

Summary
Bacillus subtilis can serve as a powerful platform for directed evolution, especially for secretory enzymes. However, cloning and transformation of a DNA mutant library in B. subtilis are not as easy as they are in Escherichia coli. For direct transformation of B. subtilis, here we developed a new protocol based on supercompetent cells prepared from the recombinant B. subtilis strain SCK6 and multimeric plasmids. This new protocol is simple (restriction enzyme‐, phosphatase‐ and ligase‐free), fast (i.e. 1 day) and of high efficiency (i.e. ∼107 or ∼104 transformants per µg of multimeric plasmid or ligated plasmid DNA respectively). Supercompetent B. subtilis SCK6 cells were prepared by overexpression of the competence master regulator ComK that was induced by adding xylose. The DNA mutant library was generated through a two‐round PCR: (i) the mutagenized DNA fragments were generated by error‐prone PCR and linearized plasmids were made using high‐fidelity PCR, and (ii) the multimeric plasmids were generated based on these two DNA templates by using overlap PCR. Both protein expression level and specific activity of glycoside hydrolase family 5 endoglucanse on regenerated amorphous cellulose were improved through this new system. To our limited knowledge, this study is the first report for enhancing secretory cellulase performance on insoluble cellulose.
Introduction
Directed enzyme evolution is a protein‐engineering method that harnesses natural selection at the molecular level and directs the evolution of proteins that are customized to meet desired specifications (Zhang et al., 2006a; Romero and Arnold, 2009). In general, it involves several sequential steps –in vitro generation of a DNA mutant library, transformation of the DNA mutant library in the host and selection/screening of protein mutants. Escherichia coli is the most used workhorse for directed enzyme evolution because of its high transformation efficiency (> 109 per µg of DNA) and simple operation, but it is difficult to screen enzymes whose substrates (e.g. cellulose and starch) cannot be transported across the cell membrane. In these cases, cell lysis must be conducted before enzyme activity is screened.
Bacillus subtilis is a good host for the production of secretory proteins (Tjalsma et al., 2004; Yamane et al., 2004; Schumann, 2007; Zhang and Zhang, 2010). Screening or selection of secretory enzymes (e.g. amylase, subtilisin and cellulase) based on B. subtilis would save a great deal of time spent on enzyme assays and characterization (Shafikhani et al., 1997; Amin et al., 2003; Zhang et al., 2006a). Different from E. coli, direct transformation of B. subtilis with a DNA mutant library prepared based on restriction enzymes and ligation is inefficient (Canosi et al., 1978). DNA mutant libraries for secretory enzymes (e.g. subtilisin, amylase) are usually constructed in E. coli and then the DNA plasmid mutant library purified from E. coli is transferred into competent B. subtilis cells (Crameri et al., 1998; Naki et al., 1998; Dunn and Handelsman, 1999; Caspers et al., 2010). As shown in Fig. 1A, the routine protocol based on the B. subtilis–E. coli shuttle vectors is time‐consuming, labour‐intensive and of low efficiency (You and Arnold, 1996; Zhao and Arnold, 1999; Caspers et al., 2010). Furthermore, the passage of a random DNA library through E. coli may introduce bias by selectively removing or amplifying certain clones (Strausberg et al., 1995; Shafikhani et al., 1997).
Figure 1.
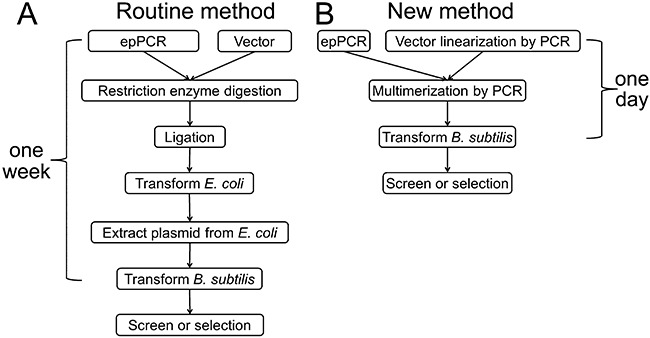
Flow schemes of the routine method and the new method for library construction in B. subtilis.
The B. subtilis competent cells are usually prepared using a two‐step procedure involving two types of minimal media (Cutting and Vander Horn, 1990). For relatively high transformation efficiencies (∼103–104 transformants per µg of DNA), the cell growth curve must be monitored carefully (Ehrlich, 1978; Cutting and Vander Horn, 1990). The reported highest transformation efficiencies of B. subtilis are ∼1–3 × 106 transformants per µg of DNA, either by using the two‐step procedure with multimeric plasmids (Shafikhani et al., 1997) or by using a high‐osmolarity electroporation method (Xue et al., 1999). But both methods are labour‐intensive, time‐consuming and hard to be operated by beginners.
