

Summary
This work demonstrates that Acinetobacter radioresistens strain S13 during the growth on medium supplemented with long‐chain alkanes as the sole energy source expresses almA gene coding for a Baeyer‐Villiger monooxygenase (BVMO) involved in alkanes subterminal oxidation. Phylogenetic analysis placed the sequence of this novel BVMO in the same clade of the prodrug activator ethionamide monooxygenase (EtaA) and it bears only a distant relation to the other known class I BVMO proteins. In silico analysis of the 3D model of the S13 BVMO generated by homology modelling also supports the similarities with EtaA by binding ethionamide to the active site. In vitro experiments carried out with the purified enzyme confirm that this novel BVMO is indeed capable of typical Baeyer‐Villiger reactions as well as oxidation of the prodrug ethionamide.
Introduction
Alkanes constitute up to 50% of crude oil, but are also produced by plants, green algae, bacteria or animals. As alkanes are apolar and chemically inert molecules (Labinger and Bercaw, 2002), their metabolism by microorganisms poses challenges related to their low water solubility, their tendency to accumulate in cell membranes, and the energy needed to activate the molecule. Nevertheless, several microorganisms, both aerobic and anaerobic, have established effective strategies involving specialized enzyme systems and metabolic pathways to access alkanes as a carbon and energy source (Berthe‐Corti and Fetzner, 2002; van Hamme et al., 2003). Aerobic alkane degraders use molecular oxygen for the activation of the alkane molecule by means of monooxygenases that are able to overcome the low chemical reactivity. A recent interesting example of such an enzyme is the flavin‐dependent monooxygenase from Geobacillus thermodenitrificans (Feng et al., 2007; Li et al., 2008). In the case of alkanes containing two or more carbon atoms, aerobic degradation usually starts by the oxidation of a terminal methyl group to give a primary alcohol, which is further oxidized to the corresponding aldehyde, and finally converted into a fatty acid. Fatty acids are conjugated to CoA and further degraded to acetyl‐CoA (Wentzel et al., 2007) as shown in Fig. 1. Subterminal oxidation of alkanes has also been reported (Kotani et al., 2007). The product generated, a secondary alcohol, is converted to the corresponding ketone and then oxidized by a Baeyer‐Villiger monooxygenase to render an ester. The ester is hydrolysed by an esterase, generating an alcohol and a fatty acid. Both terminal and subterminal oxidation can coexist in some microorganisms. Strains degrading medium‐chain‐length alkanes (C6–C18), or long‐chain‐length alkanes (> C18) with the terminal oxidation, frequently contain membrane‐bound non‐haem di‐iron monooxygenases related to the well‐characterized Pseudomonas putida GPo1 AlkB alkane hydroxylase. This system functions in complex with two electron transfer proteins, a dinuclear iron rubredoxin, and a mononuclear iron rubredoxin reductase channelling electrons from NADH to the active site of the alkane hydroxylase (van Beilen et al., 2003). Genes that are closely related to alkB of GPo1 have been identified in a large fraction of the microbial population in oil‐contaminated environments (Sotsky et al., 1994). In contrast, DNA of many strains able to grow on long‐chain alkanes does not hybridize with an alkB gene probe, suggesting that they contain alkane oxidation enzyme systems unrelated or distantly related to AlkB (Smits et al., 1999). Acinetobacter strain DSM 17874, has been found to contain a Baeyer‐Villiger monooxygenase, named AlmA, which oxidizes C20–C32 alkanes (Throne‐Holst et al., 2007). Genes homologous to almA have been identified in several other long‐chain n‐alkane‐degrading strains, including Acinetobacter sp. M1 and Acinetobacter borkumensis SK2 (Rojo, 2009).
Figure 1.
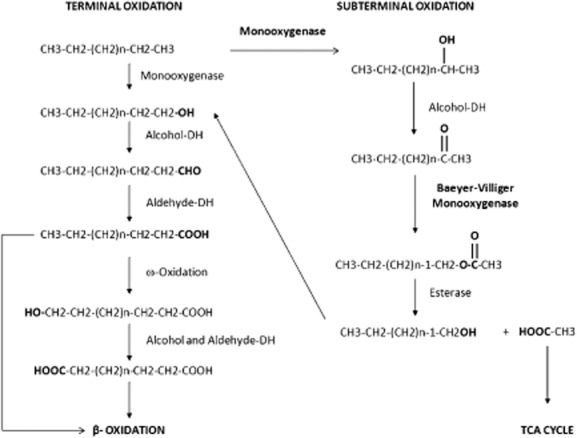
Proposed terminal and subterminal alkane degradation pathways.
Baeyer‐Villiger monooxygenases (BVMOs), produced by several bacteria and fungi, are NAD(P)H‐dependent flavoproteins that catalyse the insertion of an oxygen atom in a ketone, next to the carbonyl carbon atom (Baeyer‐Villiger oxidation). This reaction converts a ketone into an ester, a conversion that is of great value for the synthesis of fine chemicals. BVMOs can be classified in two groups: type I BVMOs contain flavin adenine dinucleotide (FAD) as cofactor, use NADPH as source for electrons and consist of identical subunits, while type II BVMOs contain flavin mononucleotide (FMN) as cofactor, use NADH as electron donor and are composed of α2β trimers. The present classification of BVMOs in the family of flavoproteins at large, places type I BVMOs in the class B and those of type II in the class C (van Berkel et al., 2006).
Several strains in the genus Acinetobacter are known as n‐alkane utilizers (Muller et al., 1989; Maeng et al., 1996; Koma et al., 2001; Maier et al., 2001; Throne‐Holst et al., 2006). Acinetobacter sp. strain S13 was isolated from soil surrounding an activated sludge pilot plant near Torino (Italy) for its fast phenol catabolism when used as the sole carbon and energy source (Pessione and Giunta, 1997). The strain was classified as radioresistens according to fatty acids and Amplified Fragment Length Polymorphysm analysis. No data concerning A. radioresistens ability to degrade alkanes are available.
This study evaluates the capability of S13 strain to utilize medium and long‐chain alkanes as growth substrates and to identify the enzymes involved in their oxidation. The in silico analysis of one of the enzymes involved leads to the interesting hypothesis of its relevance in prodrug activation.
Results
16S rRNA sequence analysis and growth of A. radioresistens strain S13 on alkanes
The taxonomic position of Acinetobacter sp. strain S13 previously elaborated (Pessione and Giunta, 1997) was confirmed by using eubacterial primers for PCR amplification of the 16S rRNA. In order to overcome any uncertainty the amplified product was sequenced: a sequence of 1490 bp was obtained and used for phylogenetic analysis that unambiguously placed S13 strain among the radioresistens species.
The ability of A. radioresistens S13 to grow in liquid mineral salts medium (MSM) cultures supplemented with alkanes of defined carbon chain lengths was investigated. The bacterial growth on MSM medium supplemented with medium length liquid (C14 and C16) and long length solid (C24 and C36) alkanes was studied (Fig. 2).
Figure 2.
