

Summary
The inducible Pm promoter together with its cognate positive transcription regulator XylS has been shown to be useful for recombinant protein production under high cell density conditions. Here we report directed evolution of XylS resulting in mutant proteins with increased ability to stimulate transcription in Escherichia coli from Pm. A first round of mutagenesis using error‐prone PCR on xylS was used to construct a library consisting of about 430 000 clones, and this library could be efficiently screened with respect to stimulation of expression from Pm due to a positive correlation between the level of expression of the reporter gene, bla (encoding β‐lactamase), and the ampicillin tolerance of the corresponding host cells. Fourteen different amino acid substitutions in XylS were found to separately lead to up to nearly a threefold stimulation of expression under induced conditions, relative to wild type. These mutations were all located in the part corresponding to the N‐terminal half of the protein. Varying combinations of the mutations resulted in further stimulation, and the best results (about 10‐fold stimulation under induced conditions) were obtained by using a random shuffling procedure followed by a new round of screening. The uninduced levels of expression for the same mutants also increased, but only about four times. Through in silico 3D modelling of the N‐terminal domain of XylS, it was observed that the evolved mutant proteins contained substitutions that were positioned in different parts of the predicted structure, including a β‐barrel putatively responsible for effector binding and a coiled coil probably important for dimerization. The total production of the host‐toxic antibody fragment scFv‐phOx expressed from Pm with the evolved XylS mutant protein StEP‐13 was about ninefold higher than with wild‐type XylS, demonstrating that directed evolution of transcription factors can be an important new tool to achieve high‐level recombinant protein production.
Introduction
Bacterial systems for heterologous gene expression are the preferred choice for high‐level production of many prokaryotic and eukaryotic proteins (Schmidt, 2004; Terpe, 2006). Suitable bacterial promoters are strong and have a low basal expression level (i.e. they are tightly regulated). This ensures high production of the heterologous protein and reduced strain on the host‐cell from expression of toxic proteins prior to induction (Hannig and Makrides, 1998; Keasling, 1999). Regulation of promoter activity is usually achieved by modulating environmental signals, which are coupled to gene expression via transcription factors that stimulate or repress transcription from the promoter. The environmental signals include pH or temperature shifts, and small ligands (inducers) added to the culture medium (Browning and Busby, 2004).
To engineer bacterial expression systems for higher expression level and lower leakage, the most common target has been the promoter sequence, e.g. the classical construction of the Escherichia coli hybrid promoter tac from the lac and trp promoters (de Boer et al., 1983), and the engineering of novel strong or tight Pm promoter mutants (Winther‐Larsen et al., 2000a; Bakke et al., 2009). Engineering transcription factors for the purpose of improved heterologous expression represents a new approach. Directed evolution, a powerful and popular technology platform for protein and nucleic acid engineering (Yuan et al., 2005), is well suited for obtaining improved transcription factors quickly by screening of reporter proteins under control of the transcription factor's cognate promoter. Collins and colleagues demonstrated the method's applicability by obtaining novel LuxR mutant proteins with a 100‐fold increased sensitivity to novel effectors. These mutant proteins maintained a wild‐type or increased response to its natural effector, although the strongest response represented only a 17% increase in expression at optimal inducer concentration, and could have been caused by increased LuxR concentration (Collins et al., 2005; 2006). To our knowledge, the use of laboratory evolved transcription factors to obtain improved heterologous expression has not previously been reported.
The AraC/XylS family of transcription factors is a large protein family consisting of more than 270 transcriptional activators and repressors (Tobes and Ramos, 2002; Brautaset et al., 2009). Family members are identified by their sequence similarity and not by their function and they regulate cellular responses as diverse as carbon metabolism, stress response and virulence (Gallegos et al., 1997). Some members have been shown experimentally to be suitable for regulation of recombinant expression, e.g. RhaS and RhaR with the rhaPBAD promoter (Haldimann et al., 1998), AraC with the PBAD promoter (Guzman et al., 1995) and XylS with the Pm promoter (Winther‐Larsen et al., 2000a;b). XylS is a transcription activator that upon induction by benzoic acid and its derivates stimulates transcription from Pm. The activator is separated into an N‐terminal domain (NTD) and C‐terminal domain (CTD). Evidence suggests that the NTD binds inducers, make contacts with the RNA polymerase α subunit and facilitates dimerization, while the CTD is the most important part for establishing contacts with the RNA polymerase α subunit. It also contains helix–turn–helix motifs required for DNA binding (Michán et al., 1992; Kessler et al., 1993; Kaldalu et al., 2000; Manzanera et al., 2000; Ruíz and Ramos, 2001; Ruíz et al., 2003; Domínguez‐Cuevas et al., 2008).
The Pm promoter with XylS forms an expression system that through the use of its ubiquitous benzoic acid inducers is suitable for expression of both prokaryotic and eukaryotic genes. Its tight regulation makes it useful for metabolic engineering and its strength allows high‐level expression of recombinant proteins, as demonstrated by industrial‐level production of the eukaryotic, therapeutic proteins hGM‐CSF and IFN‐2αb and the antibody fragment scFv‐phOx (Sletta et al., 2004; 2007). The capacity of Pm/xylS to express recombinant proteins at very high levels is shared with other well‐established systems, but the Pm/xylS system has some at least partly unique features. It can be used across species barriers, inducers do not require an uptake system and are not metabolized by most bacteria, the inducers act in a continuous concentration‐dependent manner and a large number of mutants affecting expression are available. The mutations are located either in the promoter or in the DNA region corresponding to the 5′ untranslated part of mRNA (Blatny et al., 1997; Winther‐Larsen et al., 2000a;b, Bakke et al., 2009; Berg et al., 2009). The combination of all these features in a single system makes Pm/xylS very useful for most cases in which accurate control of the expression level is important. To further expand the flexibility of this system we here report the use of directed evolution on xylS to investigate whether it is possible to obtain new XylS activators with higher induced expression levels, and retained low basal expression levels from Pm.