To decrease the labour required for cloning and obtain high B. subtilis transformation efficiencies sufficient for directed evolution of secretory enzymes, we prepared supercompetent B. subtilis cells that can be efficiently transformed by foreign DNA and multimeric plasmids. We modified B. subtilis 1A751 (Wolf et al., 1995) by overexpressing its competence master regulator ComK (Susanna et al., 2006), where the comk gene is controlled by the xylose‐inducible promoter PxylA (Hartl et al., 2001). The supercompetent cells were made by adding 1% (w/v) xylose into the cells growing in the exponential phase for 2 h. Because B. subtilis prefers to be transformed by multimeric plasmids rather than monomeric ones (Canosi et al., 1978), Shafikhani et al. have produced multimeric plasmids by using overlap PCR based on two templates: a DNA mutant library made by error‐prone PCR and a linearized plasmid prepared by digestion with a restriction enzyme (Shafikhani et al., 1997; Collier et al., 2003). Here we modified the Shafikhani protocol by generating a linear plasmid backbone by using high‐fidelity PCR so that this operation can be conducted anywhere in the plasmid (Fig. 2B). As shown in Fig. 1A, it takes about 1 week for the routine method to construct mutant libraries in B. subtilis through the shuttle vector if every step goes smoothly. In contrast, the new protocol is simple (restriction enzyme‐, phosphatase‐, and ligase‐free), fast (i.e. 1 day) and of high efficiency (i.e. 107 per µg of DNA) (Fig. 1B). In this study, the secretory expression level and specific activity of glycoside hydrolase family 5 endoglucanse were improved through this new system.
Figure 2.
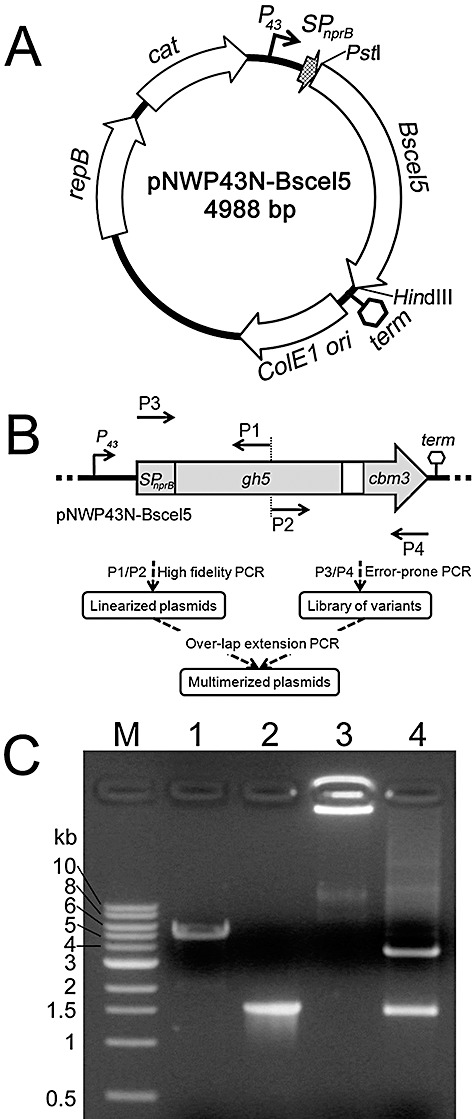
PCR‐based gene mutagenesis and plasmid multimerization. A. Relevant features of the vector pNWP43N‐Bscel5. P43, SPnprB, Bscel5 and term represent the P43 promoter, the NprB signal peptide‐encoding sequence, gene of family 5 endoglucanse and terminator of Bscel5 from B. subtilis respectively. ColE1 ori, repB and cat represent the sequences coding for the ColE1 replication origin, replicase and chloramphenicol resistance marker respectively. The arrows show the transcription directions for these genes. B. The flow scheme of the two‐step PCR procedure for the gene mutagenesis and plasmid multimerization. gh5, family 5 glycoside hydrolase‐encoding sequence; cbm3, family 3 carbohydrate‐binding module‐encoding sequence. P1, P2, P3 and P4 denote the positions of the primers for the PCR amplification. This figure was not drawn to scale. C. Plasmid multimerization by PCR. Lanes: M, DNA markers; 1, PCR‐linearized pNWP43N‐Bscel5; 2, error‐prone PCR product of SPnprB‐Bscel5; 3, multimerized plasmid; 4, multimer digested with PstI/HindIII.
Results
Mutagenesis of Bscel5 and plasmid multimerization using two‐step PCR
For the recombinant expression and secretion of B. subtilis family 5 endoglucanase BsCel5, the B. subtilis–E. coli shuttle vector pNWP43N‐Bscel5 was constructed. It contained a 1480 bp NprB signal peptide‐encoding sequence and a mature BsCel5‐encoding gene, under the control of a strong P43 promoter (Zhang et al., 2005) (Fig. 2A). First, the 1480 bp fragment in vector pNWP43N‐Bscel5 was mutagenized by error‐prone PCR with the primer pair P3/P4 (Fig. 2B). Second, the vector pNWP43N‐Bscel5 was linearized with high‐fidelity PCR by using primers P1 and P2 (Fig. 2B). The locations of P1/P2 can be randomly chosen between the locations of primers P3/P4, which is independent of the restriction enzyme cutting site. Third, multimeric plasmids were generated using high‐fidelity overlap PCR based on the two templates – a linearized vector and a mutagenized SPnprB‐Bscel5 library at a molar ratio of ∼1:100. Such a low ratio of the vector to the mutant fragment decreases the possibility of multimeric plasmid formation based on the vector itself so to decreases the possibility of background false positives. The product after overlap PCR was multimeric plasmids with a yield of ∼100 ng of DNA per µl. High‐molecular‐weight multimeric plasmids did not migrate in agarose gel using regular electrophoresis (Fig. 2C, lane 3). After the PstI and HindIII digestion, the digested product of the multimeric plasmid showed two fragments, 3572 bp and 1416 bp (Fig. 2C, lane 4), in good agreement with the PstI‐ and HindIII‐digested monomeric plasmid pNWP43N‐Bscel5.
