

Summary
Ectomycorrhiza is a mutualistic symbiosis formed between fine roots of trees and the mycelium of soil fungi. This symbiosis plays a key role in forest ecosystems for the mineral nutrition of trees and the biology of the fungal communities associated. The characterization of genes involved in developmental and metabolic processes is important to understand the complex interactions that control the ectomycorrhizal symbiosis. Agrobacterium‐mediated gene transfer (AMT) in fungi is currently opening a new era for fungal research. As whole genome sequences of several fungi are being released studies about T‐DNA integration patterns are needed in order to understand the integration mechanisms involved and to evaluate the AMT as an insertional mutagenesis tool for different fungal species. The first genome sequence of a mycorrhizal fungus, the basidiomycete Laccaria bicolor, became public in July 2006. Release of Laccaria genome sequence and the availability of AMT makes this fungus an excellent model for functional genomic studies in ectomycorrhizal research. No data on the integration pattern in Laccaria genome were available, thus we optimized a plasmid rescue approach for this fungus. To this end the transformation vector (pHg/pBSk) was constructed allowing the rescue of the T‐DNA right border (RB)–genomic DNA junctions in Escherichia coli. Fifty‐one Agrobacterium‐transformed fungal strains, picked up at random from a larger collection of T‐DNA tagged strains (about 500), were analysed. Sixty‐nine per cent were successfully rescued for the RB of which 87% were resolved for genomic integration sequences. Our results demonstrate that the plasmid rescue approach can be used for resolving T‐DNA integration sites in Laccaria. The RB was well conserved during transformation of this fungus and the integration analysis showed no clear sequence homology between different genomic sites. Neither obvious sequence similarities were found between these sites and the T‐DNA borders indicating non‐homologous integration of the transgenes. Majority (75%) of the integrations were located in predicted genes. Agrobacterium‐mediated gene transfer is a powerful tool that can be used for functional gene studies in Laccaria and will be helpful along with plasmid rescue in searching for relevant fungal genes involved in the symbiotic process.
Introduction
Ectomycorrhiza is an ecologically relevant mutualistic symbiosis formed between fine roots of higher plants, including most of forestry important trees belonging to the families Betulaceae, Dipterocarpaceae, Myrtaceae, Salicaceae and Pinaceae, and the mycelium of a broad variety of soil fungi mainly homobasidiomycetes. This symbiosis plays a key role from a trophic point of view in forest ecosystems for the mineral nutrition of trees and the biology of the fungal communities associated (Duplessis et al., 2002).
The characterization of genes involved in developmental and metabolic processes is relevant to understand the complex interactions that control the ectomycorrhizal symbiosis. To clarify this EST sequencing, gene expression analysis of cDNA arrays and proteomics have been developed for different ectomycorrhizal systems (Laurent et al., 1999; Voiblet et al., 2001; Podila et al., 2002; Peter et al., 2003; Johansson et al., 2004; Duplessis et al., 2005). In this context the development of molecular biology tools useful for large‐scale mutagenesis programmes is urgently needed in the mycorrhizal field in order to discover fungal genes relevant for the establishment and maintenance of the symbiotic organ.
Agrobacterium, the crown gall plant pathogen, is the only known example of inter‐kingdom gene transfer in nature. Besides its natural hosts, dicotyledonous plants, this Gram‐negative bacterium has also shown under laboratory conditions to be able to transfer its mobile DNA element (T‐strand) to monocotyledonous plants and other eukaryotes including yeast, filamentous fungi and mammalian cells (Bundock et al., 1995; de Groot et al., 1998; Kunik et al., 2001).
Agrobacterium‐mediated gene transfer (AMT) has become a routine procedure in plant research where it has overcome the complications of the classical gene transfer methods which in general require production of cell suspension cultures and protoplasts and frequently results in multiple integrations of the transgene. Recent finding of the usefulness of AMT in fungi is now similarly opening a new era for fungal genetic research that traditionally has suffered from the same complications as plant sciencies. Moreover, AMT has also been shown to be successful with some fungal species previously recalcitrants to genetic manipulations (Michielse et al., 2005).
In AMT the T‐DNA integration is believed to occur in a random manner via non‐homologous recombination (NHR) even though some researchers have emphasized the importance of micro‐homologies between the recipient genome and the 3′ tail of the ssT‐strand (i.e. left border, LB) in the integration site (Tinland et al., 1995; Brunaud et al., 2002; Meng et al., 2007). Generally the view of random integration has been based on the variable Southern hybridization signals obtained from independent transformed plant lines and on the fact that no extensive homology has been found either between the integration sites and T‐DNA borders or between the integration sites themselves. Furthermore, in plants even in the presence of long homologous flanks in the T‐DNA integration seems to occur via NHR referring this mechanism to be the dominant route of integration in this group of eukaryots (Offringa et al., 1990). This is also the case in filamentous fungi where the integration of new DNA into their genome occurs via NHR even when long homologous sequences are used (i.e. bigger than 0.5 kb flanks). This usually makes targeted gene deletion/replacement studies extremely difficult and unviable for large‐scale mutagenesis programmes. Even though several attempts have been carried out so far we have been unsuccessful in targeted gene knockout of Laccaria bicolor.
Surprisingly, much higher homologous recombination efficiencies have been achieved in AMT of yeast. In Saccharomyces cerevisiae only 50‐bp‐long homologous sequences can direct the transgene into a precise locus in the yeast genome (Winzeler et al., 1999). Therefore, whether the transgene integrates via homologous recombination or non‐homologous end joining (NHEJ) seems to be less dependent on the T‐DNA sequence but follows the predominant cellular mechanisms used by the host (van Attikum et al., 2001).
Like for plants (Alonso et al., 2003) and yeast (Bundock et al., 2002) AMT has also been proposed to be a powerful tool for functional genomic studies in filamentous fungi (Rogers et al., 2004; Elliott and Howlett, 2006; Blaise et al., 2007; Meng et al., 2007). The ease of producing single‐copy transgenic strains with this methodology (de Groot et al., 1998; Rho et al., 2001; Tsuji et al., 2003; LeClerque et al., 2004; Kemppainen et al., 2005; Meng et al., 2007) is far superior to earlier gene‐introduction methods traditionally used for this group of organisms. Moreover, in Cryptococcus neoformans the use of Agrobacterium for insertional mutagenesis has been shown to increase the stability of transgenes compared with biolistic methods (Idnurum et al., 2004). Besides, even less is known about the T‐DNA integration in filamentous fungi and there is no information available so far concerning higher basidiomycetes. In the few cases when AMT were conduced in homobasidiomycetes no genomic data were available to perform searches of T‐DNA–genomic DNA (gDNA) junctions. Even though the T‐DNA integration patterns in fungal genomes is less studied it is frequently assumed to be at random like in plants. In S. cerevisiae (Bundock et al., 2002) the positioning of 54 T‐DNA integration sites in the genome showed that no clear preference of integrations to certain DNA sequences or genomic regions was seen. The results showed that T‐DNA integations were present in both coding and non‐coding sequences and were distributed throughout the S. cerevisiae genome. However, Blaise and colleagues (2007) studying the pattern of Agrobacterium‐mediated transgene insertions in the genome of the phytopatogenic ascomycete Leptosphaeria maculans found that the T‐DNA preferably integrated as a single copy into gene‐rich regions of the fungal genome. This preferential insertion along with the high ratio of tagged mutants renders AMT a tool of choice for large‐scale gene discovery (Blaise et al., 2007). On the other hand, in Magnaporthe oryzaeMeng and colleagues (2007) detected a significant insertion bias, with promoters receiving twice more T‐DNA integrations than expected, and ORFs receiving three times fewer. In addition, they found that the distribution of T‐DNA inserts among the fungal chromosomes was not at random and were frequently found in sequences with micro‐homologies to the left terminus (Meng et al., 2007).