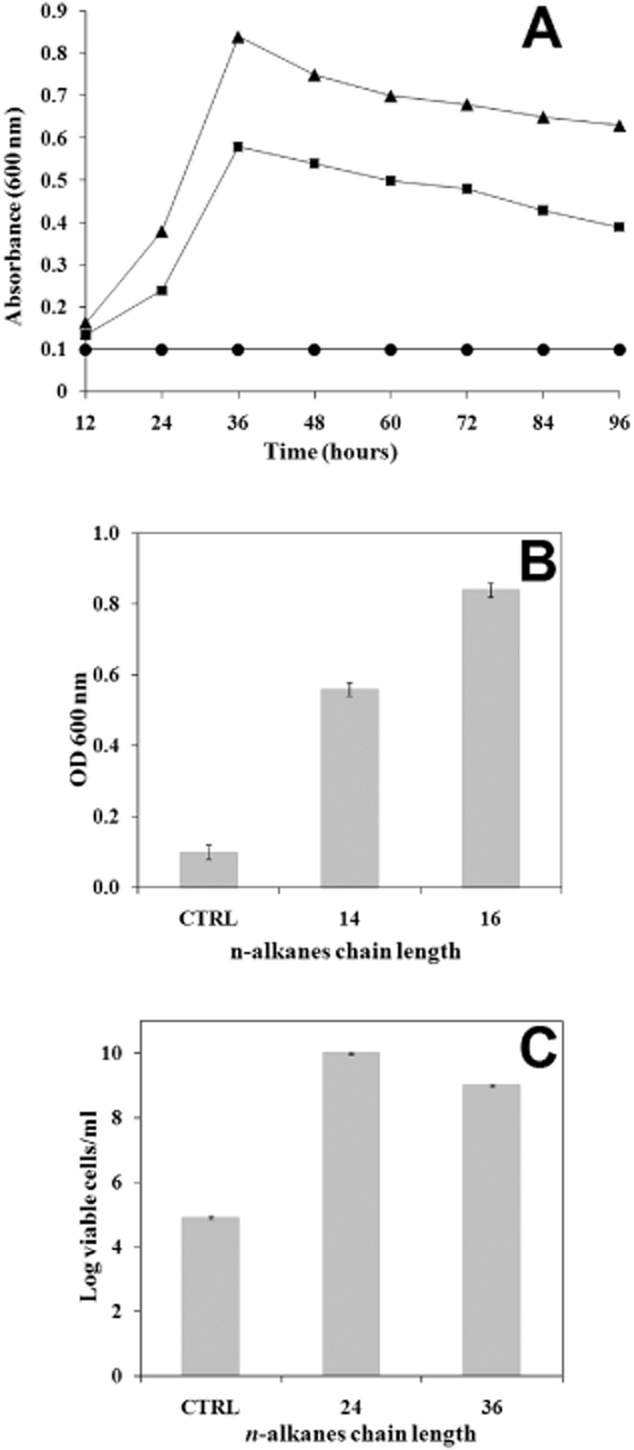
Growth of Acinetobacter radioresistens strain S13 on n‐alkanes. (A) Growth curve of Acinetobacter radioresistens strain S13 isolate in minimal medium (circle) supplemented with C14 (square) and C16 (triangle) during the first 96 h, (B) bacterial growth was followed by measuring OD at 600 nm after 36 h in the case of the liquid, medium‐length chain C14 and C16 alkanes and (C) viable cells number per millilitre of cultures when the bacteria were grown on minimal medium supplemented with the solid, long‐length chain C24 and C36 alkanes after 3 and 6 days respectively. CTRL = control experiments with bacterial culture grown on minimal medium without n‐alkanes.
In liquid cultures, growth was measured as an increase in OD600 for cultures supplemented with liquid alkanes (Fig. 2A), and for cultures supplemented with solid alkanes the number of colony‐forming units were measured on LB agar plates on which 1 ml of liquid culture containing the alkane was plated (Fig. 2B). In all cases, A. radioresistens S13 growth in the presence of C14, C16, C24 and C36 alkanes was significantly higher than that measured in control experiments. In particular, the growth of this strain on C14 and C16 alkanes reached the maximum after 36 h of incubation. The viable cell count started to increase within 1/2 days and reached a maximum of 2.36 × 1010 and 3.0 × 109 cells after 3 and 6 days of incubation at 30°C respectively.
Identification of Baeyer‐Villiger monooxygenase (BVMO) and terminal alkane 1‐monooxygenase (AlkB)
The growth of A. radioresistens strain S13 on alkanes led to the hypothesis of the presence of enzymes involved in terminal and/or subterminal alkane degradation. Therefore, PCR reactions were performed with oligonucleotide primers based on the sequence of Acinetobacter almA and alkB genes. Two PCR products of the expected size of 760 and 786 bp for the almA and alkB genes respectively, were obtained (data not shown). The translated peptide sequence of the two amplicons showed high sequence similarity to the AlmA (Throne‐Holst et al., 2007) and AlkB (Sotsky et al., 1994) gene products from other Gram‐negative bacteria. In order to obtain the full length of the two genes, a genome walking strategy was adopted. The analysis of the two entire nucleotide sequences revealed the presence of two complete open reading frames (ORFs) of 1491 and 1409 bp, hereinafter referred to as ORF1491 and ORF1409 respectively. Sequence comparison using the blast program against the non‐redundant GenBank database showed that the amino acid sequences of ORF1491 and ORF1409 had very low E‐values (the probability due to chance that there is another alignment with a similarity greater than the given score), with the large class of flavin‐dependent monooxygenases and terminal alkane hydroxylases respectively.
Analysis of the amino acid sequences of the BVMO and AlkB monooxygenases
The ORF1491 and ORF1409 encode two proteins of 496 and 402 amino acids, with a predicted molecular mass of 56 082 and 46 498 Da respectively. Amino acid sequences of the two proteins were compared with those available in the database. A clustalw multialignment of BVMO and AlkB sequences was used to identify conserved regions and to detect the degrees of sequence similarity and identity between the A. radioresistens S13 BVMO (Ar‐BVMO) and AlkB proteins and the orthologous proteins.
The sequence homology search using the amino acid sequence coded by ORF1491 revealed significant homology with several multifunctional flavin‐containing monooxygenases, the N‐hydroxylating monooxygenases and BVMO. The presence of two Rossmann folds, as evidenced by two GXGXX(G/A) motifs (Fig. > 3), which is a common sequence among FAD and NAD(P)H‐dependent oxidoreductases, discriminates the enzyme from the mechanistically related flavoprotein hydroxylases (Eppink et al., 1997). Furthermore, the presence of an identified sequence fingerprint (FXGXXXHXXXW(P/D) (Fig. 3) suggests that the protein is a flavin‐containing Baeyer‐Villiger monooxygenase (Fraaije et al., 2002). Indeed a search in the protein sequence database revealed that the closest homologue with known activity is ethionamide‐activating monooxygenase from Mycobacterium tuberculosis, sharing a 49.7% sequence identity and 71.0% sequence similarity.
Figure 3.

Multiple sequence alignment of different flavoprotein monooxygenases including several Baeyer‐Villiger monoxygenases (BVMO)s proteins. FAD‐ and NADPH‐binding domains are highlighted in yellow (Rosmann fold), the fingerprint consensus sequence is highlighted in green. Identical residues between two sequences are indicated by an asterisk (*); similar residues are indicated by two dots (high similarity) or one dot (low similarity). PAMO = phenylacetone monooxygenase from Thermobifida fusca, 1W4X; STMO = steroid monooxygenase from Rhodococcus rhodochrous, BAA24454; CPMO = cyclopentanone monooxygenase from Comamonas testosteronii NCIMB 9872, CAD10798; CHMO = cyclohexanone monooxygenase from Acinetobacter calcoaceticus NCIMB 9871, AAG10021; EtaA = ethionamide‐activating monooxygenase from Mycobacterium tuberculosis H37Rv, CAB06212; HAPMO = 4‐hydroxyacetophenone monooxygenase from Pseudomonas fluorescens ACB, AF355751; BVMO = Baeyer‐Villiger monooxygenase from Acinetobacter radioresistens strain S13, HQ685899 (this work).
The amino acid sequence coded by ORF1409 was compared with other AlkB homologues found in the GenBank database. The closest sequence similarity was found with alkane 1‐monooxygenases homologues from A. radioresistens strain SH164, with which it shares a 98.0% sequence identity and 99.2% sequence similarity.
The AlkB protein of A. radioresistens S13 possess eight histidines that are highly conserved in non‐haem iron integral membrane alkane hydroxylases and desaturases, and that are believed to be required for catalytic activity by these enzymes (Shanklin et al., 1994) (Fig. > 4). Sequences corresponding to the three histidine boxes Hist1 = HE(L/M)XHK, Hist2 = EHXXGHH and Hist3 = LQRH(S/A)DHHA are highly conserved in all bacterial alkane monooxygenases (Fig. 4). The Hist3 box is the longest and almost perfectly conserved sequence in all alkane hydroxylases, but is not well conserved in other closely related hydrocarbon monooxygenases. An additional well‐conserved histidine box NYXEHYG(L/M) containing the HYG motif that is located about 30 amino acids upstream of the Hist3 box is also present (Fig. 4) (Smits et al., 1999). This HYG motif is also well conserved in related hydrocarbon monooxygenases.