Transformation of B. subtilis SCK6
ComK of B. subtilis is the master regulator for competence development. The induction of comK combined with positive autostimulation of native comK results in an increased percentage of competent cells in the population (Maamar and Dubnau, 2005; Smits et al., 2005; Mironczuk et al., 2008). In this study, an extra copy of the comK gene was integrated at the B. subtilis 1A751 lacA locus (Fig. 3) of the chromosome and placed under the control of the xylose‐inducible promoter PxylA (Hartl et al., 2001), resulting in a new recombinant B. subtilis SCK6 that can be easily converted to a supercompetent cell by induction with 1% xylose in the Luria–Bertani (LB) medium.
Figure 3.
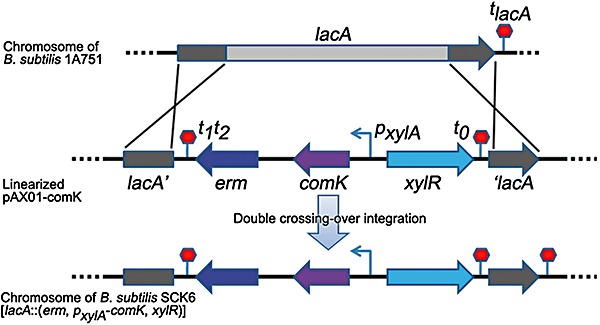
Construction of B. subtilis SCK6. The sequence between two homologous arms (lacA′ and ′lacA) was replaced by the xylose‐inducible comK expression cassette from the linearized pAX01‐comK via a double‐crossing‐over recombination. X indicates one cross‐over event. lacA′ and ′lacA, upstream and downstream sequences of the B. subtilis lacA gene respectively; erm, erythromycin resistance gene; comK, B. subtilis comK gene; pxylA, xylose‐inducible promoter of xylA gene; xylR, B. subtilis xyl operon repressor; tlacA, terminator of B. subtilis lacA gene; t1t2 and t0, terminators of lambda phage.
The induced supercompetent cells of B. subtilis strain SCK6 exhibited transformation efficiencies of ∼1 × 107 transformants per µg of multimeric plasmid DNA. The supercompetent cells also had good transformation efficiencies of ∼1 × 104 per µg of ligated plasmid DNA prepared from the regular cloning process.
Cellulase mutant screening based on insoluble cellulose substrate
The transformed cells containing vector pNWP43N‐Bscel5 were spread on LBR plates containing 0.4% regenerated amorphous cellulose (RAC) and then were incubated at 37°C for 20 h. Because secretory BsCel5 can hydrolyse insoluble cellulose on solid agar plates, it resulted in transparent halos (Fig. 4A and B). The bigger and clearer halo zones indicate that the corresponding cells can secrete a more active enzyme and/or secrete a larger quantity of cellulase. Under the tested mutation conditions, about 48.8% of the clones were estimated to harbour the inactivated enzyme mutants. In Fig. 4A, a positive mutant with a bigger and clearer halo zone compared with wild‐type or negative clones was identified from the plate. Three mutants (MT1, MT2 and MT3) with bigger and clearer halo zones were screened from ∼16 000 colonies on about 20 Petri plates.
Figure 4.
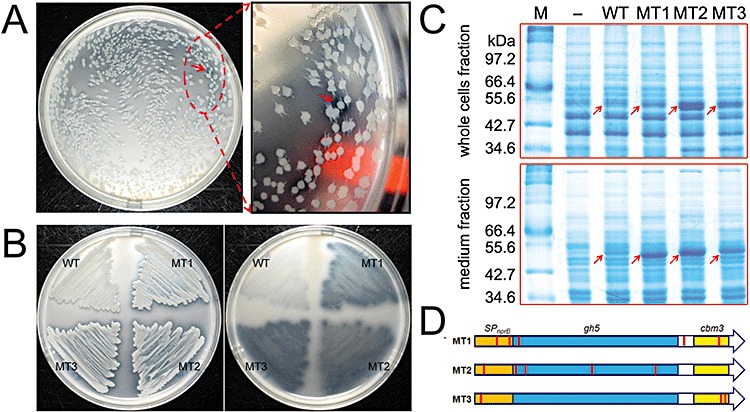
Cellulase mutant library screening for Bscel5 variants. A. A positive mutant with bigger and clearer halo zone (red arrow) was screened. B. Comparison of the performance of wild type (WT) and mutants (MT1, MT2 and MT3) based on the ability to degrade insoluble cellulose, which forms clear halo zones. The B. subtilis strains were streaked on an LBR plate and incubated for 24 h at 37°C. C. BsCel5 expression and secretion profiles of wild type and mutants. M, protein markers; −, negative control. For whole cells fraction, the protein samples corresponding to 10 µl cell culture were loaded for each lane. For medium fraction, the proteins precipitated from 100 µl of culture supernatant were loaded for each lane. The position of BsCel5 is indicated with arrows. D. Base mutations in Bscel5 variants. Red bars indicate the base mutations in the variants.
The three selected mutants and another four randomly selected clones were sequenced to estimate the mutation rates. The mutation sites in the three selected mutants were summarized in Table 1. The overall mutation rate for error‐prone PCR was ∼0.3%. There were two to seven mutations for the selected positive clones. The three positive mutants had one to two mutations in the signal peptide‐encoding region and several mutations in the catalytic module and carbohydrate‐binding module (Fig. 4D). The other randomly selected neutral or negative mutants did not contain any mutation in their signal peptide‐encoding sequence (data not shown).
