

Summary
Carboxyl esterases (CE) exhibit various reaction specificities despite of their overall structural similarity. In present study we have exploited functional metagenomics, saturation mutagenesis and experimental protein evolution to explore residues that have a significant role in substrate discrimination. We used an enzyme, designated 3A6, derived from the earthworm gut metagenome that exhibits CE and feruloyl esterase (FAE) activities with p‐nitrophenyl and cinnamate esters, respectively, with a [(kcat/Km)]CE/[(kcat/Km)]FAE factor of 17. Modelling‐guided saturation mutagenesis at specific hotspots (Lys281, Asp282, Asn316 and Lys317) situated close to the catalytic core (Ser143/Asp273/His305) and a deletion of a 34‐AA–long peptide fragment yielded mutants with the highest CE activity, while cinnamate ester bond hydrolysis was effectively abolished. Although, single to triple mutants with both improved activities (up to 180‐fold in kcat/Km values) and enzymes with inverted specificity ((kcat/Km)CE/(kcat/Km)FAE ratio of ∼0.4) were identified, no CE inactive variant was found. Screening of a large error‐prone PCR‐generated library yielded by far less mutants for substrate discrimination. We also found that no significant changes in CE activation energy occurs after any mutation (7.3 to −5.6 J mol−1), whereas a direct correlation between loss/gain of FAE function and activation energies (from 33.05 to −13.7 J mol−1) was found. Results suggest that the FAE activity in 3A6 may have evolved via introduction of a limited number of ‘hot spot’ mutations in a common CE ancestor, which may retain the original hydrolytic activity due to lower restrictive energy barriers but conveys a dynamic energetically favourable switch of a second hydrolytic reaction.
Introduction
Carboxyl‐esterases (CEs; EC 3.1.1.X) are ubiquitous α/β‐hydrolases with representatives in all three domains of life, Eukarya, Bacteria and Archaea (Bornscheuer, 2002). These proteins catalyse the cleavage and formation of ester bonds whose components differ in chain nature and length. Within the α/β‐hydrolase superfamily, CEs belong to the best structurally and functionally characterized, with more than 6016 members divided in 90 subfamilies after a comprehensive non‐redundant search in the nucleotide collections, reference genomic sequences, whole genome shotgun reads and environmental samples databases. Site‐directed mutagenesis (Manco et al., 2001; Reyes‐Duarte et al., 2005), saturation mutagenesis (Mee‐Hie Cho et al., 2006; Wang et al., 2006; Nakagawa et al., 2007) and a number of directed evolution methods (Funke et al., 2005; Ivancic et al., 2007) have successfully been applied to identify the molecular determinants of substrate specificity and enantio‐selectivity and to gain some knowledge on molecular evolution of this super‐family of proteins.
We focused in this study on esterases able to attack the ester bond between hydroxyl‐cinnamic acids, such as ferulic, p‐coumaric and sinnapic acid, and sugars present in intricate structure of the plant cell wall (Fazary and Ju, 2007). The hydrolysis of these compounds is catalysed by feruloyl esterases (FAEs; EC 3.1.1.73). About 30 FAEs have been described and based on structural and functional factors a division into four types (A–D) has been established (Crepin et al., 2004; Wong, 2006; Benoit et al., 2008). They have been purified and characterized from bacteria (Wang et al., 2004; Aurilia et al., 2007; 2008) and fungi (Shin and Chen, 2007; Kanauchi et al., 2008; Moukouli et al., 2008; Koseki et al., 2009), and showed significant variations in activities. Despite all of them having the typical α/β fold and the common CE‐like catalytic triad, Ser‐Asp/Glu‐His (Fazary and Ju, 2007), little is known about the factors determining substrate specificity of these enzymes compared with ‘common’ CEs, which has inter alia motivated present work. Only few studies have demonstrated that few mutations at the ligand‐binding cavity (Tarbouriech et al., 2005) or in insertions such as lids (Hermoso et al., 2004; Faulds et al., 2005; Koseki et al., 2005) have important effects in controlling the binding and hydrolysis of esters with m‐methoxy groups. Following on from this, Levasseur's group studied the evolutionary relationships between fungal lipases and FAEs through the study of their phylogenies (Levasseur et al., 2006). Authors concluded that lipases appear to be primitive functions from which FAE functionality could have been derived bought duplication events probably induced by environmental stresses, i.e. those caused by new substrates. Although, authors suggest that modifications within the active site might thus be positively selected, causing the shift in functionality at the organismal level (Hermoso et al., 2004; Levasseur et al., 2006), no experimental evidence for such hypothesis has been provided.
In this work, the first systematic study was performed to investigate the structural and energetic determinants for the transition between CEs and FAEs. Here, we used a novel bi‐functional CE/FAE protein (3A6) that catalyses hydrolytic cleavage of ‘common’ and ‘cinnamate’ esters, isolated from the microbial community of the gut of the earthworm Aporrectodea caliginosa. Invertebrate guts are certainly one of the most diverse environments yet studied and, in particular, the earthworm gut associated‐organisms require an efficient enzymatic machinery to cope with a continuous flux of various polymeric materials (including cinnamate esters) from the soil and from plant litter passing through intestinal tract, which make it attractive for isolating hydrolytic activities. We show here by examining saturation mutagenesis and error‐prone PCR libraries, that a reduced number of ‘hot‐spot’ mutations could alter the overall substrate hydrolysis and, more importantly, evidence is presented for the first time that suggests that the acquisition of FAE phenotype, from an ancestor CE activity, is energetically favourable. Moreover, to the best of our knowledge, this work is the first example for a successful conversion of an FAE into a common CE.
Results
General characteristics of the 3A6 protein
The fosmid carrying the 3A6 gene was isolated by screening a metagenomic library from the cellulose‐enriched microbial community from the gut of the earthworm A. caliginosa, based on its ability to hydrolyse methyl ferulate (MF). The analysis of 3081‐bp‐long subcloned DNA fragment (GC‐content 42.2%; Fig. S1) revealed the presence of two oppositely oriented open reading frames (ORFs), the first of which (1026 bp, positions 1144 to 119) encodes a putative 341‐amino‐acid protein with a predicted molecular mass of 37 502 Da that exhibited high homology (54% identity and 71% of similarity) to proteins of the esterase/lipase superfamily (pfam00135) (Table S1). The 3A6 gene was subcloned into a plasmid vector and after that, inserted into the pET41 Ek/LIC vector for expression in Escherichia coli. The protein, purified to homogeneity as a single monomer of 38 000 ± 3200 Da, was tested for its catalytic ability to hydrolyse a series of commercially available substrates (Table 1). We show that besides hydrolysing p‐nitrophenyl (pNP) esters, 3A6 efficiently catalysed the hydrolysis of cinnamates with a (kcat/Km) factor of ∼17:1 with pNP acetate (pNPC2) and MF as substrates. Overall, the enzyme was able to cleavage most efficiently methyl sinapinate, followed by MF (2.5‐fold), and methyl‐p‐coumarate (16‐fold), but not methyl caffeate. As judged by the kcat/Km values, the enzyme functions with short chain substrates from 2‐ to 1500‐fold better than the longer substrates. The enzyme was also able to hydrolyse, to some extent, p‐nitrophenyl 5‐O‐trans‐feruloyl‐α‐l‐arabinofuranoside (kcat of 1 s−1) and (5‐O‐(trans‐feruloyl)‐α‐l‐arabinofuranosyl)‐(1,3)‐β‐d‐xylopyranosyl‐(1,4)‐d‐xylopyranose (kcat of 10 s−1). According to its substrate specificity, the enzyme was classified as an FAE type A (Crepin et al., 2004). The protein showed maximal activity at 45–50°C and pH 7.5–8.5 (Fig. S2).
