

Summary
Enantiomerically pure β‐arylalkyl carboxylic acids are important synthetic intermediates for the preparation of a wide range of compounds with biological and pharmacological activities. A library of 83 enzymes isolated from the metagenome was searched for activity in the hydrolysis of ethyl esters of three racemic phenylalkyl carboxylic acids by a microtiter plate‐based screening using a pH‐indicator assay. Out of these, 20 enzymes were found to be active and were subjected to analytical scale biocatalysis in order to determine their enantioselectivity. The most enantioselective and also enantiocomplementary biocatalysts were then used for preparative scale reactions. Thus, both enantiomers of each of the three phenylalkyl carboxylic acids studied could be obtained in excellent optical purity and high yields.
Introduction
Enantiomerically pure β‐arylalkyl carboxylic acids are important synthetic intermediates for the preparation of a wide range of compounds with biological and pharmacological activities (Varadharaj et al., 1998; Kamal et al., 2007). For instance, (R)‐(−)‐3‐phenylbutyric acid was used in the synthesis of (−)‐Malyngolide (Kogure and Eliel, 1984), a δ‐lactone antibiotic of algal origin. Both enantiomers of 3‐phenylbutyric acid 1a have been used for the preparation of the four stereoisomers of β‐methylphenylalanine, as well as for the synthesis of chiral alkyl‐indanones and the corresponding dihydrocoumarins (Stephan et al., 1994). Enantiomerically pure 3‐phenylbutyric acids and their derivatives such as 3‐arylbutanol are synthetic intermediates for the preparation of aromatic sesquiterpenes of the bisabolene family (Fuganti et al., 1999; Fuganti and Serra, 2000), as well as useful building blocks in organic synthesis (Mori, 2005). The (S)‐enantiomer of 2‐phenylpropionic acid is the basic structure of profens (e.g., Ibuprofen, Naproxen), which are potent non‐steroidal anti‐inflammatory drugs (Chavda et al., 2007).
Thus, enantiopure arylalkyl carboxylic acids are attractive synthetic targets and a variety of methodologies have been developed to obtain these compounds. Chemical syntheses (Sonawane et al., 1992) like the asymmetric hydrogenation of the unsaturated derivatives (Sun et al., 2007) or Friedel‐Crafts alkylation of aromatic precursors (Piccolo et al., 1991) proved to be effective. However, a biocatalytic synthetic route would not only offer an alternative access, but also represents an environmentally more benign method. The preparation of pure enantiomers of these carboxylic acids can be performed through kinetic resolution of their esters with enantioselective hydrolases (esterases or lipases), which are still the enzyme class most widely used in organic synthesis (Faber, 2004; Bornscheuer and Kazlauskas, 2006). So far, lipases selective towards one enantiomer of the phenylalkyl carboxylic acids 1a–3a (see Fig. 1) and their derivatives have been successfully applied in kinetic resolutions (Varadharaj et al., 1998; Kamal et al., 2007) and dynamic kinetic resolutions of their thioesters (Um and Drueckhammer, 1998). Very recently a remarkable non‐enzymatical approach was published using crystallization with abrasive grinding for complete deracemization of a naproxen ester (Noorduin et al., 2009).
Figure 1.
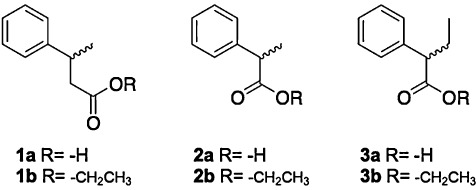
Racemic phenylalkyl carboxylic acids 1a–3a studied.
The identification of hydrolases with complementary enantioselectivity would allow a more efficient preparation of the opposite enantiomer and the required subsequent hydrolysis of the remaining substrate can be avoided.
Besides classical screening of microorganisms to find suitable enzymes, protein engineering using rational design or directed evolution is a promising and often successful tool to create the desired biocatalyst (Bornscheuer, 2002; Arnold and Georgiou, 2003). The screening of metagenomic libraries (Lorenz et al., 2003; Gabor et al., 2007) is an efficient alternative to find novel biocatalysts of diverse evolutionary origin. It has the additional advantage that the enzymes identified are already recombinantly expressed. In former studies, the usefulness of this tool for the successful identification of highly selective enzymes has been already demonstrated in our group (Kourist et al., 2007; 2008; Brüsehaber et al., 2008). Especially the fast proceeding in comparison with protein design makes metagenomic mining the method of choice where short time‐to‐market is required, which is often the case for fine chemicals or pharmaceutical intermediates.
Herein we present the screening of metagenome‐derived esterases (Schmeisser et al., 2003; Lorenz and Eck, 2005; Elend et al., 2006; Steele et al., 2009) for the preparation of both enantiomers of the phenylalkyl carboxylic acids 1a–3a by hydrolysis of the corresponding ethyl esters 