Figure 1 summarizes most important approaches used for therapeutic vaccination of chronic hepatitis.
Figure 1.
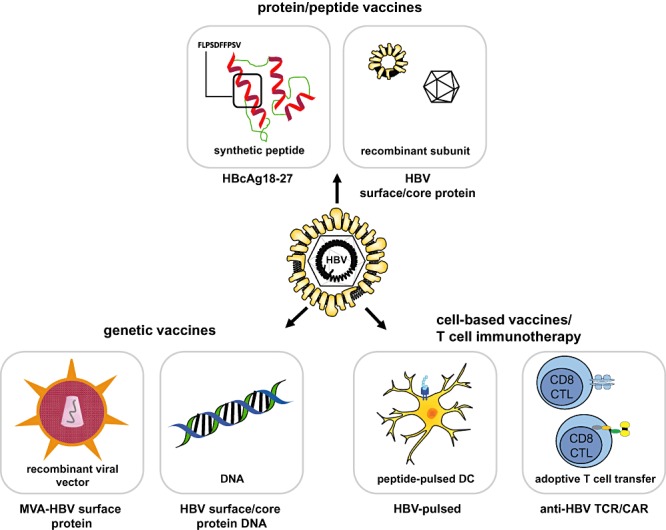
General approaches for therapeutic vaccination against HBV. Protein‐ or peptide‐based vaccine strategies include immunization with immune dominant HLA‐A2‐restricted peptide epitope HBcAg18–27 and administration of HBsAg alone or in combination with the highly immunogenic HBcAg. Genetic vaccination can be performed with recombinant, replication incompetent viral vector vaccines, like MVA or adenoviruses encoding HBV DNA, or with genetically engineered HBV–DNA. Cell‐based vaccine strategies include the transfer of peptide‐loaded antigen‐presenting, autologous DC and the transfer of functional, ex vivo expanded HBV‐specific CD8+ CTL or CTL carrying HBV‐specific T‐cell receptors or chimeric antigen receptors. HBV, hepatitis B virus; HBsAg, hepatitis B surface antigen; HBcAg, hepatitis B core antigen; anti‐HBs, antibodies against HBsAg; TCR, T‐cell receptor; CAR, chimeric antigen receptor.
Protein or peptide‐based vaccines
First attempts were based on available prophylactic vaccines, alone or in combination with IFN‐α, lamivudine or IL‐2 (Dienstag et al., 1982; Heintges et al., 2001; Dahmen et al., 2002). However, these recombinant protein‐based vaccines were not able to induce a sustained immune response sufficient to control chronic hepatitis B in larger patient cohorts (Hilleman, 2003). Failure was mainly attributed to the fact that the aluminum adjuvanted vaccines induce a pronounced Th2‐type immune response and do not stimulate a CTL response.
Hence, alternative therapeutic vaccine formulations were developed to induce a potent CTL response. The first candidate tested was a lipopeptide, Theradigm, containing an HBcAg18–27 HLA‐A2‐restricted peptide epitope and a universal tetanus toxoid T helper cell epitope (TT 830–843). Theradigm induced a CTL response against HBcAg18–27 in HLA‐A2‐positive healthy volunteers (Vitiello et al., 1995), but had no therapeutic effect in chronic hepatitis B patients (Heathcote et al., 1999).
In a more recent approach HBsAg adjuvanted with monophosphoryl lipid A (MPL) and QS21 in an oil‐in‐water emulsion induced HBsAg‐specific T cells in healthy volunteers (Vandepapeliere et al., 2008). However, despite the generation of an HBsAg‐specific T‐cell response and anti‐HBs in HBeAg‐positive patients co‐treated with the antiviral drug lamivudine, no superior clinical effect was observed in terms of HBeAg seroconversion compared with lamivudine alone (Vandepapeliere et al., 2007). Similarly, HBsAg complexed with anti‐HBs showed only minor effects in clinical studies (Xu et al., 2008). All these approaches had in common that a single HBV antigen or peptide epitope was included in the vaccine, although induction of multi‐specific CTL responses are key for resolution of HBV infection (Thimme et al., 2003; Webster et al., 2004).
Including the more immunogenic pre‐S domains of the M and L HBV envelope proteins (Milich et al., 1985) into vaccine formulations as well as using different HBsAg subtypes may be an option to induce more potent B‐ and T‐cell responses (Schirmbeck et al., 2003,Schumann et al., 2007).
Improvement of protein‐based vaccination has been achieved by combining HBsAg with HBcAg, as the latter is known to have excellent immunogenic properties. HBcAg is able to enhance priming of T cells via activation of B cells enabling them to act as potent primary antigen presenting cells (Milich et al., 1997; Lazdina et al., 2001). Additionally, it can activate Toll‐like receptor (TLR‐) signalling through nucleic acids bound to its arginine‐rich region (Aguilar et al., 2004; Storni et al., 2004). HBcAg is thus qualified for use as a potent adjuvant when aiming to generate HBV‐specific T cells as well as T cells towards heterologous antigen, e.g. HCV or tumor‐derived antigens (Chen et al., 2011).
When HBcAg is mixed with HBsAg, highly immunogenic super structures are formed. Such an HBsAg/HBcAg vaccine is able to break tolerance in HBV transgenic mice when combined with the Iscomatrix adjuvant (Buchmann and Protzer, manuscript in revision) and proved safe in phase I trials in healthy volunteers and chronic hepatitis B patients (Buchmann and Janowicz, pers. comm.). Another combination vaccine induced humoral immune responses against both antigens when applied intra‐nasally in a phase I clinical trial with HBV‐naïve healthy volunteers (Betancourt et al., 2007). In chronically HBV‐infected patients, it showed encouraging preliminary results with HBV–DNA decreasing in 11 out of 20 therapy‐naïve patients (M.A. Mamun, S.M.F. Akbar, S. Rahman, J.C.R. Aguilar, S. Mishiro, Poster presentation, The International Liver Congress 2010)
Reduction of viral and antigen load prior to vaccination using antiviral compounds provides a potential improvement of vaccine efficacy. Inhibition of viral replication through nucleos(t)ide analogues can lead to transient CD4+ T‐cell responses in chronic hepatitis B patients (Boni et al., 1998). Further evidence for sustained therapeutic effects of combined antiviral treatment and vaccination has been gained in the woodchuck model (Menne et al., 2002; Roggendorf et al., 2010). Instead of co‐administration, it seems to be rational to apply antiviral pre‐treatment prior to vaccination (Horiike et al., 2005) to lower the load of antigen most likely responsible for tolerance induction.