Figure 4.

Multiple sequence alignment of predicted amino acid sequences corresponding to alkane monooxygenase genes from various bacteria. The three conserved histidine boxes (Hist‐1, Hist‐2 and Hist‐3) are highlighted in red and the HYG‐motif is highlighted in violet. The amino acids are indicated by single‐letter codes. Gaps were introduced for optimal alignment. Identical residues between two sequences are indicated by an asterisk (*); similar residues are indicated by one dot (low similarity) or two dots (high similarity). Conserved histidine of the fingerprint motif are written in bold letters. Gordonia = alkane 1‐monooxygenase from Gordonia sp. TF6, BAD67020.1; Rhodococcus = alkane 1‐monooxygenase from Rhodococcus ruber, ACX30755.1; Mycobacterium = alkane 1‐monooxygenase from Mycobacterium sp. Spyr 1, YP_004078475.1; Streptomyces = alkane 1‐monooxygenase from Streptomyces sp. AA4, ZP_07282765.1; Thermonospora = alkane 1‐monooxygenase from Thermonospora curvata DSM 43183, YP_003298195.1; Ralstonia = alkane 1‐monooxygenase from Ralstonia sp., ZP_07673680.1; Burkholderia = alkane 1‐monooxygenase from Burkholderia phytofirmans YP_001889129; Alcanivorax = alkane 1‐monooxygenase from Alcanivorax dieselolei, AAT91722.2; SH164 = alkane hydroxylase B from Acinetobacter radioresistens SH164, ZP_06072466.1; SK 82 = alkane 1‐monooxygenase from Acinetobacter radioresistens SK82, ZP_05361594.1; Ajunii = alkane hydroxylase B from Acinetobacter junii, ZP_06066074.1; Acalcoaceticus = alkane 1‐monooxygenase from Acinetobacter calcoaceticus, CAB51020.1; Abaumannii = alkane 1‐monooxgenase from Acinetobacter baumannii, YP_001707231.1; Ahaemolyticus = alkane hydroxylase from Acinetobacter haemolyticus, AAS93604.4; Psychrobacter = alkane 1‐monooxygenase from Psychrobacter sp. PRwf‐1, YP_001280943.1; S13 = alkane 1‐monooxygenase from Acinetobacter radioresistens S13, GU145276 (this work).
Phylogenetic analysis
A clustalw multialignment was followed by visual inspection and manual editing of the A. radioresistens S13 BVMO and AlkB sequences. Phylogenetic trees of the two amino acid sequences were inferred from the selected sequences by the Distant Matrix method. Interestingly, cluster analysis places the BVMO enzyme of this study in the same branch of ethionamide monooxygenase (EtaA, Fig. 5A), a FAD‐containing monooxygenase that is responsible for activation of several thionamide prodrugs in M. tuberculosis (Vannelli et al., 2002). EtaA also clusters with other mycobacterial BVMO sequences, as well as the more recently described P. putida KT 2440 BVMO (Rehdorf et al., 2007). As expected MtmOIV, the protein responsible for the B‐V reaction in the biosynthetic pathway of mithramycin is an outlier (Fig. 5A). MtmOIV is very distinct from bacterial type I and type II BVMOs and is considered as an atypical BVMO (Beam et al., 2009).
Figure 5.

Phylogenetic relationships within Baeyer‐Villiger monooxygenases (A) and alkane monooxygenases (B). Sequences of proteins with confirmed BVMO and alkane monooxygenase activity were aligned and the un‐rooted phylogenetic trees were calculated by using clustalw and neighbour‐joining method. The numbers at the nodes are the bootstrap confidence values obtained after 1000 replicates. The scale bar indicates distance in substitutions per nucleotide. The species and proteins are: (A) CHMO A rthrobacter sp. L661 = cyclohexanone monooxygenase (ABQ10653.1); CHMO A rthrobacter sp. BP2 = cyclohexanone monooxygenase (AA N37479.1); CHMO R hodococcus sp. HI‐31 = cyclohexanone monooxygenase (BAH56670); CHMO B rachymonas petroleovorans = cyclohexanone monooxygenase (AAR99068.1); CHMO A cinetobacter = cyclohexanone monooxygenase (AF282240_5); PAMO Frankia sp. Eu1c = phenylacetone monooxygenase (YP_004016782.1); PAMO T hermobifida fusca YX = phenylacetone monooxygenase (AAZ55526.1); PAMO R hodococcus equi ATCC33707 = phenylacetone monooxygenase (ZP_ 08156844.1); CPMO R hodococcus josti RHA1 = cyclopentanone monooxygenase (YP_703208.1); CPMO C omamonas sp. NCIMB 9872 = cyclopentanone monooxygenase (BAC22652.1); CPMO P aracoccidioides brasiliensis Pb01 = cyclopentanone monooxygenase (XP_002792362.1); CPMO P yrenophora treitici‐repentis = cyclopentanone monooxygenase (XP_001942142.1); CDMO M ycobacterium avium 104 = cyclododecanone monooxygenase (YP_884322.1); CDMO R hodococcus ruber = cyclododecanone monooxygenase (AAL14233.1); CDMO H yphomonas neptunium ATCC15444 = cyclododecanone monooxygenase (YP_760004.1); CPDMO P seudomonas sp. HI‐70 = cyclopentadecanone monooxygenase (BAE93346.1); HAPMO R hodococcus equi ATCC33707 = hydroxyacetophenone monooxygenase (ZP_08156846.1); HAPMO M ycobacterium kansasii ATCC33707 = hydroxyacetophenone monooxygenase (ZP_04749205.1); HAPMO P henylobacterium zucineum HLK1 = hydroxyacetophenone monooxygenase (YP_002130647.1); Burkholderia sp. CCGE 1002 = hydroxyacetophenone monooxygenase (ADG19710.1); BVMO A cinetobacter sp. M‐1 = alkane monooxygenase (ABQ18228.1); BVMO A cinetobacter sp. DSM 17874 = Baeyer‐Villiger monooxygenase (ABQ18224.1); BVMO A cinetobacter baumannii ACICU = Baeyer‐Villiger monooxygenase (YP_001847910.1); BVMO A cinetobacter baumannii ACICU = Baeyer‐Villiger monooxygenase (YP_001847910.1); BVMO A cinetobacter baumannii AYE = Baeyer‐Villiger monooxygenase (YP_001712414); BVMO A cinetobacter sp. ADP1 = Baeyer‐Villiger monooxygenase (YP_047698.1); BVMO A cinetobacter radioresistens S13 = Baeyer‐Villiger monooxygenase (GU145276.1); PpBVMO P seudomonas putida = monooxygenase of flavin‐binding family (AAN68413.1); EtaA M ycobacterium bovis BCG = ethionamide monooxygenase (YP_979996.1); EtaA M ycobacterium tuberculosis SUMu001 = ethionamide monooxygenase (ZP_07416557.1); EtaA M ycobacterium tuberculosis H37Rv = ethionamide monooxygenase (NP_218371.1); EtaA M ycobacterium tuberculosis H37Ra = ethionamide monooxygenase (YP_001285245.1); EtaA M ycobacterium tuberculosis KZN1435 = ethionamide monooxygenase (YP_003033907); EtaA M ycobacterium tuberculosis GM1503 = ethionamide monooxygenase (ZP_03534438.1), MtmOIV S treptomyces argillaceus = mithramycin monooxygenase (3FMW_A); (B) Acinetobacter baumannii AB900 = terminal alkane‐1‐monooxygenase (ZP_04661203.1); Acinetobacter baumannii ACICU = terminal alkane‐1‐monooxygenase (YP_001846325.1); Acinetobacter sp. 6013113 = alkane‐1‐monooxygenase (ZP_06781771.1); Acinetobacter calcoaceticus = alkane‐1‐monooxygenase (CAB51020.1); Acinetobacter baumannii SDF = terminal alkane‐1‐monooxygenase (YP_001707231.1); Acinetobacter radioresistens sp. SH164 = alkane hydroxylase B (ZP_06072466.1); Acinetobacter haemolyticus = alkane hydroxylase (AAS93604.4); Acinetobacter radioresistens S13 = alkane hydroxylase B (HQ68589); Acinetobacter sp. SH164 = alkane hydroxylase B (ZP_06072466.1); Acinetobacter radioresistens SK82 = alkane‐1‐monooxygenase (ZP_05361594.1).