Table 1.
Steady‐state kinetic parameters of the enzyme 3A6 and its variant 3A6‐I.
| Substratea | 3A6 | 3A6I | ||||
|---|---|---|---|---|---|---|
| Km (mM) | kcat (s−1) | kcat/Km (s−1 mM−1) | Km (mM) | kcat (s−1) | kcat/Km (s−1 mM−1) | |
| p‐Nitrophenyl acetate | 0.18 ± 0.09 | 137.4 ± 1.5 | 763 | 3.48 ± 0.21 | 164.1 ± 1.9 | 47 |
| p‐Nitrophenyl propionate | 0.15 ± 0.09 | 56.7 ± 0.8 | 378 | 1.78 ± 0.21 | 192.2 ± 2.3 | 108 |
| p‐Nitrophenyl butyrate | 0.24 ± 0.02 | 23.1 ± 35.4 | 96 | 0.89 ± 0.07 | 553.2 ± 4.8 | 622 |
| p‐Nitrophenyl hexanoate | 1.53 ± 0.09 | 0.8 ± 0.1 | 0.5 | 1.34 ± 0.14 | 78.4 ± 0.5 | 59 |
| Methyl ferulate | 2.34 ± 0.37 | 104.3 ± 0.5 | 45 | 32.20 ± 4.41 | 5.8 × 10−3 | 1.8 × 10−4 |
| Methyl sinapinate | 0.40 ± 0.08 | 44.9 ± 0.2 | 112 | 39.12 ± 6.20 | 5.2 × 10−3 | 5.6 × 10−5 |
| Methyl p‐coumarate | 1.55 ± 0.11 | 10.4 ± 0.2 | 7 | 26.60 ± 5.20 | 3.7 × 10−4 | 1.4 × 10−5 |
| Nph‐5‐Fe‐Arafb | 0.97 ± 0.03 | 0.9 ± 0.1 | 0.9 | 28.10 ± 6.10 | 3.1 × 10−4 | 1.1 × 10−5 |
| FAXXc | 1.96 ± 0.37 | 10.3 ± 0.7 | 5 | 31.17 ± 7.70 | 6.2 × 10−4 | 2.0 × 10−5 |
Reaction conditions: [E]o = 0–12 nM, [substrate] ranging from 0 to 50 mM, 100 mM Tris‐sulfate, pH 8.5, T = 40°C.
Nph‐5‐Fe‐Araf: p‐nitrophenyl 5‐O‐trans‐feruloyl‐α‐l‐arabinofuranoside.
FAXX: 5‐O‐(trans‐feruloyl)‐α‐l‐arabinofuranosyl)‐(1,3)‐β‐d‐xylopyranosyl‐(1,4)‐d‐xylopyranose.
The substrate specificity showed strikingly different results with those reported in literature: nearly all characterized α/β hydrolases that share sequence similarity with 3A6 cleaved p‐nitrophenyl esters but not cinnamates (Table S1). Following on from this, one of the main objectives of this study was to complement the kinetic data with mechanistic and engineering protocols so as to facilitate examination the essential residues that might hinder the recognition of cinnamate esters by steric or catalytic effects in the 3A6 protein.
Saturation mutagenesis strategy for altering the enzyme's specificity
A model of the enzyme was obtained by homology using the crystal structure of carboxyl‐esterase Este1 of metagenomic origin (PDB 2C7B) as a template (Fig. S3), given the high level of identity (38% of the overall sequence, close to 70% in the active‐site region). The quality of the obtained model was judged to be adequate for our purposes as judged by the Ramachandran plots (not shown), and a putative active‐site pocket was identified (Ser143/Asp273/His305) and further confirmed by mutagenic analysis: mutations of those residues by Gly, Gln and Asn, in the same order, results in a dramatic effect on the kcat/Km for pNPC2 and MF cleavage (> 10 000‐fold). To further identify key residues that might define additional stabilization, subtraction or orientation effects on substrate preference, a detailed multiple (structural) alignment of 3A6 with homologous proteins was performed (Figs S4 and S5). The attention was focused on identifying regions that might have a fundamental role in substrate specificity and preference and that are not found in homologue proteins. The regions that were selected for mutagenesis were: Asn109‐Leu111, Lys281‐Asp282 and Tyr315‐Lys317 (Fig. S3).
Saturation mutagenesis libraries (Asn109X, Glu110X, Leu111X, Lys281X, Asp282X, Tyr315X, Asn316X and Lys317X) were initially constructed, and crude cell extracts from ∼100 colonies (found to be sufficient to ensure that the 20 AA were represented) from each library were screened for activity with both pNPC2 and MF. The results corrected for cell growth level, are shown in Fig. 1 (mutations at positions 109–111 did not affect hydrolytic proficiency and are not shown). In each case, a minor proportion of the library members were inactive with both substrates, showing that only few amino acid substitutions were not tolerated for activity. For each library some of the variants showed altered improved activity but no change in substrate preference. However, a number of particularly interesting variants were identified in the K281X, D282X, N316X and K317X libraries, suggesting that they may play important roles in controlling substrate recognition. It is particularly interesting to note that some of the subpopulations of variants in the libraries appeared to have a significant preference for the pNP substrate, with some of N316X and K317X variants having no measurable activity against MF, whereas some (i.e. K281X and D282X) showed higher overall activity for the MF screening substrate. Interestingly, pNPC2 inactive variants were not detected, suggesting that CE activity is more robust to amino acid substitutions. Detailed kinetic characterization of fourteen purified enzymes selected on the basis of their substrate preferences was carried out in order to accurately determine the importance of each of the identified amino acid substitutions in the discrimination between the pNPC2 and MF screening substrates (Table 2).
Figure 1.
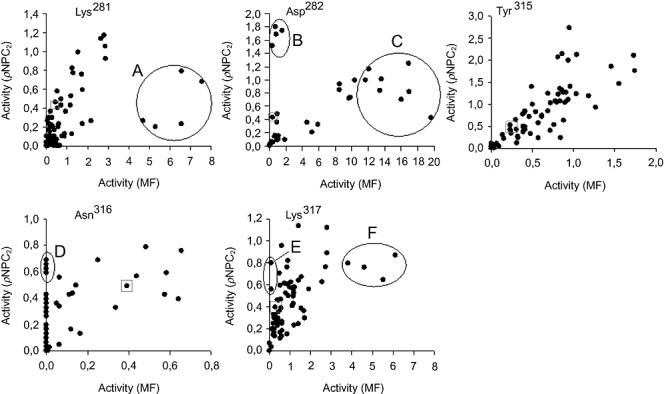
Activity screens for substrate‐specific variants of 3A6 saturation mutagenesis libraries. Identical copies of 96‐well microtiter plates containing crude cell lysates of saturation mutagenesis libraries Lys281X, Asp282X, Y315X, Asn316X and Lys317X were screened for activity with pNPC2 and MF. Activities for both substrates are plotted as rate of change of absorbance at 405 nm (for pNPC2) and 550 nm (for MF) per minute and corrected for bacterial cell growth. Subpopulations with altered substrate preference are explicitly shown: B, D and E correspond to CE‐selective mutants, whereas A, C and F correspond to FAE‐selective mutants.