Taken together, protein‐based vaccination strategies combining HBsAg and the highly immunogenic HBcAg are a promising approach to reduce viral load and induce seroconversion in chronically HBV‐infected patients. It seems, however, not be sufficient to clear HBV infection. Therefore, a subsequent Th1‐/CTL‐inducing vaccine may be necessary to induce an effective multi‐specific HBV‐specific T‐cell response finally enabling clearance of infected hepatocytes and elimination of the virus.
Genetic vaccines
In contrast to protein‐based vaccines, predominantly aiming at the induction of humoral immune responses (e.g. neutralizing antibodies), a classical approach to elicit CTL responses are genetic vaccines. DNA vaccines have been tested in numerous preventive and therapeutic settings of viral infections and malignancies (reviewed in Liu, 2011).
Chronic hepatitis B patients previously not responding to standard treatment were vaccinated with an HBsAg‐expressing DNA vaccine (encoding for S and preS2 domains of HBV envelop proteins). Although HBV–DNA levels decreased in 5 out of 10 patients, vaccine‐induced T‐cell responses were only transient (Mancini‐Bourgine et al., 2004). Rather disappointing results were obtained in a clinical trial in 30 therapy‐naïve highly replicative HBV carriers vaccinated with S/preS2 HBsAg‐based DNA vaccine (GenHevac B) although the vaccine was safe and well tolerated. No significant benefits and clinical effects (reduction of HBV viraemia or HBeAg/anti‐HBe seroconversion) were observed compared with 17 non‐treated individuals (Yalcin et al., 2003).
Enhanced efficacy of DNA‐based therapeutic vaccination was achieved by combining a poly‐domain DNA‐vaccine (encoding for S, preS1/S2, core, polymerase and X proteins) with human IL‐12 and antiviral drug treatment (Yang et al., 2006; Im et al., 2009). Besides reduction of HBV–DNA levels and HBeAg seroconversion in 50% of patients, a multi‐specific T‐cell response was induced. Importantly, in 15% of patients HBV–DNA remained undetectable 3 years after therapy was ceased. This holds some promise for genetic vaccination adjuvanted with immune stimulatory cytokines.
As shown by numerous studies immunogenicity of genetic vaccines can be enhanced by heterologous prime/boost vaccination usually applying DNA prime vaccination followed by a viral vector boost vaccination (Woodland, 2004). Genetically engineered poxviruses are among the most extensively tested viral vectors with one of the most promising being the modified vaccinia Ankara (MVA) virus (Mayr et al., 1978; Sutter and Moss, 1992). Due to its excellent safety record as a smallpox vaccine and its immunostimulatory properties MVA‐vectored vaccines are in clinical development of prophylactic vaccines against several pathogens (e.g. HIV, influenza virus) (Paris et al., 2010; Berthoud et al., 2011). Recombinant MVA has furthermore shown immunogenicity in therapeutic vaccination against HIV (Cosma et al., 2003; Kutscher et al., 2010).
In chronic hepatitis B the rationale for a DNA prime/poxviral boost vaccination stems from a chimpanzee study using HBsAg‐based DNA prime (encoding S protein) followed by canarypox boost vaccination (encoding for S, preS1/2) leading to HBV–DNA loss for more than 3 years (Pancholi et al., 2001). Based on these encouraging data a DNA/MVA prime/boost (S/preS2) regimen was designed and finally investigated in a clinical trial in The Gambia (Cavenaugh et al., 2011). Different doses and application routes were tested in a cohort of chronic hepatitis B patients with or without additional treatment with the nucleoside analogue lamivudine. The treatment was well tolerated but did not add any beneficial effects to standard antiviral treatment in terms of reducing viraemia or inducing HBsAg‐specific T‐cell responses (Cavenaugh et al., 2011). The disappointing results may be explained by the choice of antigen: vaccination with S alone has been insufficient to generate multi‐specific and strong T‐cell responses. In addition, most study participants acquired HBV infection around birth or during early childhood and likely showed profound immune tolerance.
Although so far rather disappointing, these studies help to broaden our knowledge about how therapeutic vaccines work. They show how important proper selection of patient groups as well as optimal vaccine design is.
Cell‐based immunotherapies
Transfer of peptide‐loaded antigen‐presenting, autologous DC, is a therapeutic strategy for induction of T‐cell responses currently under intensive investigation in cancer immunotherapy (Ueno et al., 2010). A DC vaccine, primed with either HBV core‐ or preS2‐derived peptides were administered to 380 patients resulting in HBV viraemia below detection levels in almost 50% of HBeAg negative, but rarely in HBeAg positive patients. DC vaccinated patients normalized ALT levels and restored antiviral immunity without major side effects (Luo et al., 2010).
Alternative approaches are still rather experimental. Dendritic cells can be effectively pulsed with subviral HBV particles and induce specific immunity in mice (Farag et al., 2010). Pulsing with HBsAg and core particles was able to break tolerance in HBV transgenic mice and induced proliferation of autologous T cells from chronic hepatitis B patients in vitro (Akbar et al., 2010). Clinical proof of these effects, however, is missing.
T cell‐based therapies are a valid alternative to vaccination strategies. Adoptive T‐cell transfer, i.e. infusion of various effector T‐cell subsets, aims at eliminating, e.g. tumour cells or virus infected cells (June, 2007). For hepatitis B, the concept is based on a clinical observation. HBV‐infected cancer patients occasionally received human progenitor cell transplants from HBV‐immune individuals. In a long‐term follow‐up study, 20 of 31 of such recipients cleared their HBV infection (Hui et al., 2005). These results encourage the development of adoptive T‐cell transfer strategies for the treatment of chronic viral hepatitis.