Results
Selection system for XylS mutant proteins with increased ability to stimulate transcription from the Pm promoter in the presence of effector
The pTA13 vector (see Supporting information) was designed for efficient selection of XylS mutant proteins. It features the bla gene for ampicillin tolerance under transcriptional control of the Pm promoter. Changes in XylS function will affect transcription from Pm and subsequently alter the host cell's ampicillin tolerance in an approximately proportional relationship (Uhlin and Nordström, 1977). Under inducing conditions (1 mM m‐toluic acid) cells with pTA13 are capable of growing at ampicillin concentrations up to about 580 µg ml−1 on solid media, in the absence of inducers they grow up to about 35 µg ml−1 of the antibiotic. Selection of cells containing XylS mutant proteins with increased ability to stimulate transcription from Pm in the presence of effector can thus be done directly by growth with ampicillin concentrations exceeding 580 µg ml−1.
Error prone PCR on xylS and selection of XylS mutant proteins with increased ability to stimulate transcription from the Pm promoter in the presence of effector
The coding region of the xylS gene was mutagenized with error‐prone PCR and the resulting variants were ligated into pTA13 and transformed into E. coli DH5α, generating a mutant library consisting of approximately 430 000 transformants. The variants originated from a pooled mixture of DNA molecules generated by five different error‐prone PCR reactions, which resulted in varying mutation frequencies (see Experimental procedures). Nine randomly selected clones from the library were also investigated by determining their xylS DNA sequences, and were found to contain an average of 1.56 point mutations per gene (corresponding to about 1.26 amino acid substitutions per gene product). The mutations appeared to be randomly distributed in xylS (data not shown).
The survival frequencies of the clones in the mutant library upon growth on solid media supplemented with effector and different ampicillin concentrations were calculated. More than 60% of the mutants had lower ampicillin tolerance than cells with parental xylS, at ampicillin concentrations two and three times the tolerance level for cells containing parental xylS; the survival frequencies were approximately 0.93% and 0.03% respectively (Fig. 1).
Figure 1.
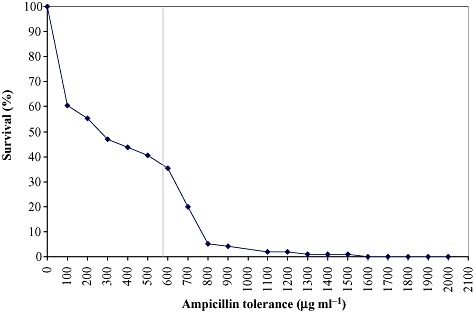
Survival frequencies of cells in the xylS mutant library. Survival rates above 10% were determined by transferring ninety‐six randomly selected colonies from the library into microtiter plate wells with LB, followed by plating (using a pin replicator) on LB agar medium with inducer and varying concentrations of ampicillin. For lower survival rates the frequencies were determined by plating on the corresponding agar media from liquid dilutions of the pooled library. The tolerance level of cells harbouring wild‐type xylS is indicated with the vertical line (580 µg ml−1). As this population is genetically homogeneous a corresponding survival rate curve would be flat up to 580 µg ml−1, and then drops rapidly with small increments in the antibiotic concentration (Bakke et al., 2009).
The mutant library was screened for increased ampicillin tolerance under inducing conditions, and 40 different xylS variants were identified. Among them 14 different xylS point mutations assumed to be at least partly responsible for the observed phenotypes were identified. These mutations were identified because of their repeated occurrence or because they were the only mutations present. The corresponding amino acid sequence changes were all positioned in the NTD of the XylS protein. To verify the assumption that these substitutions increase the ability of XylS to stimulate transcription from Pm, site‐specific mutagenesis was used to construct 14 different xylS variants containing each of the identified point mutations. All the new xylS variants increased their host cells' tolerance to ampicillin compared with cells with wild‐type xylS (Fig. 2).
Figure 2.
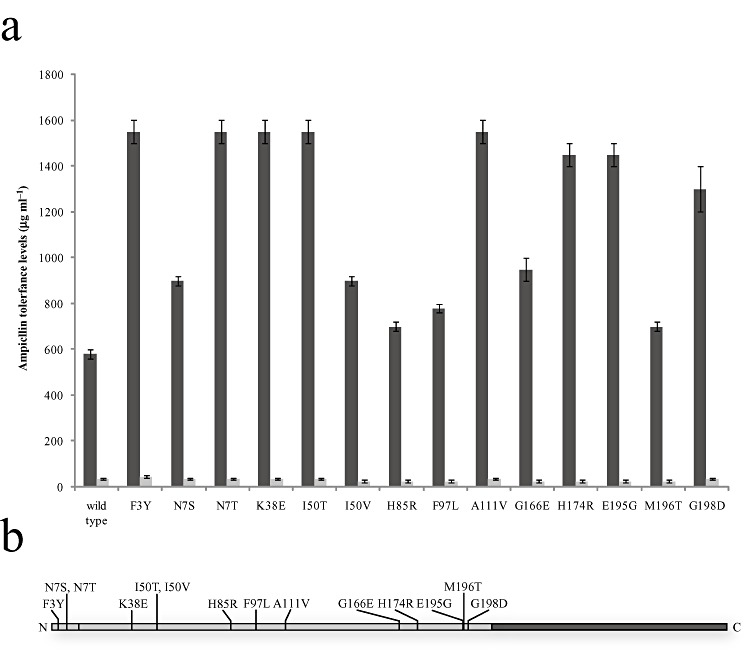
Amino acid substitutions in XylS that lead to increased transcription from Pm under induced (1 mM m‐toluic acid) conditions. A. Ampicillin tolerance levels of recombinant cells containing different xylS variants. The tolerance levels are shown as light grey bars under induced conditions and as dark grey bars under uninduced conditions. The lower error bars depict the highest ampicillin concentrations that allowed growth, while the upper error bars depict the lowest ampicillin concentrations that did not allow growth. For the uninduced conditions the difference between each step was 5 µg ml−1 of ampicillin. B. A schematic representation of the XylS primary sequence showing the NTD in light grey and the CTD in dark grey. The locations of the 14 substitutions are indicated.