The phylogenetic tree reconstructed on the basis of the AlkB amino acid sequences shows that the S13 alkane monooxygenase belongs to the same cluster of Acinetobacter AlkB enzymes (Fig. 5B).
Differential expression of the BVMO and AlkB in response to different chain length alkanes
RT‐PCR experiments were performed on the total RNA extracted from A. radioresistens S13 grown in minimal medium supplemented with C14, C16 (medium‐chain‐length alkanes) and C24, C36 (long‐chain‐length alkanes) as the sole energy and carbon source and from bacteria grown in the presence of sodium acetate. Using the two sets of alma‐ and alkB‐specific primers an amplified product of the expected size (499 and 466 bp respectively) was obtained only with RNA extracted from S13 grown with medium‐chain alkanes in the case of ALK primers (Fig. 6), and on RNA extracted from bacteria grown on long‐chain alkanes when BVMO primers were used. No amplified product was obtained from the RNA of bacteria grown with sodium acetate or from the RT‐negative controls. An amplified fragment of the expected size was also obtained using the A. radioresistens S13 16S rDNA‐specific primers on the RNA of the bacterium grown in the presence of medium‐ and long‐chain‐length alkanes and sodium acetate.
Figure 6.
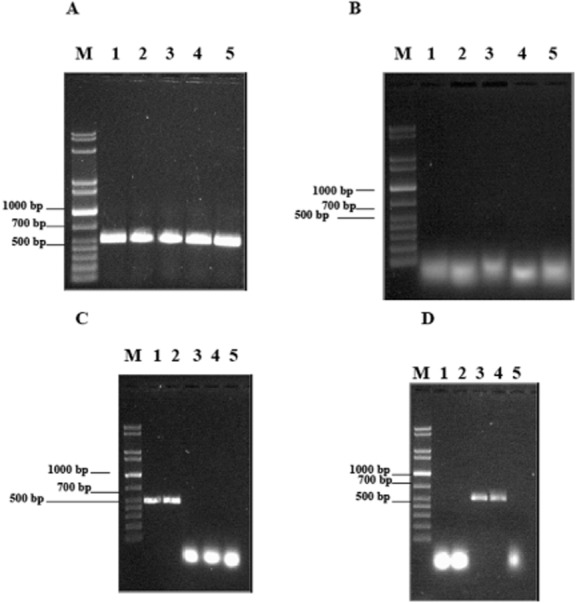
Agarose gel of RT‐PCR products amplified by using A. radioresistens S13 16S rDNA (A: experiment; B: control without RT), alkB (C) and almA genes (D) specific primers. RNA from S13 grown in minimal medium supplemented with C14 (lane 1), C16 (lane 2), C24 (lane 3) and C36 (lane 4) alkane; control experiment of RNA from S13 grown in minimal medium with sodium acetate (lane 5); lane M = DNA mass ladder (XL 1 kb, Eppendorf).
The amplicons obtained with BVMO, ALK and 16S rRNA primers were purified and sequenced. A nucleotide sequence of about 500 bp was obtained for all PCR products, showing a 100% sequence identity to almA, alkB and 16S ribosomal genes of A. radioresistens S13.
Construction of a 3D model of Ar‐BVMO
Due to the large substrate specificity of the BVMO proteins and to the absence of a published crystal structure of Ar‐BVMO, a homology model was generated by using the yasara software (Krieger and Vriend, 2002). To this end, cyclohexanone monooxygenase (CHMO) from Rhodococcus sp. was used as a template as this protein shares the highest sequence identity (20.4%) of a BVMO with a known crystal structure (PDB ID = 3GWF, Mirza et al., 2009).
A good stereochemical quality of the model was found by analysis of the geometrical parameters using the Ramachandran plot (Fig. 7A), which shows how the distribution of the ϕ and ψ angles is well comprised in the allowed regions. 86.1% of the residues are in the most favoured regions and 10.2% are in the additional allowed regions, placing the majority (96.3%) of the residues in the most favoured regions. The carbon‐α ribbon presentation of the 3D model of Ar‐BVMO is shown in Fig. 7B.
Figure 7.
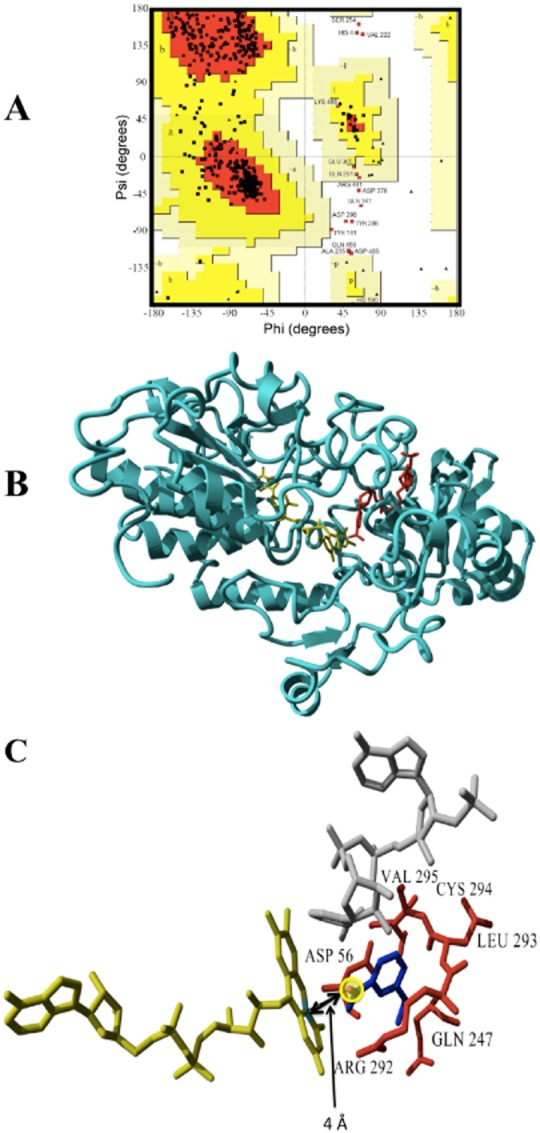
A. Ramachandran plot of the Ar‐BVMO model. The most favoured regions are represented in red, additional allowed regions in dark yellow, generously allowed regions in light yellow.
B. Ribbon representation of the 3D model for the Ar‐BVMO based on the crystal structure of cyclohexanone monooxygenase from Rhodococcus sp. (PDB ID = 3GWF). NADPH and FAD are shown in red and yellow respectively.
C. Docked ethionamide into the active site of Ar‐BVMO model. FAD is shown in yellow, NADPH in grey, the substrate is in dark blue. The S nucleophilic attack site of the ethionamide is highlighted with a yellow circle and the distance (4 Å) from the C4 of the FAD isoalloxazine ring is indicated by a double arrow. Residues interacting with the substrates are labelled and shown in red.
Since phylogenetic analysis placed Ar‐BVMO in the same clade of M. tuberculosis EtaA we investigated if ethionamide can also bind the active site of this enzyme using AutoDock 4.0 within yasara package (Krieger and Vriend, 2002).
Docking of ethionamide resulted in a complex with binding energy of 5.54 kcal mol−1. As can be seen from Fig. 7C, Arg292, Gln247, Leu293, Cys294, Val295, Asp56 residues are responsible for the stabilization of the substrate inside the Ar‐BVMO active site. The S‐oxidation site of ethionamide is located at a distance of 4 Å from the C4 of the FAD isoalloxazine ring, similar to reported distances for another Class B flavoprotein monooxygenase (Catucci et al., 2012). Ethionamide seems to interact correctly with Ar‐BVMO active site by not only resulting in a complex that is energetically favoured, but also by interacting with the enzyme in the expected binding mode for catalysis.