Table 1.
Amino acid substitutions in the selected BsCel5 mutants and the corresponding base mutations.
| Mutant | Amino acid substitution and corresponding base mutation |
|---|---|
| MT1 | T16I (ACA→ATA), S27P (TCA→CCA), K33I (AAA→ATA), I339T (ATT→ACT), A465* (GCA→GCT) |
| MT2 | S8* (TCT→TCA), A30* (GCA→GCT), T97S (ACG→TCG), S189* (TCA→TCT), Y280* (TAT→TAC) |
| MT3 | T7* (ACA→ACG), K474E (AAA→GAA), K482E (AAA→GAA) |
The asterisk (*) stands for the silent mutation.
Figure 4B shows the growth of strains harbouring one of the three mutants or wild‐type BsCel5 on an LBR plate. After washing the cells from the plate, it was clearly visible that these mutant enzymes had a significant increased ability to hydrolyse RAC compared with the wild‐type enzyme (Fig. 4B). To check the expression level of secretory BsCel5, the strains harbouring wild‐type Bscel5 and mutants were cultivated in a modified 2×L‐Mal medium for 72 h and protein profiles of whole cells fraction and medium fraction were checked with SDS‐PAGE (Fig. 4C). Compared with the wild‐type strain (Fig. 4C, WT), the much stronger bands of BsCel5 observed for all the mutants (∼52 kDa, Fig. 4C) in the whole cells fraction indicated that the expression levels of BsCel5 mutants were elevated after the directed evolution. For the medium fraction, all the BsCel5 mutants became the major extracellular protein (Fig. 4C), suggesting enhanced secretory protein expression levels as compared with the wild type. Because the signal peptide‐encoding sequences often have significant effects on secretion efficiencies of proteins (Tjalsma et al., 2000), the elevated secretory expression levels for the three positive mutants were highly likely attributed to the mutations located in signal peptide‐encoding sequences.
The wild‐type BsCel5 and mutants were overexpressed in E. coli and then were purified to homogeneity (data not shown). As shown in Fig. 5, the specific activities of mutant MT1 and MT2 were comparable to that of wild type, while mutant MT3 exhibited a ∼45% increase in the specific activity on RAC.
Figure 5.
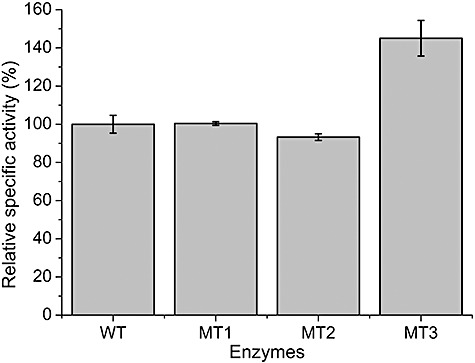
Relative specific activities of wild‐type BsCel5 (WT) and its mutants (MT1, MT2 and MT3). The specific activity of wild‐type BsCel5 under the tested condition is ∼698 U µmol−1. The error bars represent the standard deviation of the triplicate measurements.
Discussion
A combination of supercompetent cells and multimeric plasmids resulted in a simple, fast and high‐efficiency transformation system for directed evolution of secretory enzymes in B. subtilis. A new Bacillus host SCK6 from B. subtilis 1A751 (Wolf et al., 1995) was constructed, in which an extra copy of the comK gene was placed under the control of xylose‐inducible promoter on the chromosome. After a short period of xylose induction in the LB medium, this engineered strain is ready to be transformed at high efficiencies of more than 107 per µg of multimeric plasmid and ∼104 per µg of ligated plasmid. Because of the two order‐of‐magnitude cell density difference between supercompetent B. subtilis cells (∼2 × 108) and competent E. coli cells (∼2–3 × 1010 cfu ml−1) (Sambrook and Russel, 2001), much higher transformation efficiency based on the same amount of DNA is expected when the supercompetent B. subtilis cells are concentrated by centrifugation. In practice, an extra step for cell concentration is not necessary because the efficiency of 107 per µg of multimeric plasmid is high enough for directed evolution. The utilization of this supercompetent cell also enabled us to simplify cloning in B. subtilis without relying on E. coli–B. subtilis shuttle vectors any more.
In addition, it was important to construct the library for directed enzyme evolution directly in B. subtilis to avoid the risk of bias. We found that some plasmids with Bscel5 mutant obtained from B. subtilis library cannot transform E. coli for unknown reasons (data not shown).
It was long known that B. subtilis prefers to be transformed by multimeric plasmids rather than monomers (Canosi et al., 1978). In this study, we described a modified procedure for generating multimeric plasmids based on the previous work of Shafikhani et al. (Shafikhani, et al., 1997, Collier et al., 2003). The linear vector was made by PCR process so that this operation is completely independent of the use of restriction enzymes and ligase. Any region on the expression vector can be subjected to random mutagenesis, for example, the promoter, a part of a gene, the replication origin and so on.
In summary, we have developed a simple, fast and high‐efficiency method for directed evolution of secretory enzymes based on B. subtilis. The advantages of this new platform are: (i) large library (∼107 transformants per µg of DNA), (ii) no risk of bias, (iii) mutagenesis can target any region of the vector, (iv) very easy and robust transformation protocol and (v) restriction enzyme‐, phosphatase‐ and ligase‐free. A glycoside hydrolase family 5 endoglucanse mutant exhibited both enhanced secretory expression level and specific activity on RAC was obtained through this system. This new system would be very useful for efficient transformation of B. subtilis, directed evolution of secretory enzymes and knockout or insertion of genes using high‐throughput methods.