Mutants expressing XylS proteins with the solitary substitutions F3Y, N7T, K38E, I50T and A111V showed the strongest increase, about 2.8 times higher than for cells with wild‐type XylS. The increase in basal tolerance was lower than the increase in induced tolerance, at the maximum 1.29‐fold for mutant F3Y. The induction ratio (induced/basal tolerance) was increased from 17 with wild‐type XylS to more than 44 with mutant N7T. The increase in expression in the absence of inducer is generally not desired, but in this case the problem is limited as the wild‐type background expression is very low and probably do not represent a problem unless the protein to be expressed is toxic to the host.
Quantitative real‐time PCR verified that the increased ampicillin tolerances were not a consequence of altered xylS transcription, and also that they correlated well with the increases in bla mRNA transcript (data not shown). It was not technically feasible to directly measure the amounts of XylS proteins produced, as, for example, available antibodies are not sensitive enough to detect the very low levels of production.
Combinations of mutations cause apparently unpredictable induction properties of the resulting mutant XylS proteins
To test whether different substitutions in XylS can be combined to yield activators that further increase transcription from Pm, new xylS genes encoding activators containing two or three substitutions were constructed by site‐specific mutagenesis. This included combinations of substitutions identified after error‐prone PCR and the R45T substitution, which has previously been shown to increase XylS activity (Ramos et al., 1990). Eighteen different combinations were tested, and all of them gave their hosts higher induced ampicillin tolerance than mutants with single mutations in xylS (Fig. 3), verifying that the XylS substitutions can act in an additive manner. The XylS protein Syn‐15 (containing the substitutions F3Y, I50T and F97L) gave about sixfold higher induced ampicillin tolerance than wild type, and only 1.5‐fold higher basal ampicillin tolerance, thus increasing the induction ratio from 17 for wild‐type XylS to at least 56 with Syn‐15.
Figure 3.
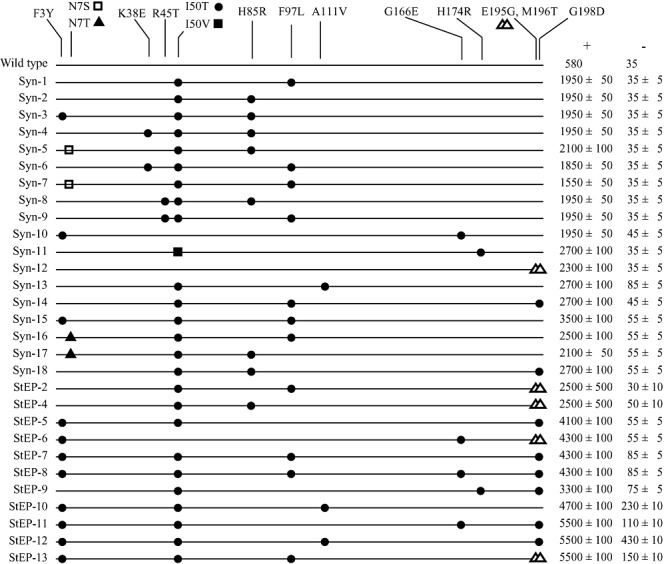
Map of the N‐terminal 200 residues of XylS mutant proteins with each substitution indicated. The figure includes mutant proteins rationally combined (designated Syn) and mutant proteins isolated after DNA shuffling (designated StEP). ‘+’ denotes the induced ampicillin tolerance of the host cell and ‘−’ denotes the basal ampicillin tolerance. In addition to those listed, Syn‐11 contained the Y96C substitution that was unintentionally introduced.
In some cases combinations of two particular substitutions resulted in improved activators, relative to activators with combinations of three substitutions (e.g. Syn‐13 compared with Syn‐9). Furthermore, some combinations resulted in better activators than others with the same number of substitutions (e.g. Syn‐15 compared with Syn‐3). These observations suggested that the optimal combinations of XylS substitutions might more easily be identified from screening of a randomly shuffled xylS mutant library.
DNA shuffling of mutant xylS genes and selection of XylS proteins with further improved activation properties
To circumvent the unpredictable phenotypes observed by combining the selected xylS mutations (see above), a StEP DNA shuffling strategy for random recombination of mutant xylS genes was employed (Zhao, 2004). A set consisting of 28 different xylS variants originating from both the error‐prone PCR study as well as from the directed combination of single mutations was used as a template for the StEP reaction. The recombined pool of xylS genes were then cloned into the screening vector pTA13 and subsequently transformed into E. coli DH5α, generating a library consisting of approximately 5000 mutants. The StEP library was screened for mutants with at least six times higher ampicillin tolerance than cells with wild‐type xylS. After further phenotypic and genotypic characterization and sequencing, 13 unique shuffled xylS genes were identified. These genes did not contain any novel point mutations originating from the shuffling process. The DNA sequences of the shuffled genes confirmed that some combinations of single mutations give rise to higher expression from Pm than others in an apparently unpredictable manner (Fig. 3).
The StEP‐11 and StEP‐13 mutant XylS proteins resulted in an ampicillin tolerance about 9.5 times higher than wild‐type XylS under induced conditions, while the uninduced levels were still low, although higher (about four‐fold) than for the wild‐type.