Now, when complete genomic sequences of several filamentous fungi are being released, studies about T‐DNA integration patterns should be conducted in order to understand the integration mechanisms involved and to evaluate the powerfulness of AMT as a mutagenesis tool for different fungal species. One of the recently sequenced genomes belongs to the homobasidiomycete L. bicolor, an ectomycorrhizal fungus that forms symbiosis with several economically and ecologically important tree species, among them Poplar, the first tree sequenced (Tuskan et al., 2006; Martin et al., 2007). The 65 Mb genome of Laccaria, public since July 2006 (http://genome.jgi‐psf.org/Lacbi1/Lacbi1.home.html), is the first mycorrhizal fungal genome sequenced (Martin et al., 2008). The access to the genome sequence is an unparallel opportunity to strength our knowledge about the mechanisms which rule the establishment and function of this symbiosis typical of forest ecosystems. The fact that this fungus has also been transformed via AMT (Kemppainen et al., 2005) makes it an excellent model for studying T‐DNA integration in symbiotic fungi and for functional genomic studies through insertional mutagenesis searching for fungal genes involved in the ontogeny of the mycorrhizal symbiosis.
Two widely used methods for obtaining sequence information about the T‐DNA–gDNA junctions in the recipient genome are TAIL‐PCR (thermal asymmetric interlaced polymerase chain reaction) and plasmid rescue. TAIL‐PCR has been used with plants and fungi but its weakness is the dependence on the conservation of T‐DNA borders. These are frequently truncated during the integration process and the degree of truncation varies between different hosts. This fact can lead to the amplification of too many PCR products lacking border sequences and most likely being unspecific (Meng et al., 2007). Besides, up to three repeated rounds of PCR are needed to produce a specific amplicon of the T‐DNA–gDNA junction. This method was used for recovering integration sequences in the ectomycorrhizal fungus Hebeloma cylindrosporum. Of 15 processed fungal transgenic strains only two right border (RB) junctions could be amplified (Combier et al., 2003). Moreover, in L. maculans agrotransformants none of the RB flanking sequences could be amplified by TAIL‐PCR (Blaise et al., 2007). It may also be rather complicated to obtain sequence information from fungal transgenic strains carrying several independent T‐DNA integrations with the TAIL‐PCR approach.
On the other hand, with the plasmid rescue method the T‐DNA–gDNA junctions are recovered via plasmid replication in Escherichia coli. The requirements are a bacterial selection marker and a replication origin in the T‐DNA used for fungal transformation. Genomic DNA of the transgenic strains is digested and self‐ligated generating plasmids carrying the integration site of the host genome for further sequencing with a T‐DNA specific primer. In the plasmid‐rescue approach there is no need for cloning PCR products. The genomic sequences are directly maintained in the rescued plasmids and the obtainment of sequence information is not strictly dependent on the degree of conservation of the T‐DNA borders. In the case of transgenic strains with multiple integrations plasmid rescue offers a straightforward way of mapping separate integration events and gives information of genetic re‐arrangements that may have happened during or after T‐DNA integration.
With no previous information about the conservation of the T‐DNA border regions in L. bicolor transgenic strains we optimized a plasmid rescue approach which allowed us to recover the RB–gDNA junctions and further analyse the T‐DNA integration pattern in Laccaria genome.
Results
We had previously shown that Agrobacterium can be used for gene transfer in L. bicolor. The T‐DNA seemed to integrate mostly as a single copy and at different places in the fungal genome suggesting this method proper for non‐targeted mutagenesis of the fungus (Kemppainen et al., 2005). These conclusions were drown from Southern hybridization analysis knowing that it was possible that integration after all could had happened in sites sharing some common features or just in non‐coding sequences. When Laccaria genome sequencing was completed mapping of precise integration sites became possible and AMT of the fungus could be further evaluated. In order to gain more information regarding the Agrobacterium T‐DNA integration in L. bicolor we constructed the plasmid rescue binary vector pHg/pBks (Fig. 1). This plasmid does not carry homologous sequences with L. bicolor genome; its T‐DNA allows the selection of fungal transgenic strains by hygromycin resistance and the rescue of the RB–gDNA junction by cutting with SacI under ampicillin selection in E. coli.
Figure 1.
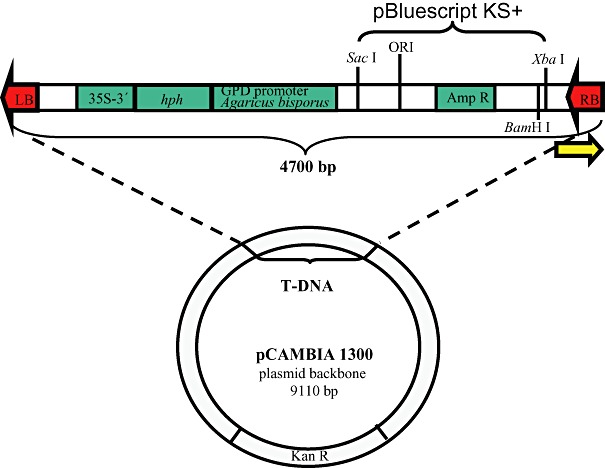
pHg/pBks rescue plasmid. The binding site for the Post‐RB primer is indicated by an arrow. GPD promoter Agaricus bisporus: glyceraldehide‐3‐phosphate dehydrogenase promoter of Agaricus bisporus. hph: hph gene of E. coli coding for an aminocyclitol phosphotransferase that confers resistance to Hygromycin B and structurally related antibiotics. 35S‐3′: cauliflower mosaic virus 35S terminator. Amp R: bla (ApR) gene of E. coli coding for a β‐lactamase that confers resistance to ampicillin. ORI: replication origin of pBluescript KS+. LB: T‐DNA left border of pCAMBIA1300. RB: T‐DNA right border of pCAMBIA1300. Kan R: aadA gene of E. coli coding for an aminoglycoside phosphotransferase that confers resistance to kanamycin. Relevant restriction sites (SacI, BamHI and XbaI) within the T‐DNA are indicated.
Laccaria bicolor dikaryotic strain S238N (heterokaryotic, the phase in the fungal life cycle which forms symbiosis with tree roots) was transformed with Agrobacterium tumefaciens AGL‐1 carrying the pHg/pBks vector. The transformation efficiency with the given plasmid was 75% calculated from the appeared hygromycin resistant growing points against the total number of fungal colonies subjected to co‐cultivation after two weeks under antibiotic selection. Properly separated resistant growing points were chosen for plasmid rescue and subcultured for mycelia harvest. In order to evaluate the T‐DNA integration pattern of pHg/pBks 12 hygromycin‐resistant fungal strains were analysed for the presence of hph and amp transgenes by PCR. Only one of the 12 samples failed to show a signal for amp transgene (data not shown). In order to evaluate further the number and the pattern of integration events genomic DNA from 11 amp‐positive transgenic fungal strains and wild type was digested with BamHI, which cuts once within the T‐DNA (Fig. 1), Southern blotted and hybridized with amp probe (Fig. 2). Southern blot analysis suggested an integration pattern at variable genomic sites and dominantly as a single copy (10 out of 11 analysed transgenic strains with a single hybridization signal) confirming previous results with a different construct (Kemppainen et al., 2005). From this sample set we concluded that the 5.1 kb T‐DNA of the rescue‐plasmid pHg/pBks integrated mostly complete and as a single copy at variable sites in Laccaria genome. Genomic DNA of 51 fungal transgenic strains picked up at random from a larger collection (about 500) was extracted and checked by PCR for the presence of both transgenes (data not shown). Forty‐seven out of 51 transgenic strains were PCR‐positive for both amp and hph and were further analysed by RB plasmid rescue.