Note: The integration vector pAX01‐comK for xylose‐inducible ComK overexpression and supercompetent B. subtilis strain SCK6 can be obtained from the Bacillus Genetic Stock Center (http://www.bgsc.org) with the Accession No. ECE222 and 1A976 respectively.
Experimental procedures
Chemicals and materials
All chemicals were reagent grade or higher, purchased from Sigma (St. Louis, MO) or Fisher Scientific (Pittsburgh, PA) unless otherwise noted. All enzymes for molecular biology experiments were purchased from New England Biolabs (Ipswich, MA). RAC was prepared from Avicel as previously described (Zhang et al., 2006b).
Bacterial strains, plasmids and oligonucleotides
The bacterial strains and plasmids used in this study are listed in Table 2. The primers (Table 3) used for the PCR amplification were synthesized by Integrated DNA Technologies. All microorganisms were grown in the LB medium, LBR medium [LB medium containing 0.4% (w/v) RAC] or the modified 2×L‐Mal medium (Ara et al., 2007). The final concentrations of antibiotics were 100 µg ml−1 ampicillin and 25 µg ml−1 chloramphenicol for E. coli and 5 µg ml−1 chloramphenicol and 1 µg ml−1 erythromycin for B. subtilis.
Table 2.
Strains and plasmids.
| Strain or plasmid | Characteristics | Reference |
|---|---|---|
| E. coli | ||
| JM109 | recA1, supE44 endA1 hsdR17 (r−k, m+k) gyrA96 relA1 thi (lac‐proAB) F'[traD36 proAB+lacIqlacZ ΔM15] | Sambrook and Russel (2001) |
| BL21 Star (DE3) | F‐ompT hsdSB (rB‐mB‐) gal dcm rne131 (DE3) | Invitrogen, Carlsbad, CA |
| B. subtilis | ||
| 168 | trpC2 | Burkholder and Giles (1947) |
| 1A751 | his nprR2 nprE18 ΔaprA3 ΔeglS102 ΔbglT bglSRV | Wolf et al. (1995) |
| SCK6 | ErmR, 1A751 derivate, lacA::PxylA‐comK | This work |
| Plasmids | ||
| pP43NMK | AmpR, KmR, E. coli–B. subtilis shuttle vector | Zhang et al. (2005) |
| pP43N‐Bscel5 | AmpR, KmR, pP43NMK derivate with Bscel5 gene cloned | This work |
| pNW33N | CmR, E. coli–B. subtilis shuttle vector | Bacillus Genetic Stock Center |
| pNWP43N‐Bscel5 | CmR, pNW33N derivate, with Bscel5 expression cassette cloned | This work |
| pAX01 | AmpR, ErmR, B. subtilis integrative vector, xylose‐inducible promoter | Hartl et al. (2001) |
| pAX01‐comK | AmpR, ErmR, pAX01 derivate with comK cloned | This work |
| pET20b | AmpR, overexpression vector containing T7‐dependent promoter | Novagen, Madison, WI |
AmpR, ampicillin resistance; CmR, chloramphenicol resistance; ErmR, erythromycin resistance; KmR, kanamycin resistance.
Table 3.
Oligonucleotides used in this study.
| Primer | Sequence |
|---|---|
| P1 | 5′‐CAAAGGAGCACCTATTTTTGTGAC‐3′ |
| P2 | 5′‐CTGAGTGCATAGTTTGCTTTATCC‐3′ |
| P3 | 5′‐GGTAAGAGAGGAATGTACACATG‐3′ |
| P4 | 5′‐GCTTAACTAATTTGGTTCTGTTC‐3′ |
| P5 | 5′‐CTAGCTGCAGCAGGGACAAAAACGCC‐3′ |
| P6 | 5′‐GATACCCGGGCACAACGCAAACCTCC‐3′ |
| P7 | 5′‐GCATACGGATCCAAAGGAGGCCATAATATGAGTCAG‐3′ |
| P8 | 5′‐CCTGATCCGCGGCTATTTTTCTAATACCGTTCC‐3′ |
| P9 | 5′‐GTGATAGCGGTACCATTATAG‐3′ |
| P10 | 5′‐ACGCAAACCTCCTATTAGATG‐3′ |
Construction of plasmids
The DNA sequence encoding mature BsCel5 (30–499 amino acids) (GenBank Accession No.: CAA82317) and its following terminator sequence were amplified from genomic DNA of B. subtilis 168 with the primer pair P5/P6. The PCR product was digested with PstI/XmaI and ligated into the corresponding sites of the vector pP43NMK (Zhang et al., 2005) to generate pP43N‐Bscel5, where the Bscel5 gene without its signal peptide‐encoding fragment was fused with B. subtilis protease NprB signal peptide‐encoding sequence. After the digestion of pP43N‐Bscel5 with BamHI/XmaI, the smaller fragment containing the P43‐SPnprB‐Bscel5‐term expression cassette was cloned into the corresponding sites of a B. subtilis–E. coli shuttle vector pNW33N to generate pNWP43N‐Bscel5 (Fig. 2A).