Use of an evolved XylS activator for increased expression of a host‐toxic single‐chain variable fragment (scFv‐phOx)
We have previously shown that vectors with Pm and xylS can be used for high‐level expression of the host‐toxic antibody fragment scFv‐phOx in E. coli (Sletta et al., 2004; 2007). By increasing the vector copy number three‐ to fourfold, from 5–7 per chromosome with the vector pJBphOx to 15–30 with the vector pJBphOx‐cop271, a proportional increase in scFv‐phOx production was observed (Sletta et al., 2004). To test scFv‐phOx production with an evolved XylS protein, the xylS gene in the vector pJBphOx was exchanged with xylS‐StEP‐13 resulting in the expression vector pJBphOx : StEP‐13.
The production of intracellular soluble, intracellular insoluble and extracellular soluble scFv‐phOx from induced (0.1 mM m‐toluic acid) and uninduced shake flask cultures of E. coli RV308 with the vectors pJBphOx‐cop271 and pJBphOx : StEP‐13 were measured (Table 1). The total scFv‐phOx production from the pJBphOx : StEP‐13 culture was found to be about ninefold higher than from the cells containing pJBphOx‐cop271. Because of the lower vector copy number of pJBphOx : StEP‐13 compared with pJBphOx‐cop271, the specific stimulation (per plasmid unit) is even higher, demonstrating that evolved XylS activators have the potential to be utilized for industrial production of heterologous proteins.
Table 1.
scFv‐phOx production in E. coli RV308 cells containing plasmids pJBphOx‐cop271 or pJBphOx‐StEP13.
| Strain | IS | ES | IIS | Total |
|---|---|---|---|---|
| pJBphOx‐cop271 | 0.5 | BD | 1.2 | 1.7 |
| pJBphOx‐StEP13 | 6.7 | 0.7 | 7.4 | 14.8 |
All values are in mg l−1. BD, below detection limit (0.1 mg l−1). IS, intracellular soluble; ES, extracellular soluble; IIS, intracellular insoluble.
Discussion
In this study we have shown that it is possible to use directed evolution of xylS to obtain novel XylS activators that cause up to about a ninefold increase in expression from Pm, compared with wild‐type XylS. This was observed both with the prokaryotic β‐lactamase reporter protein as well as the eukaryotic scFv‐phOx protein. It is well established for the Pm/xylS system that protein expression is stimulated approximately proportional to the vector copy number increase resulting from the trfAcop271 mutation, both for scFv‐phOx (Sletta et al., 2004) and for other proteins (Blatny et al., 1997). The experiments reported here therefore show that the stimulation obtained by mutating xylS is much stronger than what can be achieved by increasing the copy number of the plasmid three‐ to fourfold. The rationale for using a low inducer concentration (0.1 mM) in the experiments with scFv‐phOx is also consistent with this, as further increases in the concentration led to extensive cell toxicity (growth stops) for the xylS Step‐13 mutant (wild‐type trfA), but not to the same extent for the strain with wild‐type xylS and the trfAcop271 mutation.
As expected, we found that modification of the error‐prone PCR procedure to further increase the xylS mutation frequency resulted in reduction of the number of active proteins, although among those that were still active the frequency of mutants leading to higher expression levels increased.
The observation that all XylS substitutions that resulted in stimulation of Pm activity were located in the NTD was somewhat unexpected. It has previously been shown that substitutions in XylS that confer semi‐constitutive phenotypes can be found in the CTD (Manzanera et al., 2000) and it is plausible that substitutions that confer higher binding affinity to the regulatory DNA element may lead to increased transcription stimulation both with and without induction. Our method excluded all mutants with high basal activity, possibly explaining the observed N‐terminal bias.
Future studies of the mechanism of action of the mutants described in this paper would be significantly facilitated by access to an experimental 3D structure of XylS. In the absence of such data a homology‐based 3D model of the NTD will be useful, and we have therefore constructed such a model (Fig. 4). This was done by using DeepView (Guex and Peitsch, 1997) and the SwissModel server (Schwede et al., 2003), based on structure data from the RCSB Protein Data Bank (Berman et al., 2000). Evaluation of model quality was done with Errat (Colovos and Yeates, 1993), and the model predicts that the NTD consists of a β‐barrel connected via a loop region to two adjacent alpha helices forming a coiled coil. Based on comparison with the solved AraC structure (Soisson et al., 1997) and the observation that leucines 193 and 194 in XylS are involved in dimerization (Ruíz et al., 2003), we propose that the β‐barrel contains the inducer recognition pocket and that the coiled coil defines the dimerization surface. The substitutions identified in this work are therefore positioned in different structural sub‐motifs, and this leads to the hypothesis that they affect different functional aspects of the activator. It is difficult to conclude what specific functional role the different substituted amino acids might have, but according to the model it seems likely that R45 and A111 can interact with the inducer binding pocket. We have also carried out a separate alignment between XylS and other members of this family of transcriptional regulators (data not shown), and the results indicated that the amino acid substitutions reported here generally did not correlate with conserved residues within the family. Thus, the mutations appear to mainly represent XylS‐specific adaptations.
Figure 4.
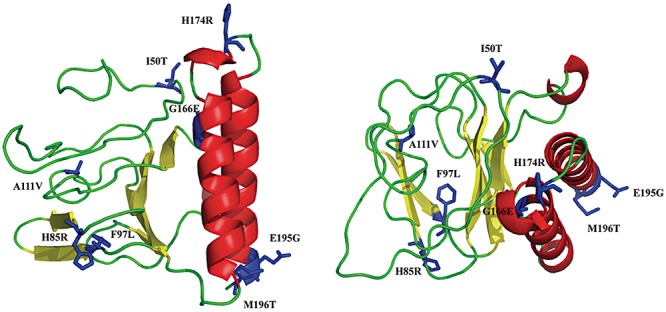
Predicted 3D model of the NTD of XylS covering residues 40–197, and shown from two different angles. The NTD consists of a β‐barrel and two α helices. The substitutions that cause increased transcription from Pm are shown in blue.