Table 2.
Steady‐state kinetic parameters of the wild‐type and variant enzymes.a
| Protein variant | Kinetic parameters for pNPC2 | Kinetic parameters for MF | Substrate discrimination | ||||
|---|---|---|---|---|---|---|---|
| Km (mM) | kcat (s−1) | kcat/Km (s−1 M−1) | Km (mM) | kcat (s−1) | kcat/Km (s−1 M−1) | (kcat/Km)pNPC2/(kcat/Km)MF | |
| 3A6 | 0.18 ± 0.09 | 137.4 ± 1.5 | 763 | 2.34 ± 0.37 | 104.3 ± 0.5 | 45 | 17 |
| K281N | 0.31 ± 0.03 | 129.2 ± 0.9 | 417 | 2.02 ± 0.14 | 337.0 ± 1.1 | 167 | 2.5 |
| K281T | 0.27 ± 0.03 | 155.3 ± 1.6 | 575 | 2.07 ± 0.26 | 296.3 ± 0.9 | 143 | 4.0 |
| K281S | 0.29 ± 0.03 | 171.8 ± 1.3 | 592 | 2.40 ± 0.60 | 277.5 ± 0.7 | 116 | 5.1 |
| K281I | 0.66 ± 0.03 | 715.2 ± 5.0 | 1084 | 0.16 ± 0.05 | 242.2 ± 0.7 | 1514 | 0.7 |
| D282E | 0.13 ± 0.03 | 147.0 ± 0.7 | 1131 | 0.93 ± 0.16 | 696.8 ± 0.4 | 749 | 1.5 |
| D282L | 0.11 ± 0.04 | 278.7 ± 2.6 | 2537 | 1.89 ± 0.10 | 69.4 ± 0.8 | 37 | 68.6 |
| N316L | 0.22 ± 0.02 | 109.9 ± 0.6 | 500 | 2.16 ± 0.48 | 235.8 ± 1.0 | 109 | 4.6 |
| N316STOP | 0.18 ± 0.04 | 161.4 ± 2.2 | 1494 | 8.79 ± 0.89 | 3.20 ± 0.05 | 0.4 | 3735.0 |
| K317N | 0.31 ± 0.04 | 88.0 ± 0.6 | 284 | 2.38 ± 0.14 | 517.4 ± 0.3 | 217 | 1.3 |
| K317G | 0.24 ± 0.02 | 151.2 ± 1.2 | 630 | 1.92 ± 0.58 | 475.9 ± 0.4 | 248 | 2.5 |
| K317L | 0.10 ± 0.01 | 138.8 ± 1.2 | 578 | 2.77 ± 0.49 | 429.8 ± 0.5 | 155 | 3.7 |
| K317D | 0.24 ± 0.03 | 136.0 ± 1.3 | 567 | 0.93 ± 0.35 | 423.5 ± 0.8 | 455 | 1.2 |
| K317H | 0.39 ± 0.12 | 129.3 ± 1.0 | 331 | 22.51 ± 3.50 | 3.6 × 10−3 | 1.6 × 10−4 | 2 × 106 |
| 3A6I | 3.48 ± 0.21 | 164.1 ± 1.9 | 47 | 32.20 ± 4.41 | 5.8 × 10−3 | 1.8 × 10−4 | 26111 |
| K281I/D282E | 0.16 ± 0.07 | 493.7 ± 3.4 | 3086 | 0.72 ± 0.08 | 3494.1 ± 2.9 | 4853 | 0.6 |
| D282L/N316STOP | 0.25 ± 0.06 | 961.5 ± 5.4 | 3844 | 5.81 ± 0.60 | 226.6 ± 1.8 | 39 | 98.6 |
| D282L/K317H | 0.12 ± 0.03 | 270.4 ± 3.1 | 2253 | 8.60 ± 1.50 | 17.2 ± 2.7 | 2 | 1126.5 |
| N316STOP/K317H | 0.07 ± 0.01 | 181.0 ± 4.7 | 2587 | 17.50 ± 1.90 | 2.3 × 10−3 | 1.6 × 10−4 | 1.6 × 106 |
| D282L/N316STOP/K317H | 0.12 ± 0.03 | 779.2 ± 3.8 | 6493 | 16.50 ± 2.51 | 93.0 ± 1.7 | 6 | 1082.2 |
| K281I/D282E/K317D | 0.48 ± 0.05 | 1611.8 ± 8.5 | 3358 | 0.39 ± 0.07 | 3171.8 ± 8.3 | 8133 | 0.4 |
| A8P4 (H26/A85P/T86P) | 2.29 ± 0.62 | 9826.7 ± 9.0 | 4279 | 2.04 ± 0.10 | 7.3 ± 0.2 | 3.6 | 1188 |
Reaction conditions: [E]o = 0–12 nM, [substrate] ranging from 0 to 50 mM, 100 mM Tris‐sulfate, pH 8.5, T = 40°C.
Construction of a CE‐selective enzyme. Screening of the saturation mutagenesis libraries (Fig. 1) identified three substitutions (populations B, D and E) that yield pNPC2 selective catalysts. In the initial screen, members of these populations showed similar or higher activity with the pNPC2 substrate than the wild‐type enzyme, but most importantly for our selection criteria, those appeared to have little or no measurable activity with MF. Sequence analysis revealed that three simple amino acid substitutions (D282L, N316STOP and K317H) are responsible for the excellent substrate discrimination: from (kcat/Km)pNPC2/(kcat/Km)MF of ∼17 for the wild‐type to 69 (for D282L) to 3735 (for N316STOP) and 2 × 106 (for K317H). This result can be explained by the up to 10‐fold greater Km values coupled with a significant (from 1.5‐ to 29 000‐fold) reduction in the kcat values for MF. This result is entirely consistent with the apparent ∼3 to > 700‐fold discrimination displayed by populations B, D and E in the initial screen (Fig. 1). Our data suggest that neither of the Y315X variants were individually responsible for discrimination of esterase and feruloyl esterase activities, although mutations at this residue equally increased both activities.
Construction of FAE‐selective enzymes. As shown in Fig. 1, populations A, C and F were identified as leading to the most MF‐selective variants. Sequencing identified 10 new mutations. The K281I mutant displayed the highest (34‐fold increase) hydrolytic efficiency for MF, mainly due to a 14‐fold reduction in binding capacity (Table 2). Overall, K281I substitution produced the only enzyme variant more selective towards MF with a (kcat/Km)pNPC2/(kcat/Km)MF ratio of 0.7 (24 times lower than wild‐type), which is in a good agreement with the apparent 16‐fold discrimination displayed by population A in the screen (Fig. 1). For the other variants (K281N, K281T, K281S, D282E, N316L, K317N, K317G, K317L and K317D) there is a large combined effect of lower kcat/Km for pNP esters and high kcat/Km for MF that led to (kcat/Km)pNPC2/(kcat/Km)MF ratios ranging from 4‐ to 14‐fold lower than wild‐type. Overall, the cleavage of MF was mostly positively affected by the mutations.