Since HBV‐specific T cells can hardly be expanded from HBV‐infected patients and bone marrow transplantation is limited by its side effects, an alternative strategy is currently developed: adoptive transfer of receptor‐modified T cells (reviewed in Hawkins et al., 2010; Protzer and Abken, 2010).
For this approach either cloned T‐cell receptor α and β chains or artificial chimeric antigen receptors (CAR) can be used. CAR are composed of an antigen‐binding domain, which is frequently a single‐chain antibody, and a signal‐transducing unit, often derived from the CD3 ζ chain optionally fused to the co‐stimulatory CD28‐signalling domain (Abken et al., 2003; Riddell and Protzer, 2010). By grafting autologous T cells with CAR or cloned T‐cell receptor α and β chains (Bohne et al., 2008; Gehring et al., 2010), HBV specificity can be genetically introduced into T cells of chronic hepatitis B patients. These grafted T cells are then transferred back into the patient (Protzer and Abken, 2010).
Although very promising, the most striking disadvantages of cell‐based vaccines and T‐cell therapies are the high costs and the complex on‐site requirements for production and application. It is therefore very unlikely that they will provide a suitable treatment for the majority of the 370 million chronically HBV‐infected patients.
Concluding remarks: implications for the design of therapeutic hepatitis B vaccines
Ideally, both innate and adaptive immune responses should be triggered by HBV‐specific therapeutic vaccination leading to a strong and multi‐specific T‐ and B‐cell immunity against several HBV antigens. Therapeutic vaccinations for chronic HBV infection aim to overcome immunosuppressive effects mediated by high antigen load, the tolerogenic liver environment, but also T‐cell dysfunction induced, e.g. by increased Treg activity.
Since T‐cell responses do not only contribute to virus control but also to disease progression, it is of importance to avoid inducing a weak T‐cell response, which does not clear the virus but may cause persistent tissue damage when stimulating the immune system by a therapeutic vaccine. Monitoring of viral parameters allows us to define successful T‐cell responses, but characteristics of such T cells are still merely defined. In addition, further research is needed in order to tailor therapeutic hepatitis B vaccines to induce an adaptive immune response that is able to control infection without shifting towards immunopathology.
For validation of any approach, patient cohorts should be selected carefully and stratified according to criteria influencing the degree of immune tolerance such as the route of transmission, age at HBV infection and duration of infection, as well as antigen levels and presence of immunosuppressive cells (e.g. regulatory or exhausted T cells). We need immunotherapeutic clinical trials in which immunologic mechanisms are monitored in detail. If vaccine immunogenicity was investigated at all in former trials, applied methods differed significantly between studies, precluding direct comparison of results. To improve immunotherapies for any chronic infection standardized and reliable immune monitoring will be essential (Janetzki et al., 2009). Detailed investigation of the different subsets of HBV‐specific CD4+ and CD8+ T cells as well as monitoring of B cell and innate immune responses is needed to advance our understanding of immunomodulatory mechanisms and tailor therapeutic strategies.
Figure 2 illustrates the rationale for development of an optimized candidate therapeutic vaccine against HBV according to current knowledge. As T cell‐inducing vaccines so far did not display efficacy in high viraemic patients or patients with high antigen load, any HBV‐specific immunotherapy scheme should aim at reducing viraemia as well as antigenaemia prior to vaccination. To reduce viraemia effective directly acting antivirals are available, but they decrease antigenaemia only after long‐term treatment – if at all. Antigenaemia is more effectively reduced by neutralizing antibodies, which can be induced by vaccination. An ideal vaccine should therefore first induce neutralizing antibodies and in a second‐step T cells. Protein‐based vaccines combined with potent adjuvants seem to be well suited for priming in prime‐boost vaccination and mainly induce antibodies. Alum‐based adjuvants, however, should be avoided since they induce a Th2 type immune response, which can hardly be shifted. Following reduction of viraemia and antigenaemia viral vector vaccines, such as MVA‐ or adenovirus‐based vaccines encoding for HBV antigens are most efficient to induce effective HBV‐specific CTL. In addition, the synergistic effect of HBsAg and HBcAg should be exploited to stimulate broad and strong HBV‐specific B‐ and T‐cell responses to decrease antigenic load and to break existing immune tolerance in chronically HBV‐infected patients.
Figure 2.
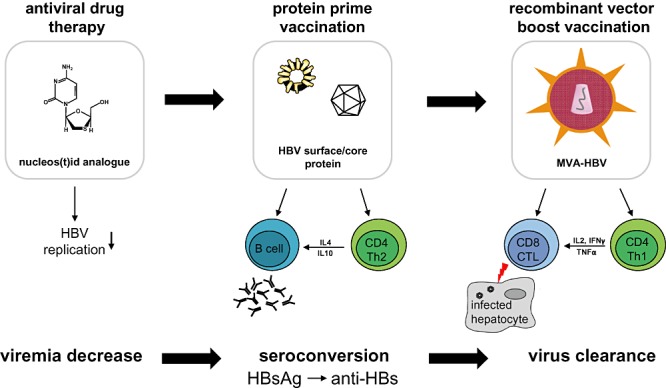
Rationale for development of an optimized candidate therapeutic vaccine against HBV. Antiviral drug treatment inhibits HBV replication and thereby efficiently reduces viraemia. HBV surface or core protein prime vaccination stimulates HBV‐specific CD4+ T cell help leading to antibody production by HBV‐specific B cells. This results in the production of antigen neutralizing antibodies and ideally in seroconversion from HBsAg to anti‐HBs. However, only vaccination with recombinant vector vaccines, encoding HBV DNA, seems to be able to induce CD8+ CTL able to kill infected hepatocytes finally resulting in virus clearance. HBV, hepatitis B virus; HBsAg, hepatitis B surface antigen; HBcAg, hepatitis B core antigen; anti‐HBs, antibodies against HBsAg; MVA, modified vaccinia virus Ankara.
Acknowledgments
The authors thank Simone Allgayer for help with figure design.