The observation that the XylS substitutions were scattered across the N‐terminal primary sequence and positioned in different structural motifs validates the use of error‐prone PCR as a method for directed evolution of transcription factors, whereas rational protein engineering, which often focuses on smaller areas of a protein as defined by solved 3D‐structures, will miss substitutions peripheral to the chosen area and hence, in this regard, be less suitable for the purpose of optimizing transcription factor function.
We therefore conclude that directed evolution can be used as a general tool to improve transcription factors with respect to recombinant protein production, an approach that to our knowledge has not been previously reported.
Experimental procedures
Bacterial strains, media and growth conditions
The bacterial strains used in this study were E. coli DH5α (Bethesda Research Laboratories) for standard laboratory procedures and E. coli RV308 (ATCC31608) for scFv‐phOx expression studies. The E. coli cells were cultivated at 37°C or 30°C in Luria broth or on L‐agar medium respectively. Kanamycin was used at 50 µg ml−1 when appropriate, ampicillin was used at concentrations between 5 and 6000 µg ml−1. The inducer was m‐toluic acid and the concentration was 1 mM unless otherwise stated.
DNA manipulations
All DNA primers are listed in Table S1 online. Standard DNA manipulations were performed as described elsewhere (Sambrook et al., 1989). Site‐specific mutagenesis was performed by using the QuikChange site‐specific mutagenesis kit from Stratagene according to the manufacturer's recommendations.
Plasmids used in this study
The screening vector pTA13 is a derivative of pJT19bla (Winther‐Larsen et al., 2000a) containing novel NcoI and AgeI sites for sub‐cloning of the xylS gene. The expression vector pJBphOx : StEP‐13 is a derivative of pJBphOx (Sletta et al., 2004) containing xylS‐StEP‐13 instead of wild‐type xylS. All plasmids used in this study and their detailed construction are presented in Table S2.
Error‐prone PCR
One single round of error‐prone PCR on the xylS gene was done according to Matsumura and Ellington (2001) by using the primers F‐epP2 xylS and R‐epP2 xylS (see Table S1) and pTA6 as template. To create a library with mutation frequencies varying from zero to 13.9 mutations per xylS gene, five separate error‐prone PCR reactions were performed with Mn2+ concentrations of 0.031, 0.063, 0.125, 0.250 and 0.500 mM (resulting in mutation frequencies of 0.0, 0.0, 1.0, 3.6 and 10.9 respectively). The lower concentrations presumably also lead to formation of mutants, but the frequency was too low to be detected with the limited number of mutants analysed. The PCR programme used the following steps: (i) 94°C for 2 min, (ii) 94°C for 30 s, (iii) 55°C for 30 s, (iv) 72°C for 1 min, (v) repeat steps (ii) to (iv) 25 times and (vi) 72°C for 7 min. The correct PCR fragments from each reaction were excised from a 0.8% agarose gel, combined and purified with the QiaQuik kit (Qiagen), before digestion with NcoI and AgeI and subsequent cloning into pTA13 (Fig. 1). The pTA13 screening vectors containing the xylS variants were then transformed into E. coli DH5α cells, generating an error‐prone PCR library.
Staggered extension process DNA shuffling
Staggered extension process (StEP) DNA shuffling was performed as described by Stemmer (Zhao, 2004). In this method a single set of primers are used together with a set of homologous genes in a PCR‐based process with repeated cycles of denaturation and very short annealing/elongation times. Partially formed products will function as primers for subsequent rounds and anneal to any of the homologous genes, thus generating recombined sequences. This is repeated until full‐length sequences are formed.
A single StEP reaction was performed with xylS variants containing single mutations and combinations of mutation originating from the error‐prone PCR library screening and rationally combined single mutations. The primers were Inner‐fw‐xylS and Inner‐rv‐xylS (see Table S1). The PCR programme used the following steps: (i) 96°C 2 min, (ii) 95°C for 30 s, (iii) 50°C for 2 min, (iv) repeat steps (ii) and (iii) 160 times and (v) 50°C for 2 min. The obtained DNA shuffling products were excised from gel, purified with QiaQuik (Qiagen) and used to substitute with wild‐type xylS (NcoI‐AgeI) in pTA13.
Screening for xylS variants –β‐lactamase assay
To screen for cells expressing novel XylS mutant proteins a β‐lactamase assay was used. This assay takes advantage of the fact that in pTA13 (see Supporting information) the bla gene is under control of the Pm promoter, which is positively regulated by XylS. Elevated expression from Pm leads to more β‐lactamase production in an approximately proportional relation to the maximum ampicillin tolerance levels of the host cells (Bakke et al., 2009; Berg et al., 2009). Here we chose to use resistance as an indirect method to monitor expression, as large numbers of mutants could more efficiently handled than by using direct enzyme activity measurements.