Gain‐of‐function by site‐directed mutagenesis
In an attempt to improve the activity and selectivity levels a number of double and triple mutants were created by Quick‐Change site‐directed mutagenesis and expressed recombinantly in E. coli. As shown in the Table 2, combination of K281I with D282E and K281I with D282E and K317D resulted in variants preferably hydrolysing MF with (kcat/Km)pNPC2/(kcat/Km)MF factors of 0.4 and 0.6, respectively, due to a 33‐fold higher kcat and sixfold lower Km for MF. However, combination of D282L with N316STOP, followed by D282L with N316STOP and K317H, D282L with K317H, and specially D282L with N316STOP, resulted in highly selective catalysts towards pNPC2. The (kcat/Km)pNPC2/(kcat/Km)MF ratio for these variants varied from 100 to 2 × 106. Here, we observed a synergistic effect, decreasing significantly the affinity (from twofold to eightfold) for MF while increasing (up to eightfold) the kcat for pNPC2.
Maximizing FAE activity of 3A6 by deletion
Although single mutations in key regions were sufficient enough to significantly alter hydrolytic activity and control substrate specificity, we attempted to delete the Phe11‐Lys29 and Gly178‐Gly211 regions, to check for their catalytic influence. Both regions, not found in homologous proteins, were selected based on structural alignments (Figs S3–S5). The first one is located on the N‐terminus of the protein and its deletion caused a complete loss of enzyme activity and will not be discussed further. The second insert is located on a long loop with extremely low sequence conservation. Circular dichroism (CD) analysis of pure protein, named 3A6‐I, revealed that the deletion variant exhibited a slightly modified CD spectrum compared with the 3A6 wild‐type protein (Fig. S6) and moreover, it showed an excellent substrate discrimination (Table 1). Interestingly, compared with parental and saturation mutants, the 3A6‐I protein was the only enzyme variant, which appeared to demonstrate a significant, 13‐fold preference for longer substrates, with pNP butyrate being the preferred substrate over pNPC2 (Table 1). This variant showed an unexpected residual activity towards the cinnamates, with a (kcat/Km)pNP/(kcat/Km)MF ratio higher than 107. It is therefore a CE‐selective enzyme. Even though we do not know the exact role in vivo of this extension, it is clear that it has a substrate recognition role.
Substrate discrimination by random mutagenesis
The mutational strategy has therefore produced a number of enzymes with high activity levels and opposite substrate preference (N316STOP/K317H, preference CE/FAE ratio ∼2 × 106; and K281I/D282E/K317D, preference CE/FAE ratio ∼0.4) compared with the wild‐type enzyme (3A6) which displayed 8‐ and 180‐fold lower CE and FAE activity levels, respectively, and a preference ratio of 17. To further prove whether other protein regions/amino acids promote substrate preference we constructed an epPCR library with a suitable number of mutations. For that, favourable conditions were first determined by amplifying the 3A6 gene at 10 different MnCl2 concentrations (not shown). A total number of 38 400 colonies were picked and inoculated overnight in 96‐well microtiter plates. Clones were then analysed for pNPC2 and MF hydrolysis, using 3A6 as control. The results indicated that most library members displayed approximately the same substrate preference as the parental 3A6 protein. Four variants, designated A8P4, E4P4, E8P3 and F2P3, were selected as displaying greater differential preferences. The four potentially positive variants from epPCR were picked, and after verification in liquid culture, only the A8P4 was selected for further analysis. This variant contains a C76AT to C76AC synonymous mutation (His26) and two other consecutive mutations Ala85Pro and Thr86Pro. We first observed that the Km value for pNPC2 was 12‐fold higher than that of the native enzyme, while maintaining that for MF (Table 1). Furthermore, mutant showed a 39‐fold increase of (kcat/Km)pNP/(kcat/Km)MF ratio (from 17 to 1200). This result is explained by the ∼14‐fold lower and ∼70‐fold higher kcat values for MF and pNPC2 respectively. These data suggest that there is a strong bias towards CE phenotype in the A8P4 mutant.
Above results unambiguously confirmed that mutations at specific hot spots of the polypeptide (i.e. K281, D282, N316 and K317) may play a more effective supportive role in substrate discrimination in the parental 3A6 protein compared with random mutations within the whole protein length, since only one mutant with substrate discrimination ability was identified by epPCR. Figure 2 summarized the effect of single to triple mutations and insert deletion on substrate preference.
Figure 2.
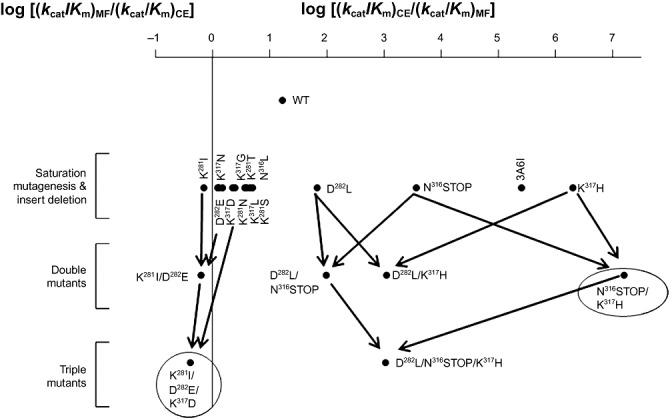
Schematic representation of the complementary substrate preference of 3A6‐like variants. The preferred substrate specificity of wild‐type and variants are shown as the logarithm of the ratio of catalytic efficiencies (kcat/Km) towards pNPC2 and MF substrates. A value of zero thus represents an enzyme, which is non‐discriminatory between the two substrates.
Substrate preference and its association with activation energies
The experimental work on the in vitro evolution of CE and FAE activities was followed by calculating the differential binding (ΔΔGB) and activation (ΔΔGA) free energy profiles for the cleavage of pNPC2 and MF, respectively, by the ‘parental’ and ‘mutant’ 3A6 variants. Difference energy diagrams (energy of the variant minus energy of the parent 3A6) are shown in Fig. 3A and B, and were used to understand the thermodynamics of the changes in enzyme reaction specificity, since the changes in substrate hydrolysis and preference are brought about by the relative changes in the free energies of the transition‐state barriers. First, we see that the changes in ΔΔGB and ΔΔGA from the native to double or triple mutants are not the sum of free energies of the single saturation mutants, either for binding or catalysis. Thus, the contribution of K281I, D282E, D282L, N316STOP, K317H and K317D is synergic rather than mutually independent for the cleavage of both substrates, each of them positively or negatively affecting free energy values.
Figure 3.