References
- Abken H., Hombach A., Heuser C. Immune response manipulation: recombinant immunoreceptors endow T‐cells with predefined specificity. Curr Pharm Des. 2003;9:1992–2001. doi: 10.2174/1381612033454289. [DOI] [PubMed] [Google Scholar]
- Addison W.R., Walters K.A., Wong W.W., Wilson J.S., Madej D., Jewell L.D., Tyrrell D.L. Half‐life of the duck hepatitis B virus covalently closed circular DNA pool in vivo following inhibition of viral replication. J Virol. 2002;76:6356–6363. doi: 10.1128/JVI.76.12.6356-6363.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar J.C., Lobaina Y., Muzio V., Garcia D., Penton E., Iglesias E. Development of a nasal vaccine for chronic hepatitis B infection that uses the ability of hepatitis B core antigen to stimulate a strong Th1 response against hepatitis B surface antigen. Immunol Cell Biol. 2004;82:539–546. doi: 10.1111/j.0818-9641.2004.01278.x. et al. [DOI] [PubMed] [Google Scholar]
- Akbar S.M., Yoshida O., Chen S., Cesar A.J., Abe M., Matsuura B. Immune modulator and antiviral potential of dendritic cells pulsed with both hepatitis B surface antigen and core antigen for treating chronic HBV infection. Antivir Ther. 2010;15:887–895. doi: 10.3851/IMP1637. et al. [DOI] [PubMed] [Google Scholar]
- Akbar S.M.F., Onji M. Preventive and therapeutic approaches against hepatitis B virus by vaccine. Hep B Annual. 2005;2:31–55. [Google Scholar]
- Bachmann M.F., Jennings G.T. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat Rev Immunol. 2010;10:787–796. doi: 10.1038/nri2868. [DOI] [PubMed] [Google Scholar]
- Bauer T., Sprinzl M., Protzer U. Immune control of hepatitis B virus. Dig Dis. 2011;29:423–433. doi: 10.1159/000329809. [DOI] [PubMed] [Google Scholar]
- Beckebaum S., Cicinnati V.R., Gerken G. DNA‐based immunotherapy: potential for treatment of chronic viral hepatitis? Rev Med Virol. 2002;12:297–319. doi: 10.1002/rmv.359. [DOI] [PubMed] [Google Scholar]
- Berthoud T.K., Hamill M., Lillie P.J., Hwenda L., Collins K.A., Ewer K.J. Potent CD8+ T‐cell immunogenicity in humans of a novel heterosubtypic influenza A vaccine, MVA‐NP+M1. Clin Infect Dis. 2011;52:1–7. doi: 10.1093/cid/ciq015. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancourt A.A., Delgado C.A., Estevez Z.C., Martinez J.C., Rios G.V., Aureoles‐Rosello S.R. Phase I clinical trial in healthy adults of a nasal vaccine candidate containing recombinant hepatitis B surface and core antigens. Int J Infect Dis. 2007;11:394–401. doi: 10.1016/j.ijid.2006.09.010. et al. [DOI] [PubMed] [Google Scholar]
- Bohne F., Chmielewski M., Ebert G., Wiegmann K., Kurschner T., Schulze A. T cells redirected against hepatitis B virus surface proteins eliminate infected hepatocytes. Gastroenterology. 2008;134:239–247. doi: 10.1053/j.gastro.2007.11.002. et al. [DOI] [PubMed] [Google Scholar]
- van Bommel F., de Man R.A., Wedemeyer H., Deterding K., Petersen J., Buggisch P. Long‐term efficacy of tenofovir monotherapy for hepatitis B virus‐monoinfected patients after failure of nucleoside/nucleotide analogues. Hepatology. 2010;51:73–80. doi: 10.1002/hep.23246. et al. [DOI] [PubMed] [Google Scholar]
- Boni C., Bertoletti A., Penna A., Cavalli A., Pilli M., Urbani S. Lamivudine treatment can restore T cell responsiveness in chronic hepatitis B. J Clin Invest. 1998;102:968–975. doi: 10.1172/JCI3731. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavenaugh J.S., Awi D., Mendy M., Hill A.V., Whittle H., McConkey S.J. Partially randomized, non‐blinded trial of DNA and MVA therapeutic vaccines based on hepatitis B virus surface protein for chronic HBV infection. PLoS ONE. 2011;6:e14626. doi: 10.1371/journal.pone.0014626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A., Ahlen G., Brenndorfer E.D., Brass A., Holmstrom F., Chen M. Heterologous T cells can help restore function in dysfunctional hepatitis C virus nonstructural 3/4A‐specific T cells during therapeutic vaccination. J Immunol. 2011;186:5107–5118. doi: 10.4049/jimmunol.1001790. et al. [DOI] [PubMed] [Google Scholar]
- Chen C.J., Yang H.I., Su J., Jen C.L., You S.L., Lu S.N. Risk of hepatocellular carcinoma across a biological gradient of serum hepatitis B virus DNA level. JAMA. 2006;295:65–73. doi: 10.1001/jama.295.1.65. et al. [DOI] [PubMed] [Google Scholar]
- Coffman R.L., Sher A., Seder R.A. Vaccine adjuvants: putting innate immunity to work. Immunity. 2010;33:492–503. doi: 10.1016/j.immuni.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornberg M., Protzer U., Dollinger M.M., Petersen J., Wedemeyer H., Berg T. The German guideline for the management of hepatitis B virus infection: short version. J Viral Hepat. 2008;15(1):1–21. doi: 10.1111/j.1365-2893.2008.01013.x. et al (Suppl. [DOI] [PubMed] [Google Scholar]
- Cornberg M., Protzer U., Petersen J., Wedemeyer H., Berg T., Jilg W. [Prophylaxis, diagnosis and therapy of hepatitis B virus infection – the german guideline] Z Gastroenterol. 2011;49:871–930. doi: 10.1055/s-2007-963714. et al. [DOI] [PubMed] [Google Scholar]