Pooled mutant libraries generated with error‐prone PCR or DNA shuffling were cultured for 18 h and then transferred to plates containing 1.0 mM m‐toluic acid or no m‐toluic acid, kanamycin and selective concentrations of ampicillin (0.75, 1 and 1.25 mg ml−1 for the error‐prone PCR library and 1.75 mg ml−1 for the DNA shuffling library). Attempts to increase the selection pressure somewhat resulted in a further reduction in the number of survivors, but the corresponding colonies with high frequencies displayed morphologies different from the wild type. Based on previous experience this is probably caused by the formation of spontaneous host mutations that lead to production of exopolysaccharides (possibly cholanic acid). Such mutations are generally known to lead to enhanced tolerance to antibiotics. Cells from colonies exhibiting high levels of ampicillin tolerance were grown in 96‐well plates with 100 µl LB broth and kanamycin, grown at 30°C for 18 h, diluted and then replica‐plated onto agar medium containing either no or 1.0 mM m‐toluic acid, kanamycin and varying ampicillin concentrations to establish the tolerance levels. The tolerance level was set to the mean value of the highest observed ampicillin concentration tolerated and the lowest observed ampicillin concentration that did not allow growth.
xylS genes from cells that reproducibly displayed elevated tolerance levels to ampicillin after replica plating were then re‐cloned into fresh background plasmids and re‐transformed into DH5α cells to eliminate potential secondary‐site effects. The mutants were then tested again to verify the observed antibiotic tolerance levels. Cells from the error‐prone PCR library screening that tolerated ampicillin concentration exceeding 50 µg ml−1 in the absence of inducers were discarded.
RNA isolation, cDNA synthesis, DNAse treatment, and two‐step quantitative real‐time PCR
Transcript amounts were determined by two‐step quantitative real‐time PCR. RNA in bacterial cell suspensions were stabilized by using RNAprotect bacteria reagent (Qiagen), and RNAqueous (Ambion) was used for total RNA isolation. Isolated RNA was treated with Turbo DNAse (Ambion). Reverse transcription was performed using a first‐strand cDNA synthesis kit (Amersham Biosciences). PCR was carried out in the presence of the power SYBR green PCR master mix (Applied Biosystems) using the 7500 real‐time PCR system (Applied Biosystems). Primers were designed using primer express software (Applied Biosystems) (see Table S1). The kanamycin‐resistance gene was used as an endogenous control to normalize all samples. Amplifications were carried out in triplicate wells for each sample.
Homology modeling of XylS
Modelling of the XylS structure was done with DeepView (Guex and Peitsch, 1997) and the SwissModel server (Schwede et al., 2003), based on structure data from the RCSB Protein Data Bank (Berman et al., 2000). The template was the NTD of AraC from E. coli (PDB code 2ARC, Soisson et al., 1997). Due to low sequence similarity the final alignment between XylS and AraC was a consensus alignment based on several alternative approaches, including model building with alternative gap lengths and positions. The sequence identity in the final alignment was 11% over 156 aligned residues. Evaluation of model quality was done with Errat (Colovos and Yeates, 1993). A detailed description of the homology‐based model building is given as Supporting information.
Analysis of single‐chain variable fragment (scFv‐phOx) production
Cultures of E. coli RV308 harbouring the plasmids pJBphOx‐cop271 or pJBphOx : StEP‐13 grown in L‐broth with 200 µg ml−1 ampicillin served as inoculum (1%) for new cultures (50 ml LB with 200 µg ml−1 ampicillin) that were grown for 2 h, induced with 0.1 mM m‐toluic acid and then grown for another 4 h. The cells were then harvested and the pellets were washed twice with 10 ml 50 mM MOPS, and finally resuspended in 3 ml sonication buffer (50 mM MOPS, 1 mM DTT, 3 mM EDTA and 1% Triton X‐100). The suspensions were sonicated and 1 ml of the lysates was centrifuged (17 000 g, 10 min). The supernatants contained the soluble protein samples and the pellets, which were resuspended in 1 ml 1% SDS, contained the insoluble proteins. Analysis of the scFv‐phOx production levels were performed with ELISA and Western blotting as described elsewhere (Sletta et al., 2004). The scFv‐phOx production levels were determined by comparing ELISA signals and Western blot band intensities from multiple ELISAs and Western blots featuring various dilutions of the samples.
Supporting information
This information can be found at the following link: http://www.biotech.ntnu.no/molgen/mbt2009
Acknowledgments
This work was funded by The Research Council of Norway, The National Program for Research in Functional Genomics in Norway and The Svanhild and Arne Must Fund for Medical Research. We would like to thank Håkon Hov at the Department of Cancer Research and Molecular Medicine, NTNU, for invaluable help with scFv‐phOx analyses.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Fig. S1. Alignment of XylS and AraC.
A. Pairwise alignment of XylS and AraC for different alignment strategies. The alignment is numbered according to XylS, and secondary structure is indicated with blue (β-strand) and red (α-helix). The predicted secondary structure for XylS is from the BioInfoBank Meta server.
B. Final consensus alignment for XylS and AraC used for model building. Conserved positions for each subfamily are shown between the sequences. Mutations discussed in the text are indicated with #.
Fig. S2. Score values for alternative alignments. Score values from DComplex, Fastcontact (kcal mol-1) and RPDock (RP score).
A. Interaction score for the XylS dimer (left) and the AraC dimer (right) at different gap lengths.
B. Interaction score for the final α-helix versus the rest of the XylS monomer (left) and same for the AraC monomer (right). See Supporting information text for details.
Fig. S3. Errat scores. Errat scores for (A) initial XylS monomer model, (B) initial XylS dimer model and (C) XylS dimer model after optimization of gap positions, corresponding to the final alignment in Fig. S1. For the dimer model only the first chain is shown.
Fig. S4. Final model. 3D representations of the final model, in two different orientations. The problematic regions from the Errat score in Fig. S3 are indicated with red in the left part of the structure, showing that this mainly affects the extreme parts of the β-barrel. The gap positions are indicated with cyan in the right part of the structure.
Fig. S5. Plasmid map of pTA13. The bla gene is under transcriptional control of the Pm promoter. xylS: gene for the transcriptional activator XylS; trfA: gene for the replicator protein TrfA; kan: gene for kanamycin resistance; oriV: origin for vegetative replication; oriT: origin for conjugational transfer. The NcoI and AgeI restriction sites for exchange of xylS are shown.