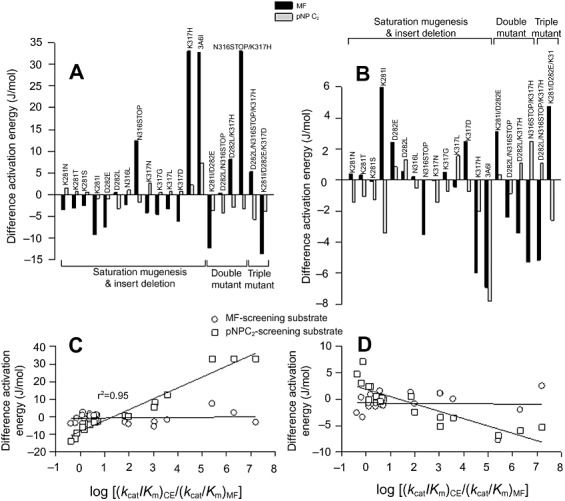
Difference activation (A) and binding (B) energy diagrams for the reactions catalysed by the 3A6 variants. The free difference energy of the binding (ΔΔGB) and activation (ΔΔGA) energies (energy of the variant minus energy of the 3A6 parent) for the pNPC2 and MF substrates were calculated for each variant from the Km values measured for the enzyme variants, assuming that Km provides an indication of the binding affinity between the enzyme and the substrate. The activation energies of the kcat/Km were also calculated to provide the heights of the transition state barriers. Plot (C) and (D) represent the difference activation and binding energies versus the logarithm of the (kcat/Km)pNPC2/ (kcat/Km)MF ratio respectively.
The effects on mutants on ΔΔGB and ΔΔGA are rather complex. As shown, major differences are observed for the activation free energies of MF compared with that shown for pNPC2 (Fig. 3A and B). This suggests that FAE phenotype is more mutation affected by introducing single point mutations in specific ‘hot spot’ residues (see also Table 2). This was further confirmed by plotting the ΔΔGA for pNPC2 and MF against the (kcat/Km)pNPC2/(kcat/Km)MF ratio for each of the variants. Surprisingly, whereas the ΔΔGA for pNPC2 do not varied significantly (−5.6 to 7.3 J mol−1), that of MF do with a sharp increase (from −13.7 to 33.1 J mol−1) at increasing the CE phenotype (Fig. 3C). Similar situation was found when analysing the ΔΔGB, although at lower level (Fig. 3D). Data suggest that mutations producing FAE‐selective enzymes, i.e. those with (kcat/Km)pNP/(kcat/Km)MF ratio lower than 17, may be more favourable energetically (decrease in ΔΔG). The low dependence of pNPC2‐associated free energy with catalytic efficiency is consistent with the fact that any of the mutations abolished the CE activity during the screening library tests (see Fig. 1 and Table 2).
Discussion
A detailed analysis of a newly identified FAE enzyme of the type A from metagenomic library is presented here. The recombinant 3A6 enzyme appeared to be a monomer of ∼37 kDa. Besides hydrolysing ester bonds, 3A6 efficiently catalysed the hydrolysis of pNP and cinnamate esters with a kcat/Km factor of ∼17. The ability to cleave both common and feruloylated esters is significantly different to that shown by homologous proteins known in bibliography and databases: those enzymes behave as common esterases and the capacity to hydrolyse cinnamates has not been reported in any of them. Here, creation of a number of complementary CE and FAE‐selective variants with improved activity phenotypes was accomplished using a combination of modelling‐guided saturation mutagenesis and site‐directed mutagenesis. Specifically, modelling and structural alignment were used to identify residues responsible for controlling discrimination of substrate targets with similar type of bonds. Most important residues for substrate preference control were K281, D282, N316 and K317. Saturation mutagenesis at those residues proved successful in the generation of CE (i.e. D282L and N316STOP) and FAE (i.e. K281I) selective hydrolytic mutants. Moreover, site‐directed mutagenesis approaches enable the accumulation of synergistic mutations D282L/N316STOP, D282L/N316STOP/K317H, D282L/K317H and N316STOP/K317H, resulting in a CE‐like mutant with virtually no FAE activity and K281I/D282E and K281I/D282E/K317D with inverted substrate preference (kcat/Km ratio lower than 1.0). Therefore, only four amino acids are required to produce both the most CE‐ and FAE‐selective enzymes. This is a remarkable result which highlights the relative ease by which specificities of esterases can be interchanged, in a good agreement with previous studies (Levasseur et al., 2006).
Since K281, D282, N316 and K317 are situated at specific loci within the catalytic core, their effect in promoting substrate promiscuity can be analysed in combination with the 3D model. K281I exerts a perturbation in the final access tunnel to the catalytic centre, generating a high (34‐fold) or mild (1.4‐fold) improvement in catalytic efficiency for FAE‐ and CE‐like substrates respectively (Fig. 4A and B). Saturation mutagenesis at the position 281 confirmed Ile as the best possible amino acid substitution for improving FAE phenotype. The Ile is a smaller residue making more accessible substrate channel that may correlate with higher values of kcat, whereas the substitution of a hydrophilic (Lys) by a hydrophobic (Ile) residue may correlate with enhanced binding of the substrates (lower Km values). This observation can also be extrapolated to the variant D282L (hydrophilic→hydrophobic); however, here, the slightly larger size of Asp than that of Leu appears to have more impact towards smaller substrates such as pNPC2 (Fig. 4C and D). Lys317His appear to have a significant negative impact on the properties of the enzyme since the mutation was associated with a complete loss of FAE activity. The effect on pNP esters was mostly associated with a twofold decrease in substrate affinity while slightly affecting the reaction rates (kcat). Since it was suggested that the presence of at least one m/p‐methoxy group in the cinnamate‐like substrates is required for binding (Tarbouriech et al., 2005), one could argue that the mutation at His317 may cause a partial unwinding of the loop and shortening the distance between binding residues and cinnamate‐like substrates while maintaining the activity with common substrates (Fig. 4E and F). The mutation Asn316STOP produced a deletion of the C‐terminal tail (25 amino acids) (Fig. 4G and H). This fragment constitutes an α‐helix located back to the catalytic cavity. Although, the deletion does not seem to be directly relevant to the catalytic core, we suggest that it may confer a higher flexibility, making catalysis more efficient, in particular, with shorter substrates. To further assess whether the higher flexibility is produced at expenses of lower structural stability, as suggested by the model, we studied the biophysical parameters of this protein variant after pre‐incubation with different concentrations of guanidinium chloride (GdmCl) and at different temperatures. We showed that this variant tends to misfold to a large degree after pre‐incubation with ≥ 0.64 M GdmCl whereas the wild‐type protein was mostly chemically stable (misfolding occurred ≥ 2.1 M GdmCl) (Fig. S7).
Figure 4.