- Cosma A., Nagaraj R., Buhler S., Hinkula J., Busch D.H., Sutter G. Therapeutic vaccination with MVA‐HIV‐1 nef elicits Nef‐specific T‐helper cell responses in chronically HIV‐1 infected individuals. Vaccine. 2003;22:21–29. doi: 10.1016/s0264-410x(03)00538-3. et al. [DOI] [PubMed] [Google Scholar]
- Crispe I.N., Giannandrea M., Klein I., John B., Sampson B., Wuensch S. Cellular and molecular mechanisms of liver tolerance. Immunol Rev. 2006;213:101–118. doi: 10.1111/j.1600-065X.2006.00435.x. [DOI] [PubMed] [Google Scholar]
- Dahmen A., Herzog‐Hauff S., Bocher W.O., Galle P.R., Lohr H.F. Clinical and immunological efficacy of intradermal vaccine plus lamivudine with or without interleukin‐2 in patients with chronic hepatitis B. J Med Virol. 2002;66:452–460. doi: 10.1002/jmv.2165. [DOI] [PubMed] [Google Scholar]
- Das A., Maini M.K. Innate and adaptive immune responses in hepatitis B virus infection. Dig Dis. 2010;28:126–132. doi: 10.1159/000282075. [DOI] [PubMed] [Google Scholar]
- Das A., Hoare M., Davies N., Lopes A.R., Dunn C., Kennedy P.T. Functional skewing of the global CD8 T cell population in chronic hepatitis B virus infection. J Exp Med. 2008;205:2111–2124. doi: 10.1084/jem.20072076. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienstag J.L., Stevens C.E., Bhan A.K., Szmuness W. Hepatitis B vaccine administered to chronic carriers of hepatitis B surface antigen. Ann Intern Med. 1982;96:575–579. doi: 10.7326/0003-4819-96-5-575. [DOI] [PubMed] [Google Scholar]
- Dunn C., Peppa D., Khanna P., Nebbia G., Jones M., Brendish N. Temporal analysis of early immune responses in patients with acute hepatitis B virus infection. Gastroenterology. 2009;137:1289–1300. doi: 10.1053/j.gastro.2009.06.054. et al. [DOI] [PubMed] [Google Scholar]
- Farag M.M., Hoyler B., Encke J., Stremmel W., Weigand K. Dendritic cells can effectively be pulsed by HBVsvp and induce specific immune reactions in mice. Vaccine. 2010;29:200–206. doi: 10.1016/j.vaccine.2010.10.056. [DOI] [PubMed] [Google Scholar]
- Finzi D., Blankson J., Siliciano J.D., Margolick J.B., Chadwick K., Pierson T. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV‐1, even in patients on effective combination therapy. Nat Med. 1999;5:512–517. doi: 10.1038/8394. et al. [DOI] [PubMed] [Google Scholar]
- Fisicaro P., Valdatta C., Massari M., Loggi E., Biasini E., Sacchelli L. Antiviral intrahepatic T‐cell responses can be restored by blocking programmed death‐1 pathway in chronic hepatitis B. Gastroenterology. 2010;138:682–693.e4. doi: 10.1053/j.gastro.2009.09.052. et al. [DOI] [PubMed] [Google Scholar]
- Ganem D., Prince A.M. Hepatitis B virus infection – natural history and clinical consequences. N Engl J Med. 2004;350:1118–1129. doi: 10.1056/NEJMra031087. [DOI] [PubMed] [Google Scholar]
- Gehring A.J., Xue S.A., Ho Z.Z., Teoh D., Ruedl C., Chia A. Engineering virus‐specific T cells that target HBV infected hepatocytes and hepatocellular carcinoma cell lines. J Hepatol. 2010;55:103–110. doi: 10.1016/j.jhep.2010.10.025. et al. [DOI] [PubMed] [Google Scholar]
- Gudmundsdotter L., Sjodin A., Bostrom A.C., Hejdeman B., Theve‐Palm R., Alaeus A. Therapeutic immunization for HIV. Springer Semin Immunopathol. 2006;28:221–230. doi: 10.1007/s00281-006-0029-0. et al. [DOI] [PubMed] [Google Scholar]
- Hawkins R.E., Gilham D.E., Debets R., Eshhar Z., Taylor N., Abken H. Development of adoptive cell therapy for cancer: a clinical perspective. Hum Gene Ther. 2010;21:665–672. doi: 10.1089/hum.2010.086. et al. [DOI] [PubMed] [Google Scholar]
- Heathcote E.J., Marcellin P., Buti M., Gane E., De Man R.A., Krastev Z. Three‐year efficacy and safety of tenofovir disoproxil fumarate treatment for chronic hepatitis B. Gastroenterology. 2011;140:132–143. doi: 10.1053/j.gastro.2010.10.011. et al. [DOI] [PubMed] [Google Scholar]
- Heathcote J., McHutchison J., Lee S., Tong M., Benner K., Minuk G. A pilot study of the CY‐1899 T‐cell vaccine in subjects chronically infected with hepatitis B virus. The CY1899 T cell vaccine study group. Hepatology. 1999;30:531–536. doi: 10.1002/hep.510300208. et al. [DOI] [PubMed] [Google Scholar]
- Heintges T., Petry W., Kaldewey M., Erhardt A., Wend U.C., Gerlich W.H. Combination therapy of active HBsAg vaccination and interferon‐alpha in interferon‐alpha nonresponders with chronic hepatitis B. Dig Dis Sci. 2001;46:901–906. doi: 10.1023/a:1010785325067. et al. [DOI] [PubMed] [Google Scholar]
- Hildesheim A., Herrero R., Wacholder S., Rodriguez A.C., Solomon D., Bratti M.C. Effect of human papillomavirus 16/18 L1 viruslike particle vaccine among young women with preexisting infection: a randomized trial. JAMA. 2007;298:743–753. doi: 10.1001/jama.298.7.743. et al. [DOI] [PubMed] [Google Scholar]
- Hilleman M.R. Critical overview and outlook: pathogenesis, prevention, and treatment of hepatitis and hepatocarcinoma caused by hepatitis B virus. Vaccine. 2003;21:4626–4649. doi: 10.1016/s0264-410x(03)00529-2. [DOI] [PubMed] [Google Scholar]
- Hoofnagle J.H., Doo E., Liang T.J., Fleischer R., Lok A.S. Management of hepatitis B: summary of a clinical research workshop. Hepatology. 2007;45:1056–1075. doi: 10.1002/hep.21627. [DOI] [PubMed] [Google Scholar]