Preparation of a supercompetent B. subtilis host – SCK6
The ComK‐encoding sequence was amplified from the genomic DNA of B. subtilis 168 with the primer pair P7/P8. The SD sequence (AAAGGAGG) of B. subtilis gsiB gene (Phan et al., 2006) was introduced upstream of the amplified comK gene. The PCR product was digested with BamHI/SacII and ligated into the corresponding sites of the vector pAX01 (Hartl et al., 2001) to generate pAX01‐comK, where the comK gene was placed under the control of xylose‐inducible PxylA promoter. Bacillus subtilis 1A751 (Wolf et al., 1995) was transformed with the PciI‐linearized pAX01‐comK. After the double‐cross‐over recombination between linearized pAX01‐comK and chromosomal DNA at the lacA locus (Fig. 3), the recombinants were selected on an LB plate with 1 µg ml−1 erythromycin. Disruption of the lacA gene and insertion of the comK gene were verified by PCR and the identified recombinant strain was designated as B. subtilis SCK6.
PCR‐based random mutagenesis
A randomly mutated library of SPnprB‐BsCel5‐encoding sequence (Fig. 2B) was generated by error‐prone PCR (0.5 ng µl−1 plasmid pNWP43N‐Bscel5 as the template, 0.2 mM dATP, 0.2 mM dGTP, 1 mM dCTP, 1 mM dTTP, 5 mM MgCl2, 0.2 mM MnCl2, 0.05 U µl−1 polymerase, and 0.4 µM each of the primers P3 and P4). The PCR reaction was conducted by using the NEB Taq DNA polymerase (94°C denaturation, 2 min; 13 cycles of 94°C denaturation, 30 s; 50°C annealing, 30 s; and 68°C extension, 1.5 min, followed by 68°C extension for 5 min).
pNWP43N‐Bscel5 linearization by PCR
The plasmid pNWP43N‐Bscel5 (4988 bp) was linearized by high‐fidelity PCR by using the primer pair P1/P2. The PCR reaction was conducted using NEB Phusion DNA polymerase (98°C denaturation, 1 min; 30 cycles of 98°C denaturation, 10 s; 56°C annealing, 20 s; and 72°C extension, 75 s, followed by 72°C extension for 5 min).
Plasmid multimerization by overlap PCR
The linearized pNWP43N‐Bscel5 and error‐prone PCR product were cleaned with a Zymo Research DNA Clean & Concentrator Kit. The multimerization process was conducted with a modified protocol of Shafikhani and colleagues (1997), using high‐fidelity PCR (dNTP, 0.2 mM for each; error‐prone PCR product, 5 ng µl−1; PCR‐linearized pNWP43N‐Bscel5, 0.15 ng µl−1; polymerase, 0.04 U µl−1). The PCR reaction was conducted using Phusion DNA polymerase (98°C denaturation, 30 s; 20 cycles of 98°C denaturation, 10 s; 72°C annealing and extension, 3 min, 15 cycles of 98°C denaturation, 10 s; 72°C annealing and extension, 6 min, followed by 72°C extension for 10 min). For the quantification of the multimerization products, the PCR product was digested with HindIII, and then was subjected to agarose gel electrophoresis and analysed with Quantity One (Version 4.6.7).
Preparation and transformation of supercompetent B. subtilis cells
The B. subtilis SCK6 strain was inoculated into 3 ml of LB medium with 1 µg ml−1 erythromycin in a test tube. The cells were cultivated at 37°C with shaking at 200 r.p.m. overnight (∼12 h). The culture was then diluted to A600 at 1.0 in a fresh LB medium containing 1% (w/v) xylose and then grown for 2 h. The resulting cell culture was ready to be transformed or divided into aliquots and stocked at −80°C with 10% (v/v) glycerol.
One microlitre of the PCR product containing plasmid multimers was mixed with 100 µl of the supercompetent cells in a plastic test tube and cultivated at 37°C with shaking at 200 r.p.m. for 90 min. An aliquot of the transformed cells was diluted by 103‐ to 104‐fold for estimating the transformation efficiency. To facilitate the further segregation of different clones in the same cell (Shafikhani et al., 1997), the rest of the transformed cells were diluted into 10 ml of M9 minimal medium containing 5 µg ml−1 chloramphenicol and grown at 37°C for 14 h. The cell cultures were then diluted by 20‐fold with the same medium and grown for an additional 8 h. The serial diluted transformants were spread on LBR plates containing 5 µg ml−1 chloramphenicol and incubated at 37°C for 20 h. Positive colonies that were surrounded with big and clear halo zones were selected for further characterization and DNA sequencing using the primer pair P9/P10.
Cellulase production and analysis in B. subtilis
The B. subtilis strains were cultivated in a modified 2×L‐Mal medium (Ara et al., 2007) at 30°C for 72 h. After centrifugation, the extracellular proteins in the supernatant were precipitated using the DOC‐TCA method (Cold Spring Harbor Protocols, 2006). Protein samples were analysed by using 12% (w/v) SDS‐PAGE.
Endoglucanase expression, purification and assays
For intracellular expression of wild‐type BsCel5 and mutants in E. coli, the wild‐type Bscel5 and its mutants were subcloned into pET20b (Novagen, Madison, WI) without their signal peptide‐encoding sequence. Escherichia coli BL21 Star (DE3) (Invitrogen, Carlsbad, CA) were transformed with the recombinant plasmids for expressing the target proteins. The recombinant BsCel5 was purified based on its ability to strongly bind RAC, followed by desorption by ethylene glycol (Hong et al., 2008; Zhang et al., 2010). The BsCel5 activity was measured in a 50 mM MES buffer (pH 6.0) containing 1 mM CaCl2 and 1% RAC (w/v) at 50°C for 30 min. The reactions were terminated by boiling at 100°C for 5 min and the concentrations of newly released soluble reducing sugars were determined by the 2,2′‐bicinchoninate (BCA) method (Zhang and Lynd, 2005). One unit of activity was defined as the amount of enzyme that released 1 µmol of reducing end per minute.