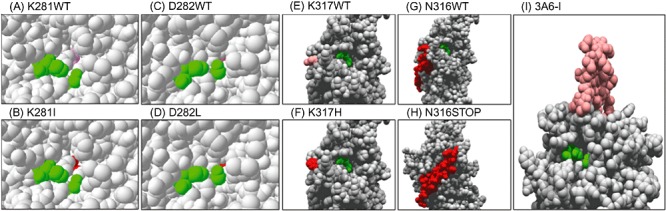
Surface representation of the substrate access pathways in the 3A6 protein. The upper panel (A, C, E, G) corresponds to the wild‐type protein whereas the bottom panel (B, D, F, H) correspond to the model containing the corresponding mutation. Panels G and H represent the wild‐type protein oriented with a difference of 90°C (in red is shown the C‐terminal part which is removed after N316STOP mutation). In all cases, the catalytic core is shown in green colour, where as the original or new introduced mutation are shown in pink or red colour. Panel I illustrates the view of the Gly178‐Gly211 insertion related to the catalytic core (green).
Our results also show that the hybrid 3A6 protein can also be converted into a common CE enzyme through the deletion of 34‐AA‐long loop. The functional cost of the Gly178‐Gly211 insert was offset by a large change in the substrate channel that enables higher efficiency of exclusion of the shorter substrates (Fig. 4I). This configuration apparently provides an effective solution to re‐channelling longer substrates to the active site. It is also likely to hamper the diffusion of shorter‐chain pNP esters to and from the active site, explaining the observed difference in hydrolytic rates and binding efficiency and the inactivity with the cinnamate substrates. We assume that this additional insertion may have been acquired as a rudimentary cap in providing considerable structural and functional substrate variability, which may also corroborate their role in the phylogenetic separation of the 3A6‐like proteins (Fig. S1).
The present study suggests that mutations at specific amino acids were most effective for promoting substrate specificity compared with the random in vitro evolution. Whatever the case, independently of the mutagenesis method applied, the CE phenotype was present in all variants, in contrast to the gain or loss of FAE activity. This view is supported by the fact that after point mutations the activity could be fully abolished towards cinnamates, but not towards pNPC2. Moreover, substrate discrimination appears to be associated to energy barriers. Free energy contribution analysis revealed that the energy barriers to affect CE‐like activity were mutation‐independent, whereas it is energetically more favourable (negative ΔΔG) to create variants with improved FAE phenotype (Fig. 3C). In this context, it is possible to assume that the 3A6 enzyme may have a CE‐like origin and that the newly evolved enzyme accumulated mutations in the vicinity of the original active site leading to an increase of its activity towards cinnamates (energetically favourable –Fig. 3C) without compromising its original esterase activity (energetically independent –Fig. 3C). We hypothesize that this scenario might be perhaps a consequence of the environmental conditions in vivo: earthworm gut microorganisms require an efficient enzymatic machinery to cope with a continuous flux of complex polymeric materials from the soil and from plant litter passing through the intestinal tract, thus demanding an instant response to adapt their enzymatic machinery to the changing environmental conditions. In this context, a substrate selection pressure applied may allow the creation/evolution of enzyme variants with broader substrate range but not necessarily high specificity (Levasseur et al., 2006). Our saturation mutagenesis experiments on 3A6 may have thus mimicked a possible scenario of natural evolution towards creating cinammate‐selective enzymes from a ‘common’ carboxyl esterase and directly evidenced the close evolutionary relationship between both enzymes. Finally, the results obtained here are intriguing from both an academic and biotechnological point of view and also highlights the importance of uncultured microbial resources (Vieites et al., 2009) to access novel functionalities.
Experimental procedures
A full description of experimental procedures is available in Supporting information.
Protein samples
All hydrolases used in the present study (wild‐type and variants) were cloned into pET‐41 Ek/LIC vector, expressed with an N‐terminal fusion to 6xHis tag and purified as described in Supporting information.
Enzyme characterization
Unlike indicated otherwise, hydrolytic activities were routinely measured and kinetic parameters determined as described by López‐Cortés and colleagues (2007). Reaction conditions were: [E]o = 0–12 nM, [substrate] ranging from 0 to 50 mM, 100 mM Tris‐HCl, pH 8.5, T = 40°C. Molecular mass of the 3A6 protein of 37.502 Da was considered for kinetic parameter calculations. Kinetic parameters were calculated from the Hanes–Woolf plot. Results shown are the average of three independent assays ± the standard deviation.
Library construction and FAE screen
Earthworms were collected at the surface level (0–20 cm) from the Ecological Soil Station of the Lomonosov Moscow State University (Solnechnogorskiy District, Moscow Region, Russia). The worms were maintained in a terrarium at 12–15°C during 3 months and were fed with sterilized birch litter. The gut contents were collected after transferring worms onto a wet filter paper and keeping them at 4°C for 1 h. Approximately 0.5 g of the gut content was inoculated into the 1 l Erlenmeyer flask containing 500 ml of Getchinson medium (K2HPO3, 1.3 g; MgSO4×7H2O, 0.3 g; CaCl2×6H2O, 0.1 g; FeCl2×6H2O, 0.01 g; NaNO3, 2.5 g; water 1 l; pH 7.2–7.4). The only carbon and energy source was the cellulose supplemented as a disk of filter paper of approximately 20 cm in diameter (Whatman 3MM), which was submerged in the medium. The enrichment was performed without shaking at 12°C until the filter paper was totally degraded (10 days). The DNA was extracted from 50 ml of this enrichment using G'NOME DNA Extraction Kit (Qbiogene). A fosmid library using CopyControl Genomic Library Production Kit was established in pCCFOS vector and E. coli EPI300‐T1R according to the protocols of the supplier (Epicentre). Fosmid clones, in total about 30 000, were picked with Qpix2 (Genetix, UK) and deposited in 384‐microtiter plates containing Luria–Bertani (LB) medium with chloramphenicol (12.5 µg ml−1) and 15% (v/v) glycerol. To screen for FAE activity the clones were replicated onto large (22.5 × 22.5 cm) square‐shaped LB agar plates containing chloramphenicol (12.5 µg ml−1) (Qtray, Genetix, UK). In total, each plate contained 2304 clones. After overnight incubation at 37°C, the plates were overlaid with 20 ml of 5 mM EPPS buffer containing 0.4% (w/v) agarose, 0.456 mM phenol red and 320 µl of MF (120 mg ml−1 in dimethylformamide). A hydrolase‐positive colony exhibiting a strong yellow halo after 2 min was picked, and the insert containing the hydrolase was sequenced after subcloning in pUC19 vector.
Rationale for mutagenesis
To understand structural backgrounds of substrate specificity we used four different mutagenesis approaches.
For site‐directed mutagenesis, mutations were introduced into the gene 3A6 using the QuikChange mutagenesis kit (Stratagene) and the plasmid pUC19‐3A6 as template with appropriate oligonucleotide pairs (s. details in Table S2). The resulting plasmids variants were then transferred into E. coli XL10 Gold and selected on the LB agar supplemented with 100 µg ml−1 ampicillin (Amp).
Fragment deletion in the 3A6 gene was carried out using a reverse PCR deletion, with pGEM‐3A6 as template, PfuTurbo DNA polymerase and the pair of primers ΔGly178‐Gly211Fwd and ΔGly178‐Gly211Rev (Table S2). Conditions were as follows: 95°C – 120 s, 30×[95°C – 45 s, 50°C – 60 s, 72°C – 120 s], 72°C – 500 s. The PCR product was purified, diluted to a concentration of 5 ng µl−1, further self‐ligated with T4 DNA ligase in a total volume of 100 µl at room temperature, and transformed into E. coli TOP10. The resulting transformants were plated onto fresh LB agar plate containing 100 µg ml−1 Amp.