- Horiike N., Fazle Akbar S.M., Michitaka K., Joukou K., Yamamoto K., Kojima N. In vivo immunization by vaccine therapy following virus suppression by lamivudine: a novel approach for treating patients with chronic hepatitis B. J Clin Virol. 2005;32:156–161. doi: 10.1016/j.jcv.2004.07.004. et al. [DOI] [PubMed] [Google Scholar]
- Hui C.K., Lie A., Au W.Y., Leung Y.H., Ma S.Y., Cheung W.W. A long‐term follow‐up study on hepatitis B surface antigen‐positive patients undergoing allogeneic hematopoietic stem cell transplantation. Blood. 2005;106:464–469. doi: 10.1182/blood-2005-02-0698. et al. [DOI] [PubMed] [Google Scholar]
- Im S.J., Yang S.H., Yoon S.K., Sung Y.C. Increase of plasma IL‐12/p40 ratio induced by the combined therapy of DNA vaccine and lamivudine correlates with sustained viremia control in CHB carriers. Immune Netw. 2009;9:20–26. doi: 10.4110/in.2009.9.1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janetzki S., Britten C.M., Kalos M., Levitsky H.I., Maecker H.T., Melief C.J. ‘MIATA’‐minimal information about T cell assays. Immunity. 2009;31:527–528. doi: 10.1016/j.immuni.2009.09.007. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- June C.H. Principles of adoptive T cell cancer therapy. J Clin Invest. 2007;117:1204–1212. doi: 10.1172/JCI31446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakimi K., Isogawa M., Chung J., Sette A., Chisari F.V. Immunogenicity and tolerogenicity of hepatitis B virus structural and nonstructural proteins: implications for immunotherapy of persistent viral infections. J Virol. 2002;76:8609–8620. doi: 10.1128/JVI.76.17.8609-8620.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knolle P.A., Gerken G. Local control of the immune response in the liver. Immunol Rev. 2000;174:21–34. doi: 10.1034/j.1600-0528.2002.017408.x. [DOI] [PubMed] [Google Scholar]
- Kosinska A.D., Zhang E., Lu M., Roggendorf M. Therapeutic vaccination in chronic hepatitis B: preclinical studies in the woodchuck. Hepat Res Treat. 2010;2010:817580. doi: 10.1155/2010/817580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutscher S., Allgayer S., Dembek C.J., Bogner J.R., Protzer U., Goebel F.D. MVA‐nef induces HIV‐1‐specific polyfunctional and proliferative T‐cell responses revealed by the combination of short‐ and long‐term immune assays. Gene Ther. 2010;17:1372–1383. doi: 10.1038/gt.2010.90. et al. [DOI] [PubMed] [Google Scholar]
- Kwon H., Lok A.S. Hepatitis B therapy. Nat Rev Gastroenterol Hepatol. 2011;8:275–284. doi: 10.1038/nrgastro.2011.33. [DOI] [PubMed] [Google Scholar]
- Lazdina U., Cao T., Steinbergs J., Alheim M., Pumpens P., Peterson D.L. Molecular basis for the interaction of the hepatitis B virus core antigen with the surface immunoglobulin receptor on naive B cells. J Virol. 2001;75:6367–6374. doi: 10.1128/JVI.75.14.6367-6374.2001. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levrero M., Pollicino T., Petersen J., Belloni L., Raimondo G., Dandri M. Control of cccDNA function in hepatitis B virus infection. J Hepatol. 2009;51:581–592. doi: 10.1016/j.jhep.2009.05.022. [DOI] [PubMed] [Google Scholar]
- Liu M.A. DNA vaccines: an historical perspective and view to the future. Immunol Rev. 2011;239:62–84. doi: 10.1111/j.1600-065X.2010.00980.x. [DOI] [PubMed] [Google Scholar]
- Loomba R., Liang T.J. Novel approaches to new therapies for hepatitis B virus infection. Antivir Ther. 2006;11:1–15. [PubMed] [Google Scholar]
- Luo J., Li J., Chen R.L., Nie L., Huang J., Liu Z.W. Autologus dendritic cell vaccine for chronic hepatitis B carriers: a pilot, open label, clinical trial in human volunteers. Vaccine. 2010;28:2497–2504. doi: 10.1016/j.vaccine.2010.01.038. et al. [DOI] [PubMed] [Google Scholar]
- McMahon B.J. The natural history of chronic hepatitis B virus infection. Hepatology. 2009;49:S45–S55. doi: 10.1002/hep.22898. [DOI] [PubMed] [Google Scholar]
- Mancini‐Bourgine M., Fontaine H., Scott‐Algara D., Pol S., Brechot C., Michel M.L. Induction or expansion of T‐cell responses by a hepatitis B DNA vaccine administered to chronic HBV carriers. Hepatology. 2004;40:874–882. doi: 10.1002/hep.20408. [DOI] [PubMed] [Google Scholar]
- Manigold T., Racanelli V. T‐cell regulation by CD4 regulatory T cells during hepatitis B and C virus infections: facts and controversies. Lancet Infect Dis. 2007;7:804–813. doi: 10.1016/S1473-3099(07)70289-X. [DOI] [PubMed] [Google Scholar]
- Manns M.P., Foster G.R., Rockstroh J.K., Zeuzem S., Zoulim F., Houghton M. The way forward in HCV treatment – finding the right path. Nat Rev Drug Discov. 2007;6:991–1000. doi: 10.1038/nrd2411. [DOI] [PubMed] [Google Scholar]
- Marcellin P., Bonino F., Lau G.K., Farci P., Yurdaydin C., Piratvisuth T. Sustained response of hepatitis B e antigen‐negative patients 3 years after treatment with peginterferon alpha‐2a. Gastroenterology. 2009;136:2169–2179.e4. doi: 10.1053/j.gastro.2009.03.006. et al. [DOI] [PubMed] [Google Scholar]
- Mayr A., Stickl H., Muller H.K., Danner K., Singer H. [The smallpox vaccination strain MVA: marker, genetic structure, experience gained with the parenteral vaccination and behavior in organisms with a debilitated defence mechanism] Zentralbl Bakteriol B. 1978;167:375–390. [PubMed] [Google Scholar]