Figure 2.
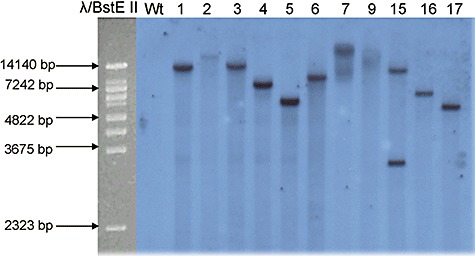
Southern blot analysis of L. bicolor transgenic and wild‐type strains. Total DNA (7 µg) was digested with BamHI which cuts once within the T‐DNA, blotted and probed with the ∼1 kb amp gene fragment. From left to right: molecular size marker λBstE II, L. bicolor wild type (Wt) and transgenic strains.
We set up a plasmid rescue protocol for L. bicolor using 1–3 µg of completely SacI‐digested and self‐ligated gDNA which proved to be enough for producing ampicillin‐resistant E. coli colonies after electroporation. The number of colonies depended largely on the rescued fungal transgenic strain and normally ranged from 10 to 100 per electroporation. We noticed that a cleaning‐up of the ligation reaction with phenol‐chloroform before electroporation was not necessary while using the enzymes and buffers detailed in Experimental procedures. Of the 47 processed fungal transgenic strains 40 produced ampicillin‐resistant bacteria and up to eight plasmids per rescued strain were analysed by linearization with SacI for a minimum size of 3 kb corresponding to pBluescript KS+. Obtained rescued plasmids ranged from 3 to 10 kb in size indicating the incorporation of genomic DNA of Laccaria (a representative set of rescued plasmids is presented in Fig. 3). Transgenic strains showing abnormalities in any of the mentioned features were excluded from further analysis. Plasmids from 35 fungal transgenic strains were subjected to sequencing of which 29 were able to be sequenced with the Post‐RB primer that binds within the T‐DNA at 43–62 bases upstream of the RB nick site. A blastnsearch was performed with the obtained sequences on the JGILaccaria genome portal (http://genome.jgi‐psf.org/Lacbi1/Lacbi1.home.html) in order to identify the T‐DNA–gDNA junctions. The exact integration sites were mapped in the genome and the RB conservation was compared between 25 rescued sequences (Fig. 4). The RB was well conserved during the integration of the T‐DNAs in Laccaria genome and 15 out of 25 sequences at their maximum possible length reached up to the original adenine RB nick site, while the rest of the sequences showed deletions of one to five bases. The samples failing in the sequencing with the primer used in this work most likely have had larger RB truncations and lack the primer binding site. Altogether 24 sequences were located in the Laccaria genome (version 1.0 as of December 2007) while one rescued sequence did not carry genomic DNA flank long enough for mapping. According to the automatized gene‐prediction models integrations had happened in both coding and non‐coding regions of the fungal genome and we were able to locate them into exons, introns, upstream and downstream elements of predicted genes or in intergenic regions. Two of the sequences could not be located in Laccaria genome and were compared with the blastn algorithm to GenBank databases to identify possible DNA contaminations. No homologies were found proposing that these sequences most probably belong to the fungus and correspond to sequencing gaps in the Laccaria genome sequence. Eighteen T‐DNA integration sites were precisely located in ORFs and their upstream or downstream elements (Table 1). Thirteen ORF interruptions were found, 11 of which were in exons and two in predicted intronic sequences. All of these interrupted ORFs have homologues in other organisms and eight have a proposed protein function (Table 1). Three integrations fall in proposed promoters and three in downstream ORFs (Table 1). Two sequences showed signs of genetic rearrangements or tandem T‐DNA integrations. One of these last sequences was obtained from the rescued fungal transgenic strain 15 that already showed a double hybridization signal in the preliminary Southern blot analysis (Fig. 2), the lower signal being rather small for representing a full‐length integration of a second T‐DNA. In this transgenic strain the RB was first followed by the inner sequence of the T‐DNA and then by a Laccaria sequence indicating a genomic reorganization during or after T‐DNA integration. Also in another rescued fungal transgenic strain the primer sequence was immediately followed by the T‐DNA LB showing a possible incomplete tandem integration (data not shown).
Figure 3.
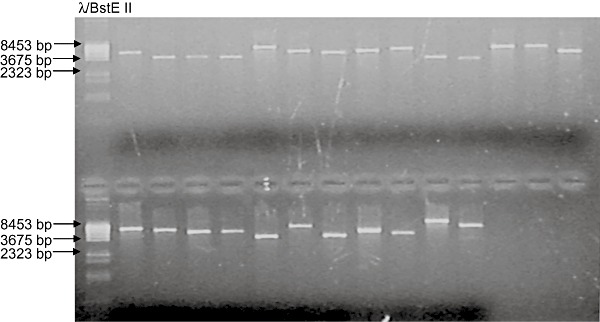
Set of rescued plasmids linearized with SacI. Genomic DNA isolated after L. bicolor transformation was self‐ligated and electroporated into E. coli. Plasmids isolated from bacterial clones were linearized with SacI and separated in an 1% agarose gel. From left to right: molecular size marker λBstE II, representative set of plasmids rescued from independent L. bicolor transgenic strains.
Figure 4.

Right border conservation of Agrobacterium‐transformed Laccaria transgenic strains. The pCAMBIA 1300 sequence is presented till the RB nick site. Letters in italics represent bases that could originate from both RB or genomic DNA.
Table 1.
T‐DNA integration sites in Laccaria genome located in ORFs and their upstream or downstream elements.