Confirmation of the Ar‐BVMO activity on 4‐phenyl‐2‐butanone and ethionamide
Following purification of Ar‐BVMO heterologously expressed in Escherichia coli BL21 (DE3) as described in Experimental procedures, its activity was tested with 4‐phenyl‐2‐butanone and the prodrug ethionamide as these compounds are known substrates of Eta A. When incubated with Ar‐BVMO both compounds led to the formation of a product as shown in Fig. 8. Analysis with commercially available standards confirmed that the peak with the retention time of 8.8 min (Fig. 8B) corresponded to the phenethyl acetate, which is the Baeyer‐Villiger product of 4‐phenyl‐2‐butanone oxidation. In the case of ethionamide, spectroscopic analysis of the peak with the retention time of 11.0 min confirmed the presence of ethionamide S‐oxide product (Vannelli et al., 2002). In both cases control experiments preformed in the absence of the enzyme (Fig. 8A and C) resulted in no product being formed.
Figure 8.

GC chromatograms of an incubation of 4‐phenyl‐2‐butanone in the absence (A) and presence (B) of Ar‐BVMO. The reaction was carried out for 30 min at 37°C with 500 μM substrate, 200 μM NADPH and 0.1 μM enzyme. HPLC chromatograms of an incubation of ethionamide in the absence (C) and presence (D) of Ar‐BVMO. The reaction was carried out for 20 min at 37°C with 500 μM substrate, 500 μM NADPH and 0.7 μM enzyme.
Discussion
Acinetobacter radioresistens strain S13 can utilize medium‐ and long‐chain alkanes as the sole carbon source. The bacterium possess genes coding for terminal alkane hydroxylase (alkB) (van Beilen et al., 1992) and Baeyer‐Villiger monooxygenase (almA) involved in the initial and final steps of terminal and subterminal oxidation respectively (Throne‐Holst et al., 2007). The two genes are differentially expressed according to the presence of medium‐ or long‐chain alkanes.
Expression of the genes involved in the initial oxidation of alkanes is tightly controlled. A specific regulator assures that the pathway genes are expressed only in the presence of the appropriate alkane. The known specific regulators that induce alkane‐degradation genes in response to alkanes belong to different families, such as the LuxR/MalT, the AraC/XylS, the GntR or other non‐related families of regulators (Rojo, 2009). For some of them there is evidence supporting that alkanes or alkanols act as effectors. Some bacterial strains contain only one alkane hydroxylase, as is the case for the well‐characterized alkane degrader P. putida GPo1. However, many other strains have several alkane‐degradation systems, each one being active on alkanes of a specific chain‐length or being expressed under specific physiological condition. A gene coding for a protein showing similarity to the transcriptional regulator of the AraC family maps close to S13 alkB gene (D. Minerdi and G. Gilardi, unpubl. data). The expression of S13 alkB and almA genes is shown to be induced by C14, C16 and C24, C36 alkanes, respectively, suggesting that the products of these genes are involved in terminal and subterminal alkane oxidation respectively. It can be hypothesized that the differential expression of alkB gene is regulated by the action of AraC‐like protein that responds to the presence of medium‐chain alkanes. almA gene codes for a BVMO that oxidizes the ketone generated by subterminal oxidation of the alkane molecule. Gene expression is induced by long‐chain alkanes through an unidentified mechanism. Liu and colleagues (2011) demonstrated that also in the marine alkane degrader, Alcanivorax dieselolei, almA mRNA levels respond to long‐chain alkanes.
The enzymes involved in alkane degradation have proven to be useful and versatile biocatalysts, opening the possibility of convert inexpensive compounds into valuable intermediates for the synthesis of pharmaceuticals or other chemicals (Furuhashi, 1992). Because of the great biotechnological interest of BVMO enzymes, we concentrated our attention to the novel BVMO of A. radioresistens S13 (Ar‐BVMO). In silico analysis of the protein showed that it belongs to the type I BVMO (van Berkel et al., 2006). In the phylogenetic tree three major branches subdivided into clusters of various sizes represent the diversity of the available BVMO sequences. The family clustering of BVMO sequences has been correlated to stereo‐preferences of some BVMOs as reported by Mihovilovic (Mihovilovic et al., 2005). Surprisingly, Ar‐BVMO sequence clusters together with ethionamide monooxygenase (EtaA) from M. tuberculosis, in a clade separated from all the other known BVMOs. EtaA is a newly identified BVMO that is responsible for the conversion of ethionamide into an active antitubercular drug in M. tuberculosis (Fraaije et al., 2004).
Often the pharmacokinetic properties of new chemical entities represent a problem in the design of new drugs, as they can affect the translation in the clinical field. A potential strategy to overcome the limitations of chemotherapeutic agents is the use of prodrugs. Prodrugs can be defined as pharmacologically inert chemical derivatives that can be converted in vivo to the active drug molecules, enzymatically or non‐enzymatically, to exert a therapeutic effect. Prodrugs can be designed to target specific enzymes by considering enzyme‐substrate specificity in order to overcome various undesirable drug properties. Recently, advances in gene cloning and controlled gene expression techniques in mammalian cells has allowed for the elucidation of the molecular nature of enzymes and carrier proteins and has made it possible to rationally design ‘targeted‐prodrugs’. In prodrug design, enzymes can be recognized as pre‐systemic metabolic sites or prodrug–drug in vivo reconversion sites.
One of the approaches used in cancer treatment is the gene‐directed enzyme prodrug therapy (GDEPT) (Singhal and Kaiser, 1998; Aghi et al., 2000; Smythe, 2000; Greco and Dachs, 2001). In this approach, genes encoding prodrug‐activating enzymes are targeted to tumour cells followed by prodrug administration. In GDEPT, non‐viral vectors that contain gene‐delivery agents, such as cationic lipids, peptides or naked DNA, are used for gene targeting.
For the appropriate combination of an enzyme and a prodrug, the choice of enzyme is very important because the appropriate prodrugs can be designed for almost any enzyme. However, enzymes that are monomeric, have low molecular weight, and lack a requirement of glycosylation would be preferable for ease of handling and possible protein modification (Anlezark et al., 1992). The best enzyme for GDEPT would appear to be a monomeric enzyme of bacterial or viral origin with wide substrate specificity (Sharma et al., 1992). One example is a bacterial nitroreductase that can convert a relatively non‐toxic monofunctional alkylating agent into a 10 000 times more cytotoxic difunctional alkylating agent (Haisma et al., 1992).
The Ar‐BVMO described in this article has a molecular weight of 56 kDa, it presents the typical NADPH binding site and it is predicted to be monomeric. The activity results confirm the hypothesis that this novel enzyme is not only a Baeyer‐Villiger monooxygenase but also an unusual class I BVMO capable of activating the prodrug ethionamide. All these properties encourage further characterizations of this novel Ar‐BVMO in order to evaluate the feasibility to candidate the protein as a potential enzyme to be studied for possible applications in GDEPT.
Experimental procedures
Culture media, bacterial growth conditions and microbial count
Acinetobacter sp. strain S13 was streaked onto Luria–Bertani (LB, 1.0% Tryptone, 0.5% Yeast Extract, 1.0% Sodium Chloride) agar medium and incubated at 30°C. For long‐term storage, bacterial culture was maintained at −80°C in liquid LB containing 20% glycerol.
Cells were grown in LB medium at 30°C on a rotary shaker at 180 r.p.m. until growth reached the stationary phase (A 600 = 3.8–4.0) and transferred to 500 ml flasks containing 100 ml of mineral salts medium (MSM) (Sakai et al., 1994). Alkanes were added either 0.35% v/v liquid alkanes (Throne‐Holst et al., 2006) (C14 and C16, 99% purity, Sigma‐Aldrich, Italy) or 3 g l−1 of solid alkanes (C24 and C36, 98% purity, Sigma‐Aldrich, Italy) as the sole carbon and energy source. Absorbance at 600 nm was adjusted to 0.1. Flasks were incubated at 30°C in a rotary shaker operating at 220 r.p.m. for 7 days. Samples of 1 ml were removed from each culture at 12 h intervals and were used to measure bacterial growth. For cultures supplemented with liquid alkanes, growth was measured spectrophotometrically (Agilent Technologies model 8453E, Santa Clara, USA) at 600 nm using a cuvette with 1 cm path length. All solid alkanes formed small micelles in the liquid medium, which made it impossible to accurately measure the OD600. Therefore, growth of cultures supplemented with solid alkanes was measured as viable cell count on LB plates at 30°C. Identical medium and MSM plus sodium acetate (5 g l−1) as carbon source in place of alkanes were used as controls. All experiments were carried out in triplicates.