- Menne S., Roneker C.A., Korba B.E., Gerin J.L., Tennant B.C., Cote P.J. Immunization with surface antigen vaccine alone and after treatment with 1‐(2‐fluoro‐5‐methyl‐beta‐l‐arabinofuranosyl)‐uracil (L‐FMAU) breaks humoral and cell‐mediated immune tolerance in chronic woodchuck hepatitis virus infection. J Virol. 2002;76:5305–5314. doi: 10.1128/JVI.76.11.5305-5314.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel M.L., Deng Q., Mancini‐Bourgine M. Therapeutic vaccines and immune‐based therapies for the treatment of chronic hepatitis B: perspectives and challenges. J Hepatol. 2011;54:1286–1296. doi: 10.1016/j.jhep.2010.12.031. [DOI] [PubMed] [Google Scholar]
- Milich D.R., Thornton G.B., Neurath A.R., Kent S.B., Michel M.L., Tiollais P., Chisari F.V. Enhanced immunogenicity of the pre‐S region of hepatitis B surface antigen. Science. 1985;228:1195–1199. doi: 10.1126/science.2408336. [DOI] [PubMed] [Google Scholar]
- Milich D.R., Chen M., Schodel F., Peterson D.L., Jones J.E., Hughes J.L. Role of B cells in antigen presentation of the hepatitis B core. Proc Natl Acad Sci USA. 1997;94:14648–14653. doi: 10.1073/pnas.94.26.14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moucari R., Korevaar A., Lada O., Martinot‐Peignoux M., Boyer N., Mackiewicz V. High rates of HBsAg seroconversion in HBeAg‐positive chronic hepatitis B patients responding to interferon: a long‐term follow‐up study. J Hepatol. 2009;50:1084–1092. doi: 10.1016/j.jhep.2009.01.016. et al. [DOI] [PubMed] [Google Scholar]
- Munir S., Saleem S., Idrees M., Tariq A., Butt S., Rauff B. Hepatitis C treatment: current and future perspectives. Virol J. 2010;7:296. doi: 10.1186/1743-422X-7-296. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pancholi P., Lee D.H., Liu Q., Tackney C., Taylor P., Perkus M. DNA prime/canarypox boost‐based immunotherapy of chronic hepatitis B virus infection in a chimpanzee. Hepatology. 2001;33:448–454. doi: 10.1053/jhep.2001.21594. et al. [DOI] [PubMed] [Google Scholar]
- Paris R.M., Kim J.H., Robb M.L., Michael N.L. Prime‐boost immunization with poxvirus or adenovirus vectors as a strategy to develop a protective vaccine for HIV‐1. Expert Rev Vaccines. 2010;9:1055–1069. doi: 10.1586/erv.10.106. [DOI] [PubMed] [Google Scholar]
- Protzer U., Abken H. Can engineered ‘designer’ T cells outsmart chronic hepatitis B? Hepat Res Treat. 2010;2010:901216. doi: 10.1155/2010/901216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehermann B., Nascimbeni M. Immunology of hepatitis B virus and hepatitis C virus infection. Nat Rev Immunol. 2005;5:215–229. doi: 10.1038/nri1573. [DOI] [PubMed] [Google Scholar]
- Riddell S.R., Protzer U. Carving the CAR. Gene Ther. 2010;17:1191–1192. doi: 10.1038/gt.2010.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roggendorf M., Yang D., Lu M. The woodchuck: a model for therapeutic vaccination against hepadnaviral infection. Pathol Biol (Paris) 2010;58:308–314. doi: 10.1016/j.patbio.2010.04.005. [DOI] [PubMed] [Google Scholar]
- Rouse B.T., Sehrawat S. Immunity and immunopathology to viruses: what decides the outcome? Nat Rev Immunol. 2010;10:514–526. doi: 10.1038/nri2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmbeck R., Dikopoulos N., Kwissa M., Leithauser F., Lamberth K., Buus S. Breaking tolerance in hepatitis B surface antigen (HBsAg) transgenic mice by vaccination with cross‐reactive, natural HBsAg variants. Eur J Immunol. 2003;33:3342–3352. doi: 10.1002/eji.200324403. et al. [DOI] [PubMed] [Google Scholar]
- Schumann A., Fiedler M., Dahmen U., Grosse‐Wilde H., Roggendorf M., Lindemann M. Cellular and humoral immune response to a third generation hepatitis B vaccine. J Viral Hepat. 2007;14:592–598. doi: 10.1111/j.1365-2893.2007.00848.x. [DOI] [PubMed] [Google Scholar]
- Seder R.A., Darrah P.A., Roederer M. T‐cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol. 2008;8:247–258. doi: 10.1038/nri2274. [DOI] [PubMed] [Google Scholar]
- Stoop J.N., van der Molen R.G., Baan C.C., van der Laan L.J., Kuipers E.J., Kusters J.G., Janssen H.L. Regulatory T cells contribute to the impaired immune response in patients with chronic hepatitis B virus infection. Hepatology. 2005;41:771–778. doi: 10.1002/hep.20649. [DOI] [PubMed] [Google Scholar]
- Storni T., Ruedl C., Schwarz K., Schwendener R.A., Renner W.A., Bachmann M.F. Nonmethylated CG motifs packaged into virus‐like particles induce protective cytotoxic T cell responses in the absence of systemic side effects. J Immunol. 2004;172:1777–1785. doi: 10.4049/jimmunol.172.3.1777. [DOI] [PubMed] [Google Scholar]
- Sutter G., Moss B. Nonreplicating vaccinia vector efficiently expresses recombinant genes. Proc Natl Acad Sci USA. 1992;89:10847–10851. doi: 10.1073/pnas.89.22.10847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang T.J., Kwekkeboom J., Mancham S., Binda R.S., de Man R.A., Schalm S.W. Intrahepatic CD8+ T‐lymphocyte response is important for therapy‐induced viral clearance in chronic hepatitis B infection. J Hepatol. 2005;43:45–52. doi: 10.1016/j.jhep.2005.01.038. et al. [DOI] [PubMed] [Google Scholar]