Table S1. Primers used in the study.
Table S2. Plasmids used in the study.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Bakke I., Berg L., Aune T.E.V., Brautaset T., Sletta H., Tøndervik A., Valla S. Random mutagenesis of the pm promoter as a powerful strategy for improvement of recombinant gene expression. Appl Env Microbiol. 2009;75:2002–2011. doi: 10.1128/AEM.02315-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg L., Lale R., Bakke I., Valla S. The expression of recombinant genes in Escherichia coli can be strongly stimulated at the transcript production level by mutating the DNA‐region corresponding to the 5′‐untranslated part of mRNA. Microb Biotechnol. 2009;2:379–389. doi: 10.1111/j.1751-7915.2009.00107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blatny J.M., Brautaset T., Winther‐Larsen H.C., Karunakaran P., Valla S. Improved broad‐host‐range RK2 vectors useful for high and low regulated gene expression levels in gram‐negative bacteria. Plasmid. 1997;38:35–51. doi: 10.1006/plas.1997.1294. [DOI] [PubMed] [Google Scholar]
- De Boer H., Comstock L.J., Vasser M. The tac promoter: a functional hybrid derived from the trp and lac promoters. Proc Nat Acad Sci USA. 1983;80:21–25. doi: 10.1073/pnas.80.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brautaset T., Lale R., Valla S. Positively regulated bacterial expresssion systems. Microb Biotechnol. 2009;2:15–30. doi: 10.1111/j.1751-7915.2008.00048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning D.F., Busby S.J.W. The regulation of bacterial transcription initiation. Nat Rev Microbiol. 2004;2:57–65. doi: 10.1038/nrmicro787. [DOI] [PubMed] [Google Scholar]
- Collins C.H., Arnold F.H., Leadbetter J.R. Directed evolution of Vibrio fischeri LuxR for increased sensitivity to a broad spectrum of acyl‐homoserine lactones. Mol Microbiol. 2005;55:712–723. doi: 10.1111/j.1365-2958.2004.04437.x. [DOI] [PubMed] [Google Scholar]
- Collins C.H., Leadbettter J.R., Arnold F.H. Dual selection enhances the signalling specificity of a variant of the quorom‐sensing transcriptional activator LuxR. Nat Biotechnol. 2006;24:708–712. doi: 10.1038/nbt1209. [DOI] [PubMed] [Google Scholar]
- Colovos C., Yeates T.O. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993;2:1511–1519. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domínguez‐Cuevas P., Marín P., Marqués S., Ramos J.L. XylS–Pm promoter interactions through two helix–turn–helix motifs: identifying XylS residues important for DNA binding and activation. J Mol Biol. 2008;375:59–69. doi: 10.1016/j.jmb.2007.10.047. [DOI] [PubMed] [Google Scholar]
- Gallegos M.‐T., Schleif R., Bairoch A., Hofmann K., Ramos J.L. AraC/XylS family of transcriptional regulators. Microbiol Mol Biol Rev. 1997;61:393–410. doi: 10.1128/mmbr.61.4.393-410.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guex N., Peitsch M.C. SWISS‐MODEL and the Swiss‐PdbViewer: An environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- Guzman L.‐M., Belin D., Carson M.J., Beckwith J. Tight regulation, modulation, and high‐level expression by vectors containing the PBAD promoter. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldimann A., Daniels L.L., Wanner B.L. Use of new methods for construction of tightly regulated arabinose and rhamnose promoter fusions in studies of the Escherichia coli phosphate regulon. J Bacteriol. 1998;180:1277–1286. doi: 10.1128/jb.180.5.1277-1286.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannig G., Makrides S.C. Strategies for optimizing heterologous protein expression in Escherichia coli. Trends Biotechnol. 1998;16:54–60. doi: 10.1016/s0167-7799(97)01155-4. [DOI] [PubMed] [Google Scholar]
- Kaldalu N., Toots U., De Lorenzo V., Ustav M. Functional domains of the TOL plasmid transcription factor XylS. J Bacteriol. 2000;182:1118–1126. doi: 10.1128/jb.182.4.1118-1126.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keasling J.D. Gene‐expression tools for the metabolic engineering of bacteria. Trends Biotechnol. 1999;17:452–460. doi: 10.1016/s0167-7799(99)01376-1. [DOI] [PubMed] [Google Scholar]
- Kessler B., Herrero M., Timmis K.N., De Lorenzo V. Genetic evidence that the XylS regulator of the Pseudomonas TOL meta operon controls the Pm promoter through weak DNA‐protein interactions. J Bacteriol. 1993;176:3171–3176. doi: 10.1128/jb.176.11.3171-3176.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzanera M., Marqués S., Ramos J.L. Mutational analysis of the highly conserved C‐terminal residues of the XylS protein, a member of the AraC family of transcriptional regulators. FEBS Lett. 2000;476:312–317. doi: 10.1016/s0014-5793(00)01749-x. [DOI] [PubMed] [Google Scholar]
- Matsumura I., Ellington A.D. Mutagenic PCR of protein‐coding genes for in vitro evolution. In: Braman J., editor. 2nd. Humana Press; 2001. pp. 259–264. [DOI] [PubMed] [Google Scholar]
- Michán C., Kessler B., De Lorenzo V., Timmis K.N., Ramos J.L. XylS domain interactions can be deduced from intraallelic dominance in double mutants of Pseudomonas putida. Mol Gen Genet. 1992;235:406–412. doi: 10.1007/BF00279387. [DOI] [PubMed] [Google Scholar]
- Ramos J.L., Michan C., Rojo F., Dwyer D., Timmis K. Signal‐regulator interactions. Genetic analysis of the effector binding site of xylS, the benzoate‐activated positive regulator of Pseudomonas TOL plasmid meta‐cleavage pathway operon. J Mol Biol. 1990;211:373–382. doi: 10.1016/0022-2836(90)90358-S. [DOI] [PubMed] [Google Scholar]