Acknowledgments
This work was supported mainly by the DOE BioEnergy Science Center. The BioEnergy Science Center is a US Department of Energy Bioenergy Research Center supported by the Office of Biological and Environmental Research in the DOE Office of Science. This work was also partially supported by the USDA Bioprocessing and Biodesign Center, the College of Agriculture and Life Sciences Biodesign and Bioprocessing Research Center at Virginia Tech and the DuPont Young Professor Award. We thank Dr Daniel Zeigler from the Bacillus Genetic Stock Center for providing bacterial strains and plasmids.
References
- Amin N.S., Wong S., Schellenberger V. Direct transformation of site‐saturation libraries in Bacillus subtilis. Biotechniques. 2003;35:1134–1136. doi: 10.2144/03356bm02. , and 11381140. [DOI] [PubMed] [Google Scholar]
- Ara K., Ozaki K., Nakamura K., Yamane K., Sekiguchi J., Ogasawara N. Bacillus minimum genome factory: effective utilization of microbial genome information. Biotechnol Appl Biochem. 2007;46:169–178. doi: 10.1042/BA20060111. [DOI] [PubMed] [Google Scholar]
- Burkholder P.R., Giles N.H., Jr Induced biochemical mutations in Bacillus subtilis. Am J Bot. 1947;34:345–348. [PubMed] [Google Scholar]
- Canosi U., Morelli G., Trautner T.A. The relationship between molecular structure and transformation efficiency of some S. aureus plasmids isolated from B. subtilis. Mol Gen Genet. 1978;166:259–267. doi: 10.1007/BF00267617. [DOI] [PubMed] [Google Scholar]
- Caspers M., Brockmeier U., Degering C., Eggert T., Freudl R. Improvement of Sec‐dependent secretion of a heterologous model protein in Bacillus subtilis by saturation mutagenesis of the N‐domain of the AmyE signal peptide. Appl Microbiol Biotechnol. 2010;86:1877–1885. doi: 10.1007/s00253-009-2405-x. [DOI] [PubMed] [Google Scholar]
- Cold Spring Harbor Protocols. Using deoxycholate and trichloroacetic acid to concentrate proteins and remove interfering substances. Cold Spring Harb Protoc. 2006 [Google Scholar]
- Collier K.D., Kean A.C., Lee E., Leiva N., Morrison T.B., Schellenberger V., Selifonova O. 2003. , and ) Phosphate limited inducible promoter and a Bacillus expression system. World patent: WO/2003/054140.
- Crameri A., Raillard S.‐A., Bermudez E., Stemmer W.P.C. DNA shuffling of a family of genes from diverse species accelerates directed evolution. Nature. 1998;391:288–291. doi: 10.1038/34663. [DOI] [PubMed] [Google Scholar]
- Cutting S.M., Vander Horn P.B., editors. John Wiley & Sons; 1990. [Google Scholar]
- Dunn A.K., Handelsman J. A vector for promoter trapping in Bacillus cereus. Gene. 1999;226:297–305. doi: 10.1016/s0378-1119(98)00544-7. [DOI] [PubMed] [Google Scholar]
- Ehrlich S.D. DNA cloning in Bacillus subtilis. Proc Natl Acad Sci USA. 1978;75:1433–1436. doi: 10.1073/pnas.75.3.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl B., Wehrl W., Wiegert T., Homuth G., Schumann W. Development of a new integration site within the Bacillus subtilis chromosome and construction of compatible expression cassettes. J Bacteriol. 2001;183:2696–2699. doi: 10.1128/JB.183.8.2696-2699.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong J., Ye X., Wang Y., Zhang Y.‐H.P. Bioseparation of recombinant cellulose binding module‐protein by affinity adsorption on an ultra‐high‐capacity cellulosic adsorbent. Anal Chim Acta. 2008;621:193–199. doi: 10.1016/j.aca.2008.05.041. [DOI] [PubMed] [Google Scholar]
- Maamar H., Dubnau D. Bistability in the Bacillus subtilis K‐state (competence) system requires a positive feedback loop. Mol Microbiol. 2005;56:615–624. doi: 10.1111/j.1365-2958.2005.04592.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironczuk A.M., Kovács Á.T., Kuipers O.P. Induction of natural competence in Bacillus cereus ATCC14579. Microb Biotechnol. 2008;1:226–235. doi: 10.1111/j.1751-7915.2008.00023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naki D., Paech C., Ganshaw G., Schellenberger V. Selection of a subtilisin‐hyperproducing Bacillus in a highly structured environment. Appl Microbiol Biotechnol. 1998;49:290–294. [Google Scholar]
- Phan T.T., Nguyen H.D., Schumann W. Novel plasmid‐based expression vectors for intra‐ and extracellular production of recombinant proteins in Bacillus subtilis. Protein Expr Purif. 2006;46:189–195. doi: 10.1016/j.pep.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Romero P.A., Arnold F.H. Exploring protein fitness landscapes by directed evolution. Nat Rev Mol Cell Biol. 2009;10:866–876. doi: 10.1038/nrm2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J., Russel D.W. Cold Spring Harbor Laboratory; 2001. [Google Scholar]