Saturation mutagenesis libraries were constructed using the QuikChange site‐directed mutagenesis protocol (Stratagene), the pGEM‐3A6 as emplate and appropriate pair of degenerated primers listed in Table S2. For each position subjected to the mutagenesis, the mutated plasmids were transformed in E. coli TOP10 (Invitrogen) and the resulting transformants plated onto fresh LB agar plate containing 100 µg ml−1 Amp.
Error‐prone PCR mutagenesis (epPCR) was carried out using Taq polymerase (Sigma Chemical Co.) and pUC19‐3A6 as template. The reaction was performed in 50 µl volume and contained 3% dimethyl sulfoxide (DMSO), 5 µM MnCl2, 1.5 mM MgCl2, 0.3 mM dNTPs, 2.5 U Taq polymerase, 5 ng of template and 4.5 pmol of oligonucleotides pet41Fwd and pet41Rev (Table S2). The concentration of template and MnCl2 was adjusted to achieve a mutation rate from 1 to 3 mutations per kb. The amplification program was as follows: 2 min at 95°C, 27 s at 94°C, 27 s at 53°C, followed by 28 cycles of 3 min at 74°C, and 10 min at 74°C. The amplified PCR products were purified from a 0.75% agarose gel using QIAEX II gel extraction kit from Qiagen, cloned into the plasmid pGEM (Promega) and transformed into E. coli TOP10 (Invitrogen) as recommended by the supplier, and the resulting transformants were plated onto LB agar plate containing 100 µg ml−1 Amp.
Clones from saturation mutagenesis and epPCR libraries were picked and growth in 96‐well microtiter plates containing LB‐100 µg ml−1 Amp. Plates were covered with gas‐permeable films (Abgene) and incubated at 37°C with shaking at 300 r.p.m. for 16–20 h. The cultures were lysed in the plate wells using 50 µl of FastBreak reagent (Novagen) and 0.1 unit ml−1 of DN aseI, at room temperature for 30 min with shaking at 300 r.p.m. These lysates were used to determine CE‐ and FAE enzymatic activities in a BioTek Synergy HT spectrophotometer, with pNPC2 and MF as substrates respectively (López‐Cortés et al., 2007).
Free energy calculations
The quantitative effects of mutations on kinetics are expressed as changes in free energy compared with that of the wild‐type enzymes and were calculated as described elsewhere (Numata and Kimura, 2001; Bethel et al., 2006; Williams et al., 2006). Differences in the free energy change (ΔΔG) caused by mutations were calculated using the equations:
where R is the gas constant (8.31 J kmol−1), T is the absolute temperature (317 K) and ΔΔGA and ΔΔGB are the differential free activation and binding energies respectively. In order to analyse the mutual interplay of amino acids involved in pNPC2 and MF binding and cleavage, the free energy barriers to substrate specificity introduced by single, double and triple mutations were analysed. When the sum of the ΔΔG values for single mutants is equal to that of the double/triple mutant [ΔΔG(X + Y) = ΔΔG(X) + ΔΔG(Y)] the sites function independently.
Acknowledgments
This research was supported by the Spanish MEC BIO2006‐11738, CSD2007‐00005 and GEN2006‐27750‐C‐4‐E projects. A.B. thanks the Spanish MEC for a FPU fellowship. O.V.G. was supported by Grant 0313751K from the Federal Ministry for Science and Education (BMBF) within the GenoMikPlus initiative. We also thank Rita Getzlaff (HZI) for protein sequence analyses.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Fig. S1. Phylogenetic affiliation of 3A6 protein with related putative and experimentally analysed hydrolytic enzymes.
Fig. S2. Temperature (A) and pH (B) optima of the wild-type 3A6 protein.
Fig. S3. Overall three-dimensional structure of the 3A6 protein, as obtained by homology modelling.
Fig. S4. Alignment of 3A6 with protein PDB 2C7B.
Fig. S5. Multiple alignment of the C-terminal part of 3A6 esterase and related enzymes.
Fig. S6. Far-UV CD spectra of 3A6 wild-type protein and 3A6-3I variant.
Fig. S7. Stability effect by a single mutation Asn316STOP.
Table S1. Annotation table of proteins homologous to the 3A6 protein sequence (blast search).
Table S2. List of primers used in this study.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Aurilia V., Parracino A., Saviano M., Rossi M., D'Auria S. The psychrophilic bacterium Pseudoalteromonas halosplanktis TAC125 possesses a gene coding for a cold‐adapted feruloyl esterase activity that shares homology with esterase enzymes from gamma‐proteobacteria and yeast. Gene. 2007;397:51–57. doi: 10.1016/j.gene.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Aurilia V., Parracino A., D'Auria S. Microbial carbohydrate esterases in cold adapted environments. Gene. 2008;410:234–240. doi: 10.1016/j.gene.2007.12.019. [DOI] [PubMed] [Google Scholar]
- Benoit I., Danchin E.G., Bleichrodt R.J., De Vries R.P. Biotechnological applications and potential of fungal feruloyl esterases based on prevalence, classification and biochemical diversity. Biotechnol Lett. 2008;30:387–396. doi: 10.1007/s10529-007-9564-6. [DOI] [PubMed] [Google Scholar]
- Bethel C.R., Hujer A.M., Hujer K.M., Thomson J.M., Ruszczycky M.W., Anderson V.E. Role of Asp104 in the SHV beta‐lactamase. Antimicrob Agents Chemother. 2006;50:4124–4131. doi: 10.1128/AAC.00848-06. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornscheuer U.T. Microbial carboxyl esterases: classification, properties and application in biocatalysis. FEMS Microbiol Rev. 2002;26:73–81. doi: 10.1111/j.1574-6976.2002.tb00599.x. [DOI] [PubMed] [Google Scholar]
- Crepin V.F., Faulds C.B., Connerton I.F. Functional classification of the microbial feruloyl esterases. Appl Microbiol Biotechnol. 2004;63:647–652. doi: 10.1007/s00253-003-1476-3. [DOI] [PubMed] [Google Scholar]
- Faulds C.B., Molina R., Gonzalez R., Husband F., Juge N., Sanz‐Aparicio J., Hermoso J.A. Probing the determinants of substrate specificity of a feruloyl esterase, AnFaeA, from Aspergillus niger. FEBS J. 2005;272:4362–4371. doi: 10.1111/j.1742-4658.2005.04849.x. [DOI] [PubMed] [Google Scholar]
- Fazary A.E., Ju Y.H. Feruloyl esterases as biotechnological tools: current and future perspectives. Acta Biochim Biophys Sin. 2007;39:811–828. doi: 10.1111/j.1745-7270.2007.00348.x. [DOI] [PubMed] [Google Scholar]