All alkanes of defined chain lengths were purchased from Sigma‐Aldrich (Italy). For simplicity, alkanes of defined chain lengths are referred to by the number of carbon atoms they contain, i.e. tetradecane will be referred to as C14, hexadecane as C16, tetracosane as C24 and hexatriacontane as C36 throughout the article.
DNA preparation and bacterial 16S rRNA gene sequencing
Acinetobacter sp. strain S13 was grown on LB medium at 30°C for 24 h and centrifuged for 10 min at 4000 r.p.m. The pellet was resuspended in 250 μl of sterile water. This suspension was heated at 95°C for 5 min to release the DNA. The heated suspension (2 μl) was then used for amplification of the 16S rRNA gene by PCR using the universal bacterial primer pair 27f/1492r (Weisburg et al., 1991). Amplification was carried out in a 20 μl volume containing 0.5 μM of each primer, 2.0 μl of 10× buffer (200 mM Tris pH 8.4 with 500 mM KCl – Promega, Madison, USA), 2.5 mM MgCl2, 250 μM each dNTP and 1 U of Taq DNA polymerase (Promega). The PCR cycling conditions were as follows: denaturation at 93°C for 3 min; 35 cycles at 94°C for 30 s, 55°C for 30 s and 72°C for 1 min; and final extension at 72°C for 7 min using a Techne TC‐312 thermal cycler.
PCR detection of almA and alkB genes
To amplify the genomic sequences of Acinetobacter sp. strain S13 almA and alkB genes, the following primers were designed on the basis of the almA gene of Acinetobacter sp. strain M‐1 and alkB gene of A. radioresistens strain SH64 sequences available in databases (almA = EF2121875; alkB = ZP_06072466.1):
ALMA‐Forward (5′‐TTCCGCTATCCTGGTATTCG‐3′),
ALMA‐Reverse (5′‐AATCACCATCTGGCACAACA‐3′),
ALKB‐Forward (5′‐GAATTAGAGCGGGGAGAACC‐3′), and
ALKB‐Reverse (5′‐GGCTTCTTCGCAGTTAGGTG‐3′).
PCR reactions were carried out in a final volume of 20 μl containing 0.5 mM of each primer, 2.0 μl of 10× buffer (200 mM Tris pH 8.4 with 500 mM KCl – Qiagen, Hilden, Germany), 2.5 mM MgCl2, 250 mM each dNTP, 2 μl of bacterial lysate preparation and 1 U of Taq DNA polymerase (Qiagen, Hilden, Germany). The PCR cycling conditions were as follows: denaturation at 95°C for 3 min; 30 cycles at 94°C for 45 s, 50°C for 1 min and 72°C for 1 min; and a final extension at 72°C for 7 min using a Techne TC‐312 thermal cycler. In order to obtain the full sequence of both almA and alkB genes a genome walking strategy was adopted using primers specifically designed on the basis of almA and alkB flanking gene sequences respectively.
PCR product purification and sequencing
The PCR products (about 760 bp for almA and 780 bp for alkB) were excised from the gel and purified using the QIAquick PCR purification kit (Qiagen, Hilden, Germany). The purified products were then directly sequenced without cloning steps by using the PCR primers described above (Eurofins MWG, Ebersberg, Germany).
Nucleotide sequences accession numbers
The 16S rRNA gene sequence obtained from Acinetobacter sp. strain S13 as well as the sequences of the almA and alkB genes were submitted to the GenBank database and assigned the following accession numbers: GU145275, GU145276 and HQ685899 respectively.
Phylogenetic analysis
almA and alkB sequences were compared with reference sequences in the GenBank databases using the NCBI blastn search tool (Altschul et al., 1997). Representative full‐length sequences of almA and alkB were selected from the GenBank database as reference sequences. Phylogenetic analysis was performed based on the amino acid sequences of AlmA and AlkB using MEGA version 4.0 (Tamura et al., 2007), after multiple sequence alignment and truncation to the same length. Distances according to the Kimura two‐parameter model (Kimura, 1980) and clustering with the neighbour‐joining method (Saitou and Nei, 1987) were determined using bootstrap values based on 1000 replications.
Semi‐quantitative reverse transcription RT‐PCR analyses
Total RNA for RT‐PCR from Acinetobacter sp. strain S13 grown in the presence of C14, C16, C24 and C36 alkanes and sodium acetate added to the minimal growth medium was isolated using RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. cDNA was synthesized in a two‐step process using Superscript II (Invitrogen). The primers used in RT‐PCR reactions consisted of the following pairs specifically designed on the basis of the sequence of Acinetobacter sp. strain S13 almA and alkB genes respectively:
| S13ALM‐Forward | 5′‐TTTCAAACCATGGCGTA‐3′, |
| S13ALM‐Reverse | 5′‐CGAAAGAAAACCACGCATTT‐3′, |
| S13ALKB‐Forward | 5′‐TCAAAAAGCAGTTTCACTGGA‐3′, |
| and | |
| S13ALKB‐Reverse | 5′‐CCCAACTTCAAACAACGTGA‐3′ |
Single‐strand cDNAs were obtained with the specific antisense primers S13ALM‐Reverse and S13ALKB‐Reverse using total RNA as the template. RNA samples were denatured at 65°C for 5 min and then reverse transcribed at 50°C for 1 h in a final volume of 20 μl containing 2 μl of total RNA, 1 μM each of specific primers, 0.5 mM dNTPs, 10 U of RNase inhibitor, 4 μl of buffer (Invitrogen), 2 μl of 0.1 M dithiothreitol and 1 μl of Superscript II (Invitrogen). The absence of undigested genomic DNA was assessed by a control PCR reaction carried out with Platinum Taq DNA polymerase (Invitrogen) and the Acinetobacter sp. strain S13 16S rRNA gene‐specific primers S1316SFOR (5′‐TTCCGAAAGGAGCGCTAATA‐3′) and S1316SREV (5′‐AGCCAACCAGTATCGAATGC‐3′) using the five total RNAs as templates and the PCR conditions described above. RT‐PCR experiments were conducted using three replicates on three independent samples. The amplified products were analysed by 1.4% agarose gel electrophoresis in a TAE running buffer (Sambrook et al., 1989).
Homology modelling of Ar‐BVMO
A homology model of Ar‐BVMO was generated using yasara (Krieger and Vriend, 2002) program. This software features a complete homology modelling module that automatically takes all the steps from an amino acid sequence to a refined high‐resolution model. The target sequence undergoes a psi‐blast against Uniprot and the results are used to search the Protein Data Bank (PDB) for potential modelling templates. The templates are ranked based on the alignment score and the structural quality so that models built using high‐resolution X‐ray templates are preferred to lower resolution X‐ray or NMR templates, even if the latter share a higher percentage of sequence identity. Models are built for the top scoring templates and are energy minimized and refined. The final model was validated by using procheck software (Laskowski et al., 1993).