| Best blast hit | Predicted functions | Cellular functional group | |
|---|---|---|---|
| Fungal transgenic strains with ORF integrations | |||
| 2. Exon integration in Protein ID: 296957 | Hypothetical protein SNOG_16556 [Phaeosphaeria nodorum SN15]. Score (Bits): 317. E‐value: 2e‐84 | Reverse transcriptase | Transposon activity |
| 4. Exon integration in Protein ID: 318751 | Predicted protein [Coprinopsis cinerea okayama7#1]. Score (Bits): 344. E‐value: 9e‐93 | Zinc finger, MYND type | Chromatin structure and dynamics |
| 23. Exon integration in Protein ID: 316402 | Copia‐type polyprotein, putative [Arabidopsis thaliana]. Score (Bits): 556. E‐value: 6e‐156 | Zinc finger Integrase core domain. Reverse transcriptase | Transposon activity |
| 24. Exon integration in Protein ID: 379393 | Hypothetical protein CC1G_06600 [Coprinopsis cinerea okayama7#130]. Score (Bits): 791. E‐value: 0.0 | Serine/Threonine protein kinase | Signal transduction |
| 28. Exon integration in Protein ID: 318121 | Predicted protein [Coprinopsis cinerea okayama7#130]. Score (Bits): 41.6. E‐value: 0.041 | Unknown | nd |
| 33. Exon integration in Protein ID: 394044 | Hypothetical protein CC1G_09476 [Coprinopsis cinerea okayama7#130]. Score (Bits): 489. E‐value: 1e‐136 | Fungal transcriptional regulatory protein | DNA‐dependent regulation of transcription |
| 34. Exon integration in Protein ID: 316757 | Hypothetical protein CC1G_03966 [Coprinopsis cinerea okayama7#130]. Score (Bits): 174. E‐value: 3e‐41 | Reverse transcriptase | Transposon activity |
| 35. Exon integration in Protein ID: 308902 | Hypothetical protein CC1G_04991 [Coprinopsis cinerea okayama7#130]. Score (Bits): 934. E‐value: 0.0 | RPAP1‐like | Global gene expression |
| 36. Intron integration in Protein ID: 318555 | Hypothetical protein CC1G_08098 [Coprinopsis cinerea okayama7#130]. Score (Bits): 156. E‐value: 2e‐36 | Protein with similarities to di‐tricarboxylate transporter | Transporter |
| 39. Intron integration in Protein ID: 385818 | Predicted protein [Coprinopsis cinerea okayama7#130]. Score (Bits): 103. E‐value: 2e‐20 | Unknown | nd |
| 43a. Exon integration in Protein ID: 306930 or in Protein ID: 304244 | Predicted protein [Coprinopsis cinerea okayama7#130]. Score (Bits): 84.7. E‐value: 1e‐14 | Unknown | nd |
| 51. Exon integration in Protein ID: 317247 | Predicted protein [Coprinopsis cinerea okayama7#130]. Score (Bits): 634. E‐value: 1e‐179 | Unknown | nd |
| Fungal transgenic strains with upstream integrations | |||
| 11. Protein ID: 333685 | Hypothetical protein RUMOBE_00303 [Ruminococcus obeum ATCC 29174]. Score (Bits): 33.5. E‐value: 3.9 Unknown | Unknown | nd |
| 12. Protein ID: 298746 | Predicted protein [Coccidioides immitis RS]. Score (Bits): 37.4. E‐value: 0.25Unknown | Unknown | nd |
| 49. Protein ID: 315197 | Predicted protein [Coprinopsis cinerea okayama7#130]. Score (Bits): 283. E‐value: 1e‐74 | Unknown | nd |
| Fungal transgenic strains with downstream integrations | |||
| 16. Protein ID: 387352 | Ubiquitin‐conjugating enzyme E2‐16 kDa [Ustilago maydis 521]. Score (Bits): 279. E‐value: 3e‐74 | Ubiquitin protein ligase | Protein modification, ubiquitin cycle |
| 18. Protein ID: 296295 | Hypothetical protein CC1G_01879 [Coprinopsis cinerea okayama7#130]. Score (Bits): 312. E‐value: 8e‐83 | RNA binding | Control of meiosis |
Transgenic strain 43 sequence was found twice in Laccaria genome in two different scaffolds.
Rescue plasmids obtained from fungal transgenic strains were subjected to sequencing with the Post‐RB primer that binds within the T‐DNA at 43–62 bases upstream from the RB nick site. A blastn search was performed with the obtained sequences on the JGI Laccaria genome portal (http://genome.jgi‐psf.org/Lacbi1/Lacbi1.home.html) and the T‐DNA–gDNA junctions identified.
nd, not determined.
The genomic zones of the T‐DNA integrations were observed for special features like repetitive sequences but no coincidence between them and integration sites was seen. Thirty bases around the exact integration sites were aligned with clustalw in order to find putative inter‐site homologies (data not shown). These integration sequences were also analysed for possible micro‐homologies shared with the T‐DNA LB or RB sequences. No obvious inter‐site similarities or micro‐homologies in comparison with RB or LB were observed supporting the NHR‐based integration in Laccaria.
None of the analysed dikaryotic fungal transgenic strains with a T‐DNA insertion showed an obvious altered phenotype such as hyphal morphology or growth rate when cultivated on P5 media. Three transgenic strains (4, 24 and 36, Table 1) with an interruption in a predicted gene were selected in order to test by reverse transcription polymerase chain reaction (RT‐PCR) whether a single integration in one of the gene copies of the dikaryotic strain can cause a detectable reduction on the gene expression level or not (Fig. 5).
Figure 5.

Reverse transcription polymerase chain reaction (RT‐PCR) expression patterns of the genes coding for Laccaria bicolor Zn finger protein and Ser/Threo protein kinase (protein ID 318751 and 379393 respectively). Total RNA from mycelium of L. bicolor wild‐type strain S238N and transgenic fungal strains 4, 24 and 36 where the gene models coding proteins 318751, 379393 and 318555 (transporter) respectively were interrupted by T‐DNA insertion was isolated and aliquots of 1 µl were used for first‐strand cDNA synthesis. A PCR was performed with 2 µl of first‐strand cDNA and between 15 and 30 cycles of amplification. The picture shows fragments amplified after 28 cycles. Laccaria glucokinase (protein ID 312018) specific primers were used as a reference to check for equal transcripts amplification in the wild‐type and the different transgenic strains. Lines 1 and 2 in each panel are L. bicolor wild‐type S238N stored at INRA‐Nancy and a subculture of it stored at University of Quilmes respectively. All the transformants were obtained using the last one as recipient strain for transformation. Lines 3–5 in each panel are L. bicolor transgenic strains 4, 24 and 36 respectively (T‐DNA insertion in protein ID 318751, Zn finger; 379393, Ser/Threo protein kinase; and 318555, transporter). No transcript could be detected for the gene interrupted in the transgenic strain 36 and coding for a putative transporter neither in the transgenic strains or in the wild type (not shown in this figure). A control with no RT in the first‐strand cDNA synthesis reaction mix was included for each strain and set of specific primers (not shown).
Semi‐quantitative RT‐PCR allowed to detect transcript levels in the wild‐type L. bicolor strain S238N for the genes interrupted in the transgenic strains 4 and 24 (i.e. zinc finger protein and Ser/Threo protein kinase encoding genes) (Fig. 5), whereas no transcript could be detected for the gene interrupted in the transgenic strain 36 and coding for a putative transporter neither in the transgenic strains or in the wild type (data not shown). A gene coding for a glucokinase known to be expressed at a detectable level under the present culture conditions (i.e. P5 medium) (Deveau A. unpubl. data) was used as reference and positive control of RT‐PCR amplification in wild‐type and transgenic strains. The reference glucokinase gene was similarly expressed in the three transgenic and wild‐type strains. We were not able to detect a strong alteration in transcript levels of the two other genes in the corresponding fungal transgenic strains where the genes were interrupted. Nevertheless, the transgenic strain 24 with an interruption in a predicted gene coding for a Ser/Threo protein kinase showed a slight gene expression reduction in RT‐PCR analysis. This observation was carried out from transcripts extracted in independent experiments with fungal strains grown on agar P5 media with (Fig. 5) or without hygromycin B (data not shown). None of these selected transgenic strains showed an altered phenotype with respect to in vitro ectomycorrhiza formation with Poplar compared with the wild type (data not shown).
Supplementary phenotyping analysis was then carried out to study closer the dikaryotic transgenic strain 24. While on P5 with ammonium this strain had not shown an altered growth, interestingly the same medium supplemented with 4 mM KNO3 as the sole N source revealed a phenotype clearly distinguishable from the wild‐type strain. On this medium the transgenic strain grew more vigorously and had at least twice more hyphae penetrating the agar than the wild type while the radial growth rate was not altered (Fig. 6). These observations finally confirm that single tagged mutagenesis of dikaryotic Laccaria indeed can generate functional mutants with detectable phenotypes when observed under variable growth conditions.
Figure 6.

Laccaria bicolor transgenic strain 24 and wild type grown on P5 medium supplemented with 4 mM KNO3 as the sole N source.
Discussion
The construct pHg/pBks was made for performing the rescue of RB–gDNA junctions in the basidiomycete L. bicolor after transformation by A. tumefaciens. Based on the information about the processing and the integration of T‐strand in plants we decided to perform the RB rescue. The processing of the T‐strand starts in Agrobacterium with the covalent binding of the VirD2 protein to the 5′ end of the RB nick and this protein stays bounded to the ssT‐strand during its transfer from Agrobacterium to the host cell and further in the host cell during its nuclear trafficking. Probably due to this protein‐protected nature the sequences close to the RB have been shown to be better conserved than the LBs after genomic integration. With no information about the conservation of the border sequences during AMT of Laccaria the RB seemed the most reliable choice. Plasmid pHg/pBks also allows the rescue of the LB with several restriction enzymes or the simultaneous rescue of both borders with restriction enzymes not cutting within the T‐DNA. The last approach was not chosen due to the risk of processing too large rescued plasmids.