- Ruíz R., Ramos J.L. Residues 137 and 153 of XylS influence contacts with the C‐terminal domain of the RNA polymerase alpha subunit. Biochem Biophys Res Commun. 2001;287:519–521. doi: 10.1006/bbrc.2001.5615. [DOI] [PubMed] [Google Scholar]
- Ruíz R., Marques S., Ramos J.L. Leucines 193 and 194 at the N‐terminal domain of the XylS protein, the positive transcriptional regulator of the TOL meta‐cleavage pathway, are involved in dimerization. J Bacteriol. 2003;185:3036–3041. doi: 10.1128/JB.185.10.3036-3041.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J., Fritsch E.F., Maniatis T. Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Schmidt F.R. Recombinant expression systems in the pharmaceutical industry. Appl Microbiol Biotechnol. 2004;65:363–372. doi: 10.1007/s00253-004-1656-9. [DOI] [PubMed] [Google Scholar]
- Schwede T., Kopp J., Guex N., Peitsch M.C. SWISS‐MODEL: an automated protein homology‐modeling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sletta H., Nedal A., Aune T.E.V., Hellebust H., Hakvåg S., Aune R. Broad‐host‐range plasmid pJB658 can be used for industrial‐level production of a secreted host‐toxic single‐chain antibody fragment in Escherichia coli. Appl Env Microbiol. 2004;70:7033–7039. doi: 10.1128/AEM.70.12.7033-7039.2004. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sletta H., Tøndervik A., Hakvåg S., Aune T.E.V., Nedal A., Aune R. The presence of N‐terminal secretion signal sequences leads to strong stimulation of the total expression levels of three tested medically important proteins during high‐cell‐density cultivations of Escherichia coli. Appl Env Microbiol. 2007;73:906–912. doi: 10.1128/AEM.01804-06. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soisson S., MacDougall‐Shackleton B., Schleif R., Wolberger C. Structural basis for ligand‐regulated oligomerization of AraC. Science. 1997;278:421–425. doi: 10.1126/science.276.5311.421. [DOI] [PubMed] [Google Scholar]
- Terpe K. Overview of bacterial expression systems for heterologous protein production: from molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol. 2006;72:211–222. doi: 10.1007/s00253-006-0465-8. [DOI] [PubMed] [Google Scholar]
- Tobes R., Ramos J.L. AraC‐XylS database: a family of positive transcription regulators in bacteria. Nucleic Acids Res. 2002;30:318–321. doi: 10.1093/nar/30.1.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlin B., Nordström K. R plasmid gene dosage in Escherichia coli K‐12: copy mutants of the R plasmid R1drd‐19. Plasmid. 1977;1:1–7. doi: 10.1016/0147-619x(77)90003-8. [DOI] [PubMed] [Google Scholar]
- Winther‐Larsen H.C., Blatny J.M., Valand B., Brautaset T., Valla S. Pm promoter expression mutants and their use in broad‐host‐range RK2 plasmid vectors. Metab Eng. 2000a;2:92–103. doi: 10.1006/mben.1999.0143. [DOI] [PubMed] [Google Scholar]
- Winther‐Larsen H.C., Josefsen K.D., Brautaset T., Valla S. Parameters affecting gene expression from the Pm promoter in gram‐negative bacteria. Metab Eng. 2000b;2:79–91. doi: 10.1006/mben.1999.0142. [DOI] [PubMed] [Google Scholar]
- Yuan L., Kurek I., English J., Keenan R. Laboratory‐directed protein evolution. Microbiol Mol Biol Rev. 2005;69:373–392. doi: 10.1128/MMBR.69.3.373-392.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H. Staggered extension process in vitro DNA recombination. Methods Enzymol. 2004;388:42–49. doi: 10.1016/S0076-6879(04)88005-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Alignment of XylS and AraC.
A. Pairwise alignment of XylS and AraC for different alignment strategies. The alignment is numbered according to XylS, and secondary structure is indicated with blue (β-strand) and red (α-helix). The predicted secondary structure for XylS is from the BioInfoBank Meta server.
B. Final consensus alignment for XylS and AraC used for model building. Conserved positions for each subfamily are shown between the sequences. Mutations discussed in the text are indicated with #.
Fig. S2. Score values for alternative alignments. Score values from DComplex, Fastcontact (kcal mol-1) and RPDock (RP score).
A. Interaction score for the XylS dimer (left) and the AraC dimer (right) at different gap lengths.
B. Interaction score for the final α-helix versus the rest of the XylS monomer (left) and same for the AraC monomer (right). See Supporting information text for details.
Fig. S3. Errat scores. Errat scores for (A) initial XylS monomer model, (B) initial XylS dimer model and (C) XylS dimer model after optimization of gap positions, corresponding to the final alignment in Fig. S1. For the dimer model only the first chain is shown.
Fig. S4. Final model. 3D representations of the final model, in two different orientations. The problematic regions from the Errat score in Fig. S3 are indicated with red in the left part of the structure, showing that this mainly affects the extreme parts of the β-barrel. The gap positions are indicated with cyan in the right part of the structure.
Fig. S5. Plasmid map of pTA13. The bla gene is under transcriptional control of the Pm promoter. xylS: gene for the transcriptional activator XylS; trfA: gene for the replicator protein TrfA; kan: gene for kanamycin resistance; oriV: origin for vegetative replication; oriT: origin for conjugational transfer. The NcoI and AgeI restriction sites for exchange of xylS are shown.
Table S1. Primers used in the study.
Table S2. Plasmids used in the study.