Protein–ligand interaction
In order to investigate the active site of Ar‐BVMO and its substrate specificity, protein–ligand interaction analysis was performed. AutoDock 4.0 (Goodsell et al., 1996) embedded into the yasara Structure package, was used to dock ethionamide to the refined model. Ethionamide was obtained from the DrugBank database (Knox et al., 2011). The optimized ligand molecule was docked into the refined protein model by running 25 of Global Docking centring a 15 × 15 × 15 Å simulation cell around the FAD group. In yasara, docking runs of the ligand to receptor yield results sorted by binding energy where more positive energies indicate stronger binding and negative energies equate to no binding. After global docking the best binding mode (pose) was selected based on the best binding energy. The complexes were then subjected to 999 runs of Local Docking yielding the final docked binding modes. The global docking experiment, in which the drug is originally outside the simulation box and is placed inside the cell by exploiting the AutoDock algorithm, resulted in a series of binding modes classified by the binding energy outputs. Among these binding modes the complex protein–ligand bearing the highest binding energy calculated by yasara (Krieger and Vriend, 2002) as the mechanical energy required for disassembling a whole into separate parts, was selected and refined by local docking. In local docking experiments the ligand is within the simulation cell and the possible conformations are assayed for a maximum of 999 runs and the results are again sorted by binding energy.
Cloning, expression and purification of Ar‐BVMO
The Ar‐BVMO gene was cloned in the pT7 expression vector using EcoRI and BamHI restriction enzymes. A tag of six histidines was also introduced at the C‐terminus of the gene for ease of purification. The Ar‐BVMO enzyme was expressed in E. coli BL21 (DE3) cells transformed with pT7‐Ar‐BVMO in a 5 l fermentor (Biostat A plus, Sartorius AG, Goettingen, Germany) grown at 28°C in LB medium supplemented with ampicillin (100 μg ml−1) and riboflavin (20 mg l−1). The cells were harvested 24 h post induction by centrifugation at 4000 r.p.m. at 4°C. Subsequently, the bacterial pellet was disrupted by sonication and the clarified lysate was loaded on a nickel chelating sepharose Fast‐Flow column (GE Healthcare Europe, Italy). The protein was eluted with 40 mM histidine, exchanged with 100 mM phosphate buffer pH 7.4 and stored at −20°C until use.
Ar‐BVMO enzymatic activity: HPLC and GC analysis
The HPLC analysis was performed with an Agilent 1200 quaternary pump HPLC System (Agilent Technologies, Italy) using a C18 reverse phase column (LiChroCART 250‐4 column packed with Lichrosphere RP C18 5 μM). The ethionamide and ethionamide‐SO separation method consisted of a mobile phase of 85% v/v 25 mM KH2PO4 and 15% v/v acetonitrile with a flow rate of 1 ml min−1. The DAD (Diode Array Detector) was set to 350 nm. The retention times for ethionamide and ethionamide‐SO were 16.5 min and 11.0 min respectively.
The GC analysis was performed with a 7890A GC instrument (Agilent Technologies, Italy) with a DB‐Wax column (Length 30 m; Diameter 0.320 mm; PEG film 0.25 μM). The method to separate the 4‐phenyl‐2‐butanone and phenethyl acetate consisted of the following steps: oven start at 50°C; increase temperature to 80°C at 10°C min−1; to 230°C at 25°C min−1; to 250°C at 20°C min−1; final hold 5 min at 250°C. The carrier gas used was He (3 ml min−1). The Flame Ionization Detector was set at 275°C. The retention times for 4‐phenyl‐2‐butanone and phenethyl acetate were 9.2 min and 8.8 min respectively. The reactions were carried out for 30 min at 37°C with 500 μM substrate, 200 μM NADPH with an enzyme concentration of 0.1 μM.
References
- Aghi M., Hochberg F., Breakefield X.O. Prodrug activation enzymes in cancer gene therapy. J Gene Med. 2000;2:148–164. doi: 10.1002/(SICI)1521-2254(200005/06)2:3<148::AID-JGM105>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. Gapped blast and psi‐blast: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anlezark G.M., Melton R.G., Sherwood R.F., Coles B., Friedlos F., Knox R.J. The bioactivation of 5‐(aziridin‐1‐yl)‐2,4‐dinitrobenzamide (CB 1954) – I. Purification and properties of a nitroreductase enzyme from Escherichia coli – a potential enzyme for antibody‐directed enzyme prodrug therapy (ADEPT) Biochem Pharmacol. 1992;44:2289–2295. doi: 10.1016/0006-2952(92)90671-5. [DOI] [PubMed] [Google Scholar]
- Beam M.P., Bosserman M.A., Noinaj N., Wehenkel M., Rohr J. Crystal structure of Baeyer‐Villiger monooxygenase MtmOIV, the key enzyme of the mithramycin biosynthetic pathway. Biochemistry. 2009;48:4476–4487. doi: 10.1021/bi8023509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Beilen J.B., Penninga D., Witholt B. Topology of the membrane‐bound alkane hydroxylase of Pseudomonas oleovorans. J Biol Chem. 1992;267:9194–9201. [PubMed] [Google Scholar]
- van Beilen J.B., Li Z., Duetz W.A., Smits T.H.M., Witholt B. Diversity of alkane hydroxylase systems in the environment. Oil Gas Sci Technol. 2003;58:427–440. [Google Scholar]
- van Berkel W.J.H., Kamerbeek N.M., Fraaije M.W. Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J Biotechnol. 2006;124:670–689. doi: 10.1016/j.jbiotec.2006.03.044. [DOI] [PubMed] [Google Scholar]
- Berthe‐Corti L., Fetzner S. Bacterial metabolism of alkanes and ammonia under oxygen depleted conditions. Acta Biotechnol. 2002;22:299–336. [Google Scholar]
- Catucci G., Gilardi G., Jeuken L., Sadeghi S.J. In vitro drug metabolism by C‐terminally truncated human flavin‐containing monooxygenase 3. Biochem Pharmacol. 2012;83:551–558. doi: 10.1016/j.bcp.2011.11.029. [DOI] [PubMed] [Google Scholar]
- Eppink M.H.M., van Jacobs D., Berkel W.J.H. Involvement of His162 in NADPH binding of p‐hydroxybenzoate hydroxylase. In: Mas‐sey K., Williams V., editors. Calgary: University Press; 1997. pp. 315–318. [Google Scholar]
- Feng L., Wang W., Cheng J., Ren Y., Zhao G., Gao C. Genome and proteome of long‐chain alkane degrading Geobacillus thermodenitrificans NG80‐2 isolated from a deep‐subsurface oil reservoir. Proc Natl Acad Sci USA. 2007;104:5602–5607. doi: 10.1073/pnas.0609650104. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraaije M.W., van Kamerbeek N.M., Berkel W.J.H., Janssen D.B. Identification of a Baeyer–Villiger monooxygenase sequence motif. FEBS Lett. 2002;518:43–47. doi: 10.1016/s0014-5793(02)02623-6. [DOI] [PubMed] [Google Scholar]
- Fraaije M.W., Kamerbeek N.M., Heidekamp A.J., Fortin R., Janssen D.B. The prodrug activator EtaA from Mycobacterium tuberculosis is a Baeyer‐Villiger monooxygenase. J Biol Chem. 2004;279:3354–3360. doi: 10.1074/jbc.M307770200. [DOI] [PubMed] [Google Scholar]
- Furuhashi K. Biological routes to optically active epoxides. In: Collins A.N., Sheldrake G.N., Crosby J., editors. London, UK: John Wiley & Sons; 1992. pp. 167–186. [Google Scholar]