With the presented protocol we were able to rescue 69% of processed fungal transgenic strains and 57% could be sequenced with the Post‐RB primer for resolving T‐DNA–gDNA junctions. The RB sequence was shown to be well conserved in the sequenced samples but sequencing may have failed in the case of integrations with longer truncations in the RB. Truncations of variable length are frequently reported in AMT studies in plants and other fungi (Mayerhofer et al., 1991; Kumar and Fladung, 2000; Combier et al., 2003; Kim et al., 2003; Meng et al., 2007). No obvious homology was noticed between different genomic integration sites or clear micro‐homologies between the integration sites and the right or LB sequences. The T‐DNAs integrated in coding and non‐coding regions of different scaffolds of the Laccaria genome assembly, 54% corresponding to ORFs, 13% to upstream sequences, 8% to downstream sequences and 25% to presumed intergenic regions. Signs of genome rearrangements during or after the T‐DNA integration were also observed with some of the fungal transgenic strains studied, a phenomena also reported in AMT of plants and fungi, especially in samples giving multiple signals in Southern blot hybridizations (Hiei et al., 1994; Michielse et al., 2005). Our results suggest that T‐DNA integration in Laccaria genome occurs with a bias towards genes. As Laccaria genome contains a large number of predicted genes compared with other fungal genomes sequenced so far (F. Martin, unpubl. results) it is difficult at present to conclude whether the T‐DNA insertion is targeted to gene‐rich zones of the genome or only reflect the important proportion of these elements in L. bicolor genome.
Two models of integration processes in plants are double‐stranded break repair (DSB) and single‐stranded gap repair, the latter being the classic model based on micro‐homologies between the T‐strand and the target genomic sequences (Mayerhofer et al., 1991; Tinland et al., 1995; Tzfira et al., 2004). Both models also suggest a role for VirD2 (a protein that detects the RB sequence, nicks it and covalently binds to the 5′ end of the ssT‐strand directing it to the recipient nucleus) in recognition of nicks and gaps in the genome by interacting with host factors. The micro‐homologies postulated by the classic model for understanding the integration events are restricted to only one to three conserved bases between the integration site and the LB of the T‐strand. Similarly these have not shown conservation between different studies and now more data are pointing towards the DSB model to be the mechanism for T‐DNA integration at least in plants. Furthermore, it seems that the ssT‐strand becomes double‐stranded before integration while T‐DNAs were shown to be directed into introduced specific double‐stranded breaks in the plant genome (van Attikum et al., 2001).
An analysis of AMT in Arabidopsis has revealed that T‐DNAs mainly integrate in the vicinity of T‐rich regions in this plant (Brunaud et al., 2002). A recent work on Agrobacterium protein‐assisted T‐DNA integration in HeLa cells demonstrated a tendency of integration in AT repeats in mammalian cells as well (Pelczar et al., 2004). In yeast, even though the general conclusion was that no strong preference of T‐DNA integration to certain positions in the genome was demonstrated, integrations in Ty‐transposable elements and rDNA repeats were also detected. Most important, an increased integration efficiency was shown to certain chromosomal regions that interestingly also harboured clusters of transposable elements (Bundock et al., 2002). Moreover, in L. maculans T‐DNA preferentially integrated in gene‐rich regions of the fungal genome (Blaise et al., 2007). Also in the analysis of T‐DNA insertion site sequences in Arabidopsis indicated that the density of T‐DNA insertion events correlated with gene density along Arabidopsis chromosomes (Alonso et al., 2003).
These findings do not fit with a random integration pattern. Finally, the long known fact that in plants, two T‐DNAs originating from different Agrobacterium strains used in the same co‐cultivation experiment frequently are found to be integrated in tandem at the same genomic position (de Block and Debrouwer, 1991; De Neve et al., 1997; Kumar and Fladung, 2000) strongly argues with the random integration pattern that traditionally presents AMT as a perfect random mutagenesis tool. Some hot spots of integration towards which T‐DNAs may be guided could be present in eukaryotic genomes (Kumar and Fladung, 2000). Nevertheless, before the Laccaria genome is not fully annotated into its at least 10 chromosomes (F. Martin, unpubl. results) it is not possible to conclude whether these integration hot spots for T‐DNAs exist in L. bicolor or not.
In the present study, we also tested whether the transcript concentration of genes interrupted by T‐DNA integration was affected or not. Semi‐quantitative RT‐PCR analysis did not allow to detect complete repression of gene expression for the genes tested. This result can be explained by the activity of a second allele of the gene in the second nucleus of L. bicolor dikaryon. This allele may also express the gene at the same level when the other allele is interrupted. In order to obtain an important number of mutants by T‐DNA insertion the use of monokaryotic strains could be an approach to counteract transcript expression by a second allele of the interrupted gene in the dikaryon. However, monokaryotic (homokaryotic) mycelia of L. bicolor shows rather poor or no capacity at all to form ectomycorrhiza with partner trees in vitro and is unable to compete with dikaryotic (heterokaryotic) mycelia in nature. This makes monokaryotic strains poor and artificial candidates to test the symbiotic lifestyle. Nevertheless, T‐DNA integration in Laccaria monokaryons could be an interesting alternative to evaluate cellular functions related to other stages of the fungal lifestyle (i.e. mating interaction).
In the expression analysis of three transgenic strains we were only able to detect a slight decrease in transcript concentration in the case of a gene encoding a Ser/Threo protein kinase in the fungal transgenic strain where this gene was interrupted by T‐DNA insertion. A further characterization of the phenotype of this mutant with a single T‐DNA insertion showed an alteration in hyphal growth rate and number of hyphae compared with the wild‐type L. bicolor strain S238N grown on a medium with nitrate as the only N source. This last observation confirms that this transformation approach could be useful to generate dikaryotic mutants affected in a given function even though a second allele could partly recover the expression level of the gene. Agrobacterium‐mediated gene transfer is a powerful tool that can be used for functional gene studies in Laccaria and will be helpful along with plasmid rescue and RNA silencing (M. Kemppainen, unpubl. results) in searching for relevant fungal genes involved in the symbiotic process.
Experimental procedures
Fungal and bacterial strains
Laccaria bicolor (Maire) Orton dikaryotic strain S238N (Di Battista et al., 1996) was used in this study. The vegetative mycelium was maintained at 22°C in the dark on Pachlewski (P5)‐modified agar medium with the following composition (per litre): di‐ammonium tartrate, 0.5 g; glucose, 20 g; maltose, 5 g; KH2PO4, 1 g; MgSO4, 7H2O, 0.5 g; thiamine‐HCl, 0.1 g; MnCl2·4H2O, 5 mg; BO3H3, 8.5 mg; Na2MoO4, 0.3 mg; FeCl3, 6 mg; CuSO4·5H2O, 0.6 mg; agar, 20 g, pH 5.5. Escherichia coli TOP10 (Invitrogen) was used for plasmid cloning. Agrobacterium tumefaciens AGL‐1 was employed for Laccaria transformation. Voucher specimens are kept in the Universidad Nacional de Quilmes Culture Collection (Argentina).
Construction of the binary vector
The plasmid‐rescue binary vector was constructed using pBGgHg (Chen et al., 2000). The egfp box was removed from the T‐DNA with SacI and XbaI (Promega) and the resulting fragment was ligated with pBluescript KS+ (Invitrogen) complete plasmid cut with the same restriction enzymes creating the ampicillin rescue‐plasmid pHg/pBks (Fig. 1). The vector was electroporated into A. tumefaciens according to Pardo and colleagues (2002).