- Schumann W. Production of recombinant proteins in Bacillus subtilis. Adv Appl Microbiol. 2007;62:137–189. doi: 10.1016/S0065-2164(07)62006-1. [DOI] [PubMed] [Google Scholar]
- Shafikhani S., Siegel R.A., Ferrari E., Schellenberger V. Generation of large libraries of random mutants in Bacillus subtilis by PCR‐based plasmid multimerization. Biotechniques. 1997;23:304–310. doi: 10.2144/97232rr01. [DOI] [PubMed] [Google Scholar]
- Smits W.K., Eschevins C.C., Susanna K.A., Bron S., Kuipers O.P., Hamoen L.W. Stripping Bacillus: ComK auto‐stimulation is responsible for the bistable response in competence development. Mol Microbiol. 2005;56:604–614. doi: 10.1111/j.1365-2958.2005.04488.x. [DOI] [PubMed] [Google Scholar]
- Strausberg S.L., Alexander P.A., Gallagher D.T., Gilliland G.L., Barnett B.L., Bryan P.N. Directed evolution of a subtilisin with calcium‐independent stability. Biotechnology (N Y) 1995;13:669–673. doi: 10.1038/nbt0795-669. [DOI] [PubMed] [Google Scholar]
- Susanna K.A., Fusetti F., Thunnissen A.‐M.W.H., Hamoen L.W., Kuipers O.P. Functional analysis of the competence transcription factor ComK of Bacillus subtilis by characterization of truncation variants. Microbiology. 2006;152:473–483. doi: 10.1099/mic.0.28357-0. [DOI] [PubMed] [Google Scholar]
- Tjalsma H., Bolhuis A., Jongbloed J.D., Bron S., van Dijl J.M. Signal peptide‐dependent protein transport in Bacillus subtilis: a genome‐based survey of the secretome. Microbiol Mol Biol Rev. 2000;64:515–547. doi: 10.1128/mmbr.64.3.515-547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjalsma H., Antelmann H., Jongbloed J.D., Braun P.G., Darmon E., Dorenbos R. Proteomics of protein secretion by Bacillus subtilis: separating the ‘secrets’ of the secretome. Microbiol Mol Biol Rev. 2004;68:207–233. doi: 10.1128/MMBR.68.2.207-233.2004. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf M., Geczi A., Simon O., Borriss R. Genes encoding xylan and beta‐glucan hydrolysing enzymes in Bacillus subtilis: characterization, mapping and construction of strains deficient in lichenase, cellulase and xylanase. Microbiology. 1995;141:281–290. doi: 10.1099/13500872-141-2-281. [DOI] [PubMed] [Google Scholar]
- Xue G.‐P., Johnson J.S., Dalrymple B.P. High osmolarity improves the electro‐transformation efficiency of the gram‐positive bacteria Bacillus subtilis and Bacillus licheniformis. J Microbiol Methods. 1999;34:183–191. [Google Scholar]
- Yamane K., Bunai K., Kakeshita H. Protein traffic for secretion and related machinery of Bacillus subtilis. Biosci Biotechnol Biochem. 2004;68:2007–2023. doi: 10.1271/bbb.68.2007. [DOI] [PubMed] [Google Scholar]
- You L., Arnold F.H. Directed evolution of subtilisin E in Bacillus subtilis to enhance total activity in aqueous dimethylformamide. Protein Eng. 1996;9:77–83. doi: 10.1093/protein/9.1.77. [DOI] [PubMed] [Google Scholar]
- Zhang X.‐Z., Zhang Y.‐H.P. One‐step production of biocommodities from lignocellulosic biomass by recombinant cellulolytic Bacillus subtilis: opportunities and challenges. Eng Life Sci. 2010;10:398–406. [Google Scholar]
- Zhang X.‐Z., Cui Z.‐L., Hong Q., Li S.‐P. High‐level expression and secretion of methyl parathion hydrolase in Bacillus subtilis WB800. Appl Environ Microbiol. 2005;71:4101–4103. doi: 10.1128/AEM.71.7.4101-4103.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.‐Z., Zhang Z., Zhu Z., Sathitsuksanoh N., Yang Y., Zhang Y.H.P. The noncellulosomal family 48 cellobiohydrolase from Clostridium phytofermentans ISDg: heterologous expression, characterization, and processivity. Appl Microbiol Biotechnol. 2010;86:525–533. doi: 10.1007/s00253-009-2231-1. [DOI] [PubMed] [Google Scholar]
- Zhang Y.‐H.P., Lynd L.R. Determination of the number‐average degree of polymerization of cellodextrins and cellulose with application to enzymatic hydrolysis. Biomacromolecules. 2005;6:1510–1515. doi: 10.1021/bm049235j. [DOI] [PubMed] [Google Scholar]
- Zhang Y.‐H.P., Himmel M., Mielenz J.R. Outlook for cellulase improvement: screening and selection strategies. Biotechnol Adv. 2006a;24:452–481. doi: 10.1016/j.biotechadv.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Zhang Y.‐H.P., Cui J.‐B., Lynd L.R., Kuang L.R. A transition from cellulose swelling to cellulose dissolution by o‐phosphoric acid: evidences from enzymatic hydrolysis and supramolecular structure. Biomacromolecules. 2006b;7:644–648. doi: 10.1021/bm050799c. [DOI] [PubMed] [Google Scholar]
- Zhao H., Arnold F.H. Directed evolution converts subtilisin E into a functional equivalent of thermitase. Protein Eng. 1999;12:47–53. doi: 10.1093/protein/12.1.47. [DOI] [PubMed] [Google Scholar]