- Goodsell D.S., Morris G.M., Olson A.J. Automated docking of flexible ligands: applications of autodock. J Mol Recognit. 1996;9:1–5. doi: 10.1002/(sici)1099-1352(199601)9:1<1::aid-jmr241>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Greco O., Dachs G.U. Gene directed enzyme/prodrug therapy of cancer: historical appraisal and future prospectives. J Cell Physiol. 2001;187:22–36. doi: 10.1002/1097-4652(2001)9999:9999<::AID-JCP1060>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Haisma H.J., van Boven E., de Muijen M., van der Jong J., Vijgh W.J., Pinedo H.M. A monoclonal antibody‐beta‐glucuronidase conjugate as activator of the prodrug epirubicin‐glucuronide for specific treatment of cancer. Br J Cancer. 1992;66:474–478. doi: 10.1038/bjc.1992.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- Knox C., Law V., Jewison T., Liu P., Ly S., Frolkis A. DrugBank 3.0: a comprehensive resource for ‘omics’ research on drugs. Nucleic Acids Res. 2011;39:D1035–D1041. doi: 10.1093/nar/gkq1126. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koma D., Hasumi F., Yamamoto E., Ohta T., Chung S.‐Y., Kubo M. Biodegradation of long‐chain n‐paraffins from waste oil of car engine by Acinetobacter sp. J Biosci Bioeng. 2001;91:94–96. doi: 10.1263/jbb.91.94. [DOI] [PubMed] [Google Scholar]
- Kotani T., Yurimoto H., Kato N., Sakai Y. Novel acetone metabolism in a propane‐utilizing bacterium, Gordonia sp. strain TY‐5. J Bacteriol. 2007;189:886–889. doi: 10.1128/JB.01054-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger E., Vriend A. Model@Home: distributed computing in bioinformatics using a screensaver based approach. Bioinformatics. 2002;18:315–318. doi: 10.1093/bioinformatics/18.2.315. [DOI] [PubMed] [Google Scholar]
- Labinger J.A., Bercaw J.E. Understanding and exploiting C–H bond activation. Nature. 2002;417:507–514. doi: 10.1038/417507a. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. procheck: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- Li L., Liu X., Yang W., Xu F., Wang W., Feng L. Crystal structure of long‐chain alakane monooxygenase (LadA) in complex with coenzyme FMN: unveiling the long‐chain alane hydroxylase. J Mol Biol. 2008;376:453–465. doi: 10.1016/j.jmb.2007.11.069. et al. [DOI] [PubMed] [Google Scholar]
- Liu C., Wang W., Wu Y., Zhou Z., Lai Q. Multiple alkane hydroxylase systems in a marine alkane degrader, Alcanivorax dieselolei B‐5. Environ Microbiol. 2011;13:1168–1178. doi: 10.1111/j.1462-2920.2010.02416.x. [DOI] [PubMed] [Google Scholar]
- Maeng J.H., Sakai Y., Tani Y., Kato N. Isolation and characterization of a novel oxygenase that catalyzes the first step of n‐alkane oxidation in Acinetobacter sp. strain M‐1. J Bacteriol. 1996;178:3695–3700. doi: 10.1128/jb.178.13.3695-3700.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier T., Förster H.H., Asperger O., Hahn U. Molecular characterization of the 56‐kDA CYP153 from Acinetobacter sp. EB104. Biochem Biophys Res Commun. 2001;286:652–658. doi: 10.1006/bbrc.2001.5449. [DOI] [PubMed] [Google Scholar]
- Mihovilovic M.D., Rudroff F., Grtzl B., Kapitan P., Snajdrova R., Rydz J., Mach R. Family clustering of Baeyer‐Villiger monooxygenases based on protein sequence and stereopreference. Angew Chem Int Ed Engl. 2005;44:3609–3613. doi: 10.1002/anie.200462964. [DOI] [PubMed] [Google Scholar]
- Mirza I.A., Yachnin B.J., Wang S., Grosse S., Bergeron H., Imura A. Crystal structures of cyclohexanone monooxygenase reveal complex domain movements and a sliding cofactor. J Am Chem Soc. 2009;131:8848–8854. doi: 10.1021/ja9010578. et al. [DOI] [PubMed] [Google Scholar]
- Muller R., Asperger O., Kleber H.P. Purification of cytochrome P‐450 from n‐hexadecane‐grown Acinetobacter calcoaceticus. Biomed Biochim Acta. 1989;48:243–254. [PubMed] [Google Scholar]
- Pessione E., Giunta C. Acinetobacter radioresistens metabolizing aromatic compounds. Biochemical and microbiological characterization of the strain. Microbios. 1997;89:105–117. [PubMed] [Google Scholar]
- Rehdorf J., Kirschner A., Bornscheuer U.T. Cloning, expression and characterization of a Baeyer‐Villiger monooxygenase from Pseudomonas putida KT2440. Biotechnol Lett. 2007;29:1393–1398. doi: 10.1007/s10529-007-9401-y. [DOI] [PubMed] [Google Scholar]
- Rojo F. Degradation of alkanes by bacteria. Environ Microbiol. 2009;11:2477–2490. doi: 10.1111/j.1462-2920.2009.01948.x. [DOI] [PubMed] [Google Scholar]
- Saitou N., Nei M. The neighbor‐joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Sakai Y., Maeng J.H., Tani Y., Kato N. Use of long‐chain n‐alkanes (C13–C44) by an isolate, Acinetobacter sp. M‐1. Biosci Biotechnol Biochem. 1994;58:2128–2130. [Google Scholar]
- Sambrook J., Fritsch E.F., Maniatis T. Cold Spring Harbor, NY, USA: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Shanklin J., Whittle E., Fox B.G. Eight histidine residues are catalytically essential in a membrane associated iron enzyme, stearoyl‐CoA desaturase and are conserved in alkane hydroxylase and xylene monooxygenase. Biochemistry. 1994;33:12787–12794. doi: 10.1021/bi00209a009. [DOI] [PubMed] [Google Scholar]
- Sharma S.K., Bagshawe K.D., Melton R.G., Sherwood R.F. Human immune response to monoclonal antibody‐enzyme conjugates in ADEPT pilot clinical trial. Cell Biophys. 1992;21:109–120. doi: 10.1007/BF02789482. [DOI] [PubMed] [Google Scholar]
- Singhal S., Kaiser L.R. Cancer chemotherapy using suicide genes. Surg Oncol Clin N Am. 1998;7:505–536. [PubMed] [Google Scholar]
- Smits T.H.M., Röthlisberger M., van Witholt B., Beilen J.B. Molecular screening for alkane hydroxylase genes in Gram‐negative and Gram‐positive strains. Environ Microbiol. 1999;1:307–318. doi: 10.1046/j.1462-2920.1999.00037.x. [DOI] [PubMed] [Google Scholar]
- Smythe W.R. Prodrug/drug sensitivity gene therapy: current status. Curr Oncol Rep. 2000;2:17–22. doi: 10.1007/s11912-000-0006-z. [DOI] [PubMed] [Google Scholar]
- Sotsky J.B., Greer C.W., Atlas R.M. Frequency of genes in aromatic and aliphatic hydrocarbon biodegradation pathways within bacterial populations from Alaskan sediments. Can J Microbiol. 1994;40:981–985. doi: 10.1139/m94-157. [DOI] [PubMed] [Google Scholar]
- Tamura K., Dudley J., Nei M., Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Throne‐Holst M., Markussen S., Winnberg A., Ellingsen T.E., Kotlar H.K., Zotchev S.B. Utilization of n‐alkanes by a newly isolated strain of Acinetobacter venetianus: the role of two AlkB‐type alkane hydroxylases. Appl Microbiol Biotechnol. 2006;72:353–360. doi: 10.1007/s00253-005-0262-9. [DOI] [PubMed] [Google Scholar]
- Throne‐Holst M., Wentzel A., Ellingsen T.E., Kotlar H.K., Zotchev S.B. Identification of novel genes involved in long‐chain n‐alkane degradation by Acinetobacter sp. strain DSM 17874. Appl Environ Microbiol. 2007;73:3327–3332. doi: 10.1128/AEM.00064-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hamme J.D., Singh A., Ward O.P. Recent advances in petroleum microbiology and molecular biology. Microbiology. 2003;67:503–549. doi: 10.1128/MMBR.67.4.503-549.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannelli T., Dykman A., Ortiz de Montellano P. The antituberculosis drug ethionamide is activated by a flavoprotein monooxygenase. J Biol Chem. 2002;277:12824–12829. doi: 10.1074/jbc.M110751200. [DOI] [PubMed] [Google Scholar]
- Weisburg W.G.S., Barns M., Pelletier D.A., Lane D.J. 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol. 1991;173:697–703. doi: 10.1128/jb.173.2.697-703.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wentzel A., Ellingsen T.E., Kotlar H.K., Zotchev S.B., Throne‐Holst M. Bacterial metabolism of long‐chain n‐alkanes. Appl Microbiol Biotechnol. 2007;76:1209–1221. doi: 10.1007/s00253-007-1119-1. [DOI] [PubMed] [Google Scholar]