- Funke S.A., Otte N., Eggert T., Bocola M., Jaeger K.E., Thiel W. Combination of computational prescreening and experimental library construction can accelerate enzyme optimization by directed evolution. Protein Eng Des Sel. 2005;18:509–514. doi: 10.1093/protein/gzi062. [DOI] [PubMed] [Google Scholar]
- Hermoso J.A., Sanz‐Aparicio J., Molina R., Juge N., González R., Faulds C.B. The crystal structure of feruloyl esterase A from Aspergillus niger suggests evolutive functional convergence in feruloyl esterase family. J Mol Biol. 2004;338:495–506. doi: 10.1016/j.jmb.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Ivancic M., Valinger G., Gruber K., Schwab H. Inverting enantioselectivity of Burkholderia gladioli esterase EstB by directed and designed evolution. J Biotechnol. 2007;129:109–122. doi: 10.1016/j.jbiotec.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Kanauchi M., Watanabe S., Tsukada T., Atta K., Kakuta T., Koizumi T. Purification and characteristics of feruloyl esterase from Aspergillus awamori G‐2 strain. J Food Sci. 2008;73:C458–463. doi: 10.1111/j.1750-3841.2008.00839.x. [DOI] [PubMed] [Google Scholar]
- Koseki T., Takahashi K., Fushinobu S., Lefuji H., Iwano K., Hasihuze K., Matsuzawa H. Mutational analysis of a feruloyl esterase from Aspergillus awamori involved in substrate discrimination and pH dependence. Biochim Biophys Acta. 2005;1722:200–208. doi: 10.1016/j.bbagen.2004.12.016. [DOI] [PubMed] [Google Scholar]
- Koseki T., Hori A., Seki S., Murayama T., Shiono Y. Characterization of two distinct feruloyl esterases, AoFaeB and AoFaeC, from Aspergillus oryzae. Appl Microbiol Biotechnol. 2009;83:689–696. doi: 10.1007/s00253-009-1913-z. [DOI] [PubMed] [Google Scholar]
- Levasseur A., Gouret P., Lesage‐Meessen L., Asther M., Record E., Pontarotti P. Tracking the connection between evolutionary and functional shifts using the fungal lipase/feruloyl esterase A family. BMC Evol Biol. 2006;6:92. doi: 10.1186/1471-2148-6-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López‐Cortés N., Reyes‐Duarte D., Beloqui A., Polaina J., Ghazi I., Golyshina O.V. Catalytic role of conserved HQGE motif in the CE6 carbohydrate esterase family. FEBS Lett. 2007;581:4657–4662. doi: 10.1016/j.febslet.2007.08.060. et al. [DOI] [PubMed] [Google Scholar]
- Manco G., Mandrich L., Rossi M. Residues at the active site of the esterase 2 from Alicyclobacillus acidocaldarius involved in substrate specificity and catalytic activity at high temperature. J Biol Chem. 2001;276:37482–37490. doi: 10.1074/jbc.M103017200. [DOI] [PubMed] [Google Scholar]
- Mee‐Hie Cho C., Mulchandani A., Chen W. Functional analysis of organophosphorus hydrolase variants with high degradation activity towards organophosphate pesticides. Protein Eng Des Sel. 2006;19:99–105. doi: 10.1093/protein/gzj007. [DOI] [PubMed] [Google Scholar]
- Moukouli M., Topakas E., Christakopoulous P. Cloning, characterization and functional expression of an alkalitolerant type C feruloyl esterase from Fusarium oxysporum. Appl Microbiol Biotechnol. 2008;79:245–254. doi: 10.1007/s00253-008-1432-3. [DOI] [PubMed] [Google Scholar]
- Nakagawa Y., Hasegawa A., Hiratake J., Sakata K. Engineering of Pseudomonas aeruginosa lipase by directed evolution for enhanced amidase activity: mechanistic implication for amide hydrolysis by serine hydrolases. Protein Eng Des Sel. 2007;20:339–346. doi: 10.1093/protein/gzm025. [DOI] [PubMed] [Google Scholar]
- Numata T., Kimura M. Contribution of Gln9 and Phe80 to substrate binding in ribonuclease MC1 from bitter gourd seeds. J Biochem. 2001;130:621–626. doi: 10.1093/oxfordjournals.jbchem.a003027. [DOI] [PubMed] [Google Scholar]
- Reyes‐Duarte D., Polaina J., López‐Cortés N., Alcalde M., Plou F.J., Elborough K. Conversion of a carboxylesterase into a triacylglycerol lipase by a random mutation. Angew Chem Int Ed Engl. 2005;44:7553–7557. doi: 10.1002/anie.200502461. et al. [DOI] [PubMed] [Google Scholar]
- Shin H.D., Chen R.R. A type B feruloyl esterase from Aspergillus nidulans with broad pH applicability. Appl Microbiol Biotechnol. 2007;73:1323–1330. doi: 10.1007/s00253-006-0612-2. [DOI] [PubMed] [Google Scholar]
- Tarbouriech N., Prates J.A.M., Fintes C.M.G.A., Davis G.J. Molecular determinants of substrate specificity in the feruloyl esterase module of xylanase 10B from Clostridium thermocellum. Acta Cryst. 2005;61:194–197. doi: 10.1107/S0907444904029695. [DOI] [PubMed] [Google Scholar]
- Vieites J.M., Guazzaroni M.E., Beloqui A., Golyshin P.N., Ferrer M. Metagenomics approaches in systems microbiology. FEMS Microbiol Rev. 2009;33:236–255. doi: 10.1111/j.1574-6976.2008.00152.x. [DOI] [PubMed] [Google Scholar]
- Wang Q., Yang G., Liu Y., Feng Y. Discrimination of esterase and peptidase activities of acylaminoacyl peptidase from hyperthermophilic Aeropyrum pernix K1 by a single mutation. J Biol Chem. 2006;281:18618–18625. doi: 10.1074/jbc.M601015200. [DOI] [PubMed] [Google Scholar]
- Wang X., Geng X., Egashira Y., Sanada H. Purification and characterization of a feruloyl esterase from the intestinal bacterium Lactobacillus acidophilus. Appl Environ Microbiol. 2004;70:2367–2372. doi: 10.1128/AEM.70.4.2367-2372.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams G.J., Woodhall T., Fanrswirth L.M., Nelson A., Berry A. Creation of a pair of stereochemically complementary biocatalysts. J Am Chem Soc. 2006;128:16238–16247. doi: 10.1021/ja065233q. [DOI] [PubMed] [Google Scholar]
- Wong D.W. Feruloyl esterase: a key enzyme in biomass degradation. Appl Biochem Biotechnol. 2006;133:87–112. doi: 10.1385/abab:133:2:87. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Phylogenetic affiliation of 3A6 protein with related putative and experimentally analysed hydrolytic enzymes.
Fig. S2. Temperature (A) and pH (B) optima of the wild-type 3A6 protein.
Fig. S3. Overall three-dimensional structure of the 3A6 protein, as obtained by homology modelling.
Fig. S4. Alignment of 3A6 with protein PDB 2C7B.
Fig. S5. Multiple alignment of the C-terminal part of 3A6 esterase and related enzymes.
Fig. S6. Far-UV CD spectra of 3A6 wild-type protein and 3A6-3I variant.
Fig. S7. Stability effect by a single mutation Asn316STOP.
Table S1. Annotation table of proteins homologous to the 3A6 protein sequence (blast search).
Table S2. List of primers used in this study.