Agrobacterium‐based transformation of L. bicolor
Laccaria bicolorS238N was transformed with A. tumefaciens AGL‐1 carrying the pHg/pBks according to a previously established protocol (Kemppainen et al., 2005). Obtained hygromycin transgenic fungal strains were passed through three selection rounds on P5 medium with 300 µg ml−1 hygromycin B (Invitrogen) before growing mycelia for genomic DNA extraction.
PCR analysis of fungal transgenic strains
Mycelia of wild‐type and transgenic Laccaria S238N strains were grown on cellophane membranes on P5 medium for DNA extraction. With transgenic strains selection pressure of 150 µg ml−1 hygromycin B (Invitrogen) was maintained. Total genomic DNA was extracted with the DNeasy® Plant Mini Kit (Qiagen) according to manufacturer's protocol and analysed by PCR for the presence of hph and amp transgenes with primers HpH‐For/HpH‐Rev (5′‐AAGCCTGAACTCACC GCGAC‐3′/5′‐CTATTCCTTTGCCCTCGGAC‐3′) and Amp‐For/Amp‐Rev (5′‐CCCAAGGTTTGCAAGCAGCAGATTA CGCG‐3′/5′‐CGCGGATCCGCTCATGAGACAATAACCC‐3′) (Invitrogen) respectively. Polymerase chain reaction conditions used in both cases were: 1 µM primers, 200 µM dNPTs, 1.5 mM MgCl, 0.2 U Taq DNA polymerase (Fermentas) in 1× buffer with (NH4)2SO4 (Fermentas). About 50 ng of genomic DNA and ~1 ng of plasmid pHg/pBks DNA respectively were used as template. The PCR programme used for both amplifications was as follows: initial denaturation at 94°C for 2 min, 35 cycles of 94°C for 30 s, 60°C for 30 s and 72°C for 1 min, and final extension at 72°C for 5 min. Polymerase chain reaction products were separated in an 1% agarose gel and stained with ethidium bromide.
Southern blotting
Approximately 7 µg of fungal genomic DNA per sample was digested overnight with BamHI (Promega) which cuts once within the T‐DNA, separated in 1% agarose gel and transferred by alkaline capillarity blotting to a Hygrobond‐ N+ nylon membrane (Amershamn Biosciences) according to the manufacturer's protocol. A ~1 kb amp probe was generated from pHg/pBks by PCR using Amp‐For/Amp‐Rev primers as previously described and purified for labelling with the Wizard® PCR Prep DNA Purification System (Promega). The labelling of the probe, hybridization and detection were performed with the Gene Images AlkPhos Direct Labelling and Detection System (Amersham Biosciences) according to manufacturer's protocol by hybridizing at 55°C overnight and detecting after 4 h of exposure to a film (Agfa new medical X‐ray film).
Plasmid rescue
One to 3 µg of genomic DNA, purified with the DNeasy® Plant Mini Kit (Qiagen) and eluted with ddH2O, was digested with SacI (Promega) (20–30 U µg−1 DNA) overnight. Restriction reactions were heat inactivated, precipitated with ammonium acetate and ethanol and dissolved in ddH2O (Sambrook et al., 1989). Cut genomic DNA was self‐ligated with 1 U of T4 DNA ligase (Promega) in 70 µl final volume at 4°C overnight and thereafter precipitated with ammonium acetate and ethanol (Sambrook et al., 1989). Samples were dissolved in 10 µl of ddH2O and 5 µl were used for electroporating E. coli by a standard protocol (Sambrook et al., 1989). Electroporated bacteria were plated onto LB agar media supplemented with 100 µg ml−1 ampicillin. Up to eight ampicillin‐resistant bacterial colonies were picked up from each plasmid‐rescued Laccaria strain and cultured in LB liquid media overnight for plasmid purification under the same selection conditions. A plasmid linearization test was carried out with SacI (Promega) which should cut once within the rescued plasmids from the transgenic L. bicolor strains. Restriction products were separated in an 1% agarose gel and stained with ethidium bromide. If a fungal strain failed to produce resistant bacterial colonies at the first rescue attempt this was repeated twice by variating the concentration of genomic DNA.
Sequencing and analysis of T‐DNA–gDNA junctions
Rescued plasmids were purified for sequencing with the QIAprep® Spin Miniprep Kit (Qiagen). The sequencing of the RB–gDNA junctions was performed with the Post‐RB primer (5′‐AGCAGCTTGAGCTTGGATC‐3′) (Invitrogen), which binds to the T‐DNA near the RB sequence of pHg/pBks, the CEQ Dye Terminator Cycle sequencing kit and the CEQ2000XL automated sequencer (Beckman Coulter). In the obtained sequences the T‐DNA–gDNA junctions were identified in the Laccaria genome sequence by using the blastn algorithm on the JGI genome portal (http://genome.jgi‐psf.org/Lacbi1/Lacbi1.home.html). The exact integration sites were mapped in Laccaria genome and the T‐DNA RB conservation was compared between the sequences. The T‐DNA integration sites were located into the predicted genes (in exons or introns), their upstream (up to 1500 bp upstream from ATG) and downstream (up to 500 bp downstream from TAG) elements or in intergenic regions. The genomic zones of the T‐DNA integrations were observed for special features like repetitions and signs of transpositions. The 30 nucleotides sequences around the exact integration sites were aligned with clustalw in order to find putative inter‐site homologies. Integration site sequences were also individually analysed for micro‐homologies with the T‐DNA LB and RB sequences.
Semi‐quantitative RT‐PCR
For RNA isolation L. bicolor wild‐type and transgenic strains 4, 24 and 36 were grown for up to 3 weeks on agar P5 medium covered with a cellophane membrane to facilitate mycelium harvest. Laccaria bicolor transgenic strains were also grown on agar P5 medium supplemented with 150 µg ml−1hygromycin B (Invitrogen). Only the 5 mm fringe of the fungal colonies corresponding to actively growing mycelium was harvested and frozen in liquid nitrogen. Total RNA extraction was performed with the RNeasy Plant Mini Kit (Qiagen) from 100 mg of frozen mycelium with the RLC buffer, and DNase I (Qiagen) treatment was included in the RNA extraction procedure according the manufacturer's instructions to eliminate traces of genomic DNA. Quality and quantity of RNA samples were checked both in agarose gel and by spectrometry.
To allow the amplification of specific transcripts by semi‐quantitative RT‐PCR we designed primers for the L. bicolor gene models interrupted by T‐DNA integration in transgenic strains 4, 24 and 36 (coding protein ID 318751, 379393 and 318555 respectively). The primers were designed in the coding sequence around the T‐DNA integration site and were synthesized (Eurogentec, Belgium) in the way that the amplified fragments were about 200 bp in length. Primers amplifying a fragment of a L. bicolor glucokinase transcript (protein ID 312018; kindly provided by A. Deveau, INRA Nancy) were used for positive control amplification. The primers list is detailed in Table 2. A blastnagainst the L. bicolor genome sequence was performed for each primer sequence to verify the absence of cross‐annealing in other regions of the genome sequence. First‐strand cDNAs were synthesized from 1 µg DNase‐treated total RNA using the iScript cDNA Synthesis Kit (Bio‐Rad) in a total volume of 20 µl according to manufacturer's instructions. Two microlitres of RT products were amplified by PCR with the Eppendorf MasterMix (Eppendorf) and appropriate primers (10 µM each) in 50 µl reaction volume (95°C for 5 min; then 95°C for 30 s, 60°C for 30 s and 72°C for 30 s for 15–30 cycles; and a final extension at 72°C for 10 min). Aliquots of 10 µl of cDNA were sampled after 15, 20, 25, 28 and 30 cycles of amplification, separated by electrophoresis in an 1% agarose gel and stained with ethidium bromide to determine the quantity of the amplified fragments.
Table 2.
Specific forward and reverse primers designed for RT‐PCR amplification of Laccaria transcripts.
| Gene name/predicted function | Laccaria transgenic strain | Laccaria protein ID | Forward primer | Reverse primer |
|---|---|---|---|---|
| Zinc finger protein, MYND type | 4 | 318751 | 5′‐TGAGCGATCGCTTTGTTCCTC‐3′ | 5′‐GACGCTCGGAATTTTGCGTTG‐3′ |
| Ser/Threo protein kinase | 24 | 379393 | 5′‐GCTTGGATAGCGCTTGGAACG‐3′ | 5′‐GTATGGAGTTGGTACTTTGCT‐3′ |
| Di‐ tri‐carboxylate and phosphate transporter | 36 | 318555 | 5′‐TGGCAACGGAGATCGGTCGTG‐3′ | 5′‐GGTCTTGTTGGGGCAGATGAA‐3′ |
| Glucokinase | – | 312018 | 5′‐CTCAAGTCTGGGGAAGCAAC‐3′ | 5′‐CTTGGTAGTCGCGTGAGTGA‐3′ |
Protein ID numbers of gene models in Laccaria bicolor genome sequence (http://genome.jgi‐psf.org/Lacbi1/Lacbi1.home.html) used to design the primers are indicated.
Acknowledgments
This work was supported by Universidad Nacional de Quilmes, CONICET, Agencia Nacional de Promocion Científica y Tecnologica (ANPCyT), Argentina, and the Argentina‐France ECOS‐Sud grants. The support of an INRA innovating grant from EFPA Deparment to Sébastien Duplessis is acknowledged. We kindly thank Aurélie Deveau at INRA Nancy for providing the primers for amplification of L. bicolor glucokinase transcripts. The Laccaria genome sequence data were produced by the US Department of Energy Joint Genome Institute http://www.jgi.doe.gov/. The present work was partly supported by INRA Innovating Grants ‘Genome Sequencing 2005–2006’, Institut Fédérateur de Recherche 110 ‘Génomique, Ecophysiologie et Ecologie Fonctionnelles’ and Région Lorraine for the DNA sequencing and functional genomics facilities. Finally we would like to acknowledge two anonymous reviewers and editorial board who contributed to improve this work.
References
- Alonso J.M., Stepanova A.N., Leisse T.J., Kim C.J., Chen H., Shinn P. Genome‐wide insertional mutagenesis of Arabidopsis thaliana. Science. 2003;301:653–657. doi: 10.1126/science.1086391. et al. [DOI] [PubMed] [Google Scholar]
- Blaise F., Remy E., Meyer M., Zhou L., Narcy J.P., Roux J. A critical assessment of Agrobacterium tumefaciens‐mediated transformation as a tool for pathogenicity gene discovery in the phytopathogenic fungus Leptosphaeria maculans. Fungal Genet Biol. 2007;44:123–138. doi: 10.1016/j.fgb.2006.07.006. et al. [DOI] [PubMed] [Google Scholar]
- Brunaud V., Balzergue S., Dubreucq B., Aubourg S., Samson F., Chauvin S. T‐DNA integration into the Arabidopsis genome depends on sequences of pre‐insertions site. EMBO Rep. 2002;3:1152–1157. doi: 10.1093/embo-reports/kvf237. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bundock P., Dulk‐Ras A., Beijersbergen A., Hooykaas P.J. Trans‐kingdom T‐DNA transfer from Agrobacterium tumefaciens to Saccharomyces cerevisiae. EMBO J. 1995;14:3206–3214. doi: 10.1002/j.1460-2075.1995.tb07323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bundock P., Van Atticum H., Dulk‐Ras A., Hooykaas P.J. Insertional mutagenesis in yeast using T‐DNA from Agrobacterium tumefaciens. Yeast. 2002;19:529–536. doi: 10.1002/yea.858. [DOI] [PubMed] [Google Scholar]
- Chen X., Stone M., Schlagnhaufer C., Romaine C.P. A fruiting body tissue method for efficient Agrobacterium‐mediated transformation of Agaricus bisporus. Appl Environ Micorbiol. 2000;66:4510–4513. doi: 10.1128/aem.66.10.4510-4513.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combier J.P., Melayah D., Raffier C., Gay G., Marmeisse R. Agrobacterium tumefaciens‐mediated transformation as a tool for insertional mutagenesis in the symbiotic ectomycorrhizal fungus Hebeloma cylindrosporum. FEMS Microbiol Lett. 2003;220:141–148. doi: 10.1016/S0378-1097(03)00089-2. [DOI] [PubMed] [Google Scholar]
- De Block M., And Debrouwer D. Two T‐DNAs co‐transformed into Brassica napus by a double Agrobacterium infection are mainly integrated at the same locus. Theor Appl Genet. 1991;82:257–263. doi: 10.1007/BF02190610. [DOI] [PubMed] [Google Scholar]
- De Groot M.J., Bundock P., Hooykaas P.J., Beijersbergen A.G. Agrobacterium tumefaciens‐mediated transformation of filamentous fungi. Nat Biotechnol. 1998;16:839–842. doi: 10.1038/nbt0998-839. [DOI] [PubMed] [Google Scholar]
- De Neve M., De Bruck S., Jacobs A., Van Montagu M., Depicker A. T‐DNA integration patterns in co‐transformed plant cells suggest that T‐DNA repeats originate from co‐integration of separate T‐DNAs. Plant J. 1997;11:15–29. doi: 10.1046/j.1365-313x.1997.11010015.x. [DOI] [PubMed] [Google Scholar]
- Di Battista C., Selosse M.A., Bouchard D., Stenström E., Le Tacon F. Variation in symbiotic efficiency, phenotypic characters and ploidy level among different isolates of the ectomycorrhizal basidiomycete Laccaria bicolor strain S238. Mycol Res. 1996;100:1315–1324. [Google Scholar]
- Duplessis S., Tagu D., Martin F. Living together underground: a molecular glimpse of the ectomycorrhizal symbiosis. In: Osiewaccz H.D., editor. Dekker & Dekker; 2002. pp. 297–323. [Google Scholar]
- Duplessis S., Courty P., Tagu D., Martin F. Transcript patterns associated with ectomycorrhiza development in Eucalyptus globulus and Pisolithus microcarpus. New Phytol. 2005;165:599–611. doi: 10.1111/j.1469-8137.2004.01248.x. [DOI] [PubMed] [Google Scholar]
- Elliott C.E., Howlett B.J. Overexpression of a 3‐ketoacyl‐CoA‐thiolase in Leptosphaeria maculans causes reduced pathogenicity on Brassica napus. Mol Plant Microbe Interact. 2006;19:588–596. doi: 10.1094/MPMI-19-0588. [DOI] [PubMed] [Google Scholar]
- Hiei Y., Ohta S., Komari T., Kumashiro T. Efficient transformation of rice (Oryza sativa L.) mediated by Agrobacterium and sequence analysis of the boundaries of the T‐DNA. Plant J. 1994;6:271–282. doi: 10.1046/j.1365-313x.1994.6020271.x. [DOI] [PubMed] [Google Scholar]
- Idnurum A., Reedy J.L., Nuusbaum J.C., Heitman J. Cryptococcus neoformans virulence gene discovery through insertional mutagenesis. Eukaryot Cell. 2004;3:420–429. doi: 10.1128/EC.3.2.420-429.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson T., Le Quéré A., Ahren D., Söderström B., Erlandsson R., Lundeberg J. Transcriptional responses of Paxillus involutus and Betula pendula during formation of ectomycorrhizal root tissue. Mol Plant Microbe Interact. 2004;17:202–215. doi: 10.1094/MPMI.2004.17.2.202. et al. [DOI] [PubMed] [Google Scholar]