- Thimme R., Wieland S., Steiger C., Ghrayeb J., Reimann K.A., Purcell R.H., Chisari F.V. CD8(+) T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J Virol. 2003;77:68–76. doi: 10.1128/JVI.77.1.68-76.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson A.W., Knolle P.A. Antigen‐presenting cell function in the tolerogenic liver environment. Nat Rev Immunol. 2010;10:753–766. doi: 10.1038/nri2858. [DOI] [PubMed] [Google Scholar]
- Ueno H., Schmitt N., Klechevsky E., Pedroza‐Gonzalez A., Matsui T., Zurawski G. Harnessing human dendritic cell subsets for medicine. Immunol Rev. 2010;234:199–212. doi: 10.1111/j.0105-2896.2009.00884.x. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandepapeliere P., Lau G.K., Leroux‐Roels G., Horsmans Y., Gane E., Tawandee T. Therapeutic vaccination of chronic hepatitis B patients with virus suppression by antiviral therapy: a randomized, controlled study of co‐administration of HBsAg/AS02 candidate vaccine and lamivudine. Vaccine. 2007;25:8585–8597. doi: 10.1016/j.vaccine.2007.09.072. et al. [DOI] [PubMed] [Google Scholar]
- Vandepapeliere P., Horsmans Y., Moris P., Van Mechelen M., Janssens M., Koutsoukos M. Vaccine adjuvant systems containing monophosphoryl lipid A and QS21 induce strong and persistent humoral and T cell responses against hepatitis B surface antigen in healthy adult volunteers. Vaccine. 2008;26:1375–1386. doi: 10.1016/j.vaccine.2007.12.038. et al. [DOI] [PubMed] [Google Scholar]
- Virgin H.W., Wherry E.J., Ahmed R. Redefining chronic viral infection. Cell. 2009;138:30–50. doi: 10.1016/j.cell.2009.06.036. [DOI] [PubMed] [Google Scholar]
- Vitiello A., Ishioka G., Grey H.M., Rose R., Farness P., LaFond R. Development of a lipopeptide‐based therapeutic vaccine to treat chronic HBV infection. I. Induction of a primary cytotoxic T lymphocyte response in humans. J Clin Invest. 1995;95:341–349. doi: 10.1172/JCI117662. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F.S., Zhang Z. Host immunity influences disease progression and antiviral efficacy in humans infected with hepatitis B virus. Expert Rev Gastroenterol Hepatol. 2009;3:499–512. doi: 10.1586/egh.09.50. [DOI] [PubMed] [Google Scholar]
- Webster G.J., Reignat S., Maini M.K., Whalley S.A., Ogg G.S., King A. Incubation phase of acute hepatitis B in man: dynamic of cellular immune mechanisms. Hepatology. 2000;32:1117–1124. doi: 10.1053/jhep.2000.19324. et al. [DOI] [PubMed] [Google Scholar]
- Webster G.J., Reignat S., Brown D., Ogg G.S., Jones L., Seneviratne S.L. Longitudinal analysis of CD8+ T cells specific for structural and nonstructural hepatitis B virus proteins in patients with chronic hepatitis B: implications for immunotherapy. J Virol. 2004;78:5707–5719. doi: 10.1128/JVI.78.11.5707-5719.2004. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland S.F., Spangenberg H.C., Thimme R., Purcell R.H., Chisari F.V. Expansion and contraction of the hepatitis B virus transcriptional template in infected chimpanzees. Proc Natl Acad Sci USA. 2004;101:2129–2134. doi: 10.1073/pnas.0308478100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodland D.L. Jump‐starting the immune system: prime‐boosting comes of age. Trends Immunol. 2004;25:98–104. doi: 10.1016/j.it.2003.11.009. [DOI] [PubMed] [Google Scholar]
- Wu J., Lu M., Meng Z., Trippler M., Broering R., Szczeponek A. Toll‐like receptor‐mediated control of HBV replication by nonparenchymal liver cells in mice. Hepatology. 2007;46:1769–1778. doi: 10.1002/hep.21897. et al. [DOI] [PubMed] [Google Scholar]
- Xu D.Z., Zhao K., Guo L.M., Li L.J., Xie Q., Ren H. A randomized controlled phase IIb trial of antigen‐antibody immunogenic complex therapeutic vaccine in chronic hepatitis B patients. PLoS ONE. 2008;3:e2565. doi: 10.1371/journal.pone.0002565. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalcin K., Danis R., Degertekin H., Alp M.N., Tekes S., Budak T. The lack of effect of therapeutic vaccination with a pre‐S2/S HBV vaccine in the immune tolerant phase of chronic HBV infection. J Clin Gastroenterol. 2003;37:330–335. doi: 10.1097/00004836-200310000-00012. [DOI] [PubMed] [Google Scholar]
- Yang S.H., Lee C.G., Park S.H., Im S.J., Kim Y.M., Son J.M. Correlation of antiviral T‐cell responses with suppression of viral rebound in chronic hepatitis B carriers: a proof‐of‐concept study. Gene Ther. 2006;13:1110–1117. doi: 10.1038/sj.gt.3302751. et al. [DOI] [PubMed] [Google Scholar]
- Zhang Y.Y., Zhang B.H., Theele D., Litwin S., Toll E., Summers J. Single‐cell analysis of covalently closed circular DNA copy numbers in a hepadnavirus‐infected liver. Proc Natl Acad Sci USA. 2003;100:12372–12377. doi: 10.1073/pnas.2033898100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y., Yamamoto T., Cullen J., Saputelli J., Aldrich C.E., Miller D.S. Kinetics of hepadnavirus loss from the liver during inhibition of viral DNA synthesis. J Virol. 2001;75:311–322. doi: 10.1128/JVI.75.1.311-322.2001. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]