- Kemppainen M., Circosta A., Tagu D., Martin F., Pardo A.G. Agrobacterium‐mediated transformation of the ectomycorrhizal symbiont Laccaria bicolor S238N. Mycorrhiza. 2005;16:19–22. doi: 10.1007/s00572-005-0008-7. [DOI] [PubMed] [Google Scholar]
- Kim S.R., Lee J., Jun S.H., Park S., Kang H.G., Kwon S., An G. Transgene structures in T‐DNA inserted rice plants. Plant Mol Biol. 2003;52:761–773. doi: 10.1023/a:1025093101021. [DOI] [PubMed] [Google Scholar]
- Kumar S., Fladung M. Transgene repeats in aspen: molecular characterization suggest simultaneous integration of independent T‐DNAs into receptive hotspots in the host genome. Mol Genet Genomics. 2000;264:20–28. doi: 10.1007/s004380000296. [DOI] [PubMed] [Google Scholar]
- Kunik T., Tzfira T., Kapulnik Y., Gafni Y., Dingwall C., Citovsky V. Genetic transformation of HeLa cells by Agrobacterium. Proc Natl Acad Sci USA. 2001;98:1871–1876. doi: 10.1073/pnas.041327598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent P., Voilet C., Tagu D., De Carvalho D., Nehls U., De Bellis R. A novel class of ectomycorrhiza‐regulated cell wall polypeptides in Pisolithus tinctorius. Mol Plant Microbe Interact. 1999;12:862–871. doi: 10.1094/MPMI.1999.12.10.862. et al. [DOI] [PubMed] [Google Scholar]
- LeClerque A., Wang H., Abschutz A., Chen S., Mitina G.V., Zimmerman G., Schairer H.U. Agrobacterium‐mediated insertional mutagenesis (AIM) of the entomopathogenic fungus Beauveria bassiana. Curr Genet. 2004;45:111–119. doi: 10.1007/s00294-003-0468-2. [DOI] [PubMed] [Google Scholar]
- Martin F., Kohler A., Duplessis S. Living in harmony in the wood underground: ectomycorrhizal genomics. Curr Opin Plant Biol. 2007;10:204–210. doi: 10.1016/j.pbi.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Martin F., Aerts A., Ahnén D., Brun A., Danchin E.G.J., Duchaussoy F. The genome of Laccaria biocolor provides insights into mycorrhizal symbiosis. Nature. 2008;452:88–92. doi: 10.1038/nature06556. et al. [DOI] [PubMed] [Google Scholar]
- Mayerhofer R., Koncz‐Kalman Z., Nawrath C., Bakkeren G., Crameri A., Angelis K. T‐DNA integration: a mode of illegimate recombination in plants. EMBO J. 1991;10:697–704. doi: 10.1002/j.1460-2075.1991.tb07999.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Y., Patel G., Heist M., Betts M., Tucker S.L., Galadima N. A systematic analysis of T‐DNA insertion events in Magnaporthe oryzae. Fungal Genet Biol. 2007;44:1050–1064. doi: 10.1016/j.fgb.2007.04.002. et al. [DOI] [PubMed] [Google Scholar]
- Michielse C.B., Hooykaas P.J., Hondel C.A.M., Ram A.F.J. Agrobacterium‐mediated transformation as a tool for functional genomics in fungi. Curr Genet. 2005;48:1–17. doi: 10.1007/s00294-005-0578-0. [DOI] [PubMed] [Google Scholar]
- Offringa R., De Groot M.J.A., Haagsman H.J., Does M.P., Elzner P.J.M., Hooykaas P.J. Extrachromosomal homologous recombination and gene targeting in plant cells after Agrobacterium mediated transformation. EMBO J. 1990;9:3077–3084. doi: 10.1002/j.1460-2075.1990.tb07504.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo A.G., Hanif M., Raudaskoski M., Gorfer M. Genetic transformation of ectomycorrhizal fungi mediated by Agrobacterium tumefaciens. Mycol Res. 2002;106:132–137. [Google Scholar]
- Pelczar P., Kalck V., Gomes D., Hohn B. Agrobacterium proteins VirD2 and VirE2 mediate precise integration of synthetic T‐DNA complexes in mammalian cells. EMBO Rep. 2004;6:632–637. doi: 10.1038/sj.embor.7400165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter M., Courty P., Kohler A., Delaruelle C., Martin D., Tagu D. Analysis of expressed sequence tags from the ectomycorrhizal basidiomycetes Laccaria bicolor and Pisolithus microcarpus. New Phytol. 2003;159:117–129. doi: 10.1046/j.1469-8137.2003.00796.x. et al. [DOI] [PubMed] [Google Scholar]
- Podila G., Zheng J., Balasubramanian S., Sundaram S., Hieremath S., Brand J., Hymes M. Molecular interactions in ectomycorrhizas: identification of fungal genes involved in early symbiotic interactions between Laccaria bicolor and red pine. Plant and Soil. 2002;244:117–128. [Google Scholar]
- Rho H.S., Kang S., Lee Y.H. Agrobacterium tumefaciens‐mediated transformation of the plant pathogenic fungus Magnaporthe grisea. Mol Cells. 2001;12:407–411. [PubMed] [Google Scholar]
- Rogers C.W., Challen M.P., Green J.R., Whipps J.M. Use of REMI and Agrobacterium‐mediated transformation to identify pathogenicity mutants of the biocontrol fungus Coniothyrium minitans. FEMS Microbiol Lett. 2004;241:207–214. doi: 10.1016/j.femsle.2004.10.022. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritsch E.F., Maniatis T. Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- Tinland B., Schoumacher F., Cloeckler V., Bravo‐Angel A.M., Hohn B. The Agrobacterium tumefaciens virulence protein D2 is responsible for precise integration of T‐DNA into the plant genome. EMBO J. 1995;14:3585–3595. doi: 10.1002/j.1460-2075.1995.tb07364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji G., Fujii S., Fujihara N., Hirose C., Tsuge S., Shiraishi T., Kubo Y. Agrobacterium tumefaciens‐mediated transformation for random insertional mutagenesis in Colletotrichum lagenarium. J Gen Plant Pathol. 2003;69:230–239. [Google Scholar]
- Tuskan G.A., DiFazio S., Jansson S., Bohlmann J., Grigoriev I., Hellsten U. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray) Science. 2006;313:1596–1604. doi: 10.1126/science.1128691. et al. [DOI] [PubMed] [Google Scholar]
- Tzfira T., Li J., Lacroix B., Citovsky V. Agrobacterium T‐DNA integration: molecules and models. Trends Genet. 2004;20:375–383. doi: 10.1016/j.tig.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Van Attikum H., Bundock P., Hooykaas P.J. Non‐homologous end‐joining proteins are required for Agrobacterium T‐DNA integration. EMBO J. 2001;20:6550–6558. doi: 10.1093/emboj/20.22.6550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voiblet C., Duplessis S., Encelot N., Martin F. Title Identification of symbiosis‐regulated genes in Eucalyptus globulus–Pisolithus tinctorius ectomycorrhiza by differential hybridization of arrayed cDNAs. Plant J. 2001;25:181–191. doi: 10.1046/j.1365-313x.2001.00953.x. [DOI] [PubMed] [Google Scholar]
- Winzeler E.A., Shoemaker D.D., Astromoff A., Liang H., Anderson K., Andre B. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–906. doi: 10.1126/science.285.5429.901. et al. [DOI] [PubMed] [Google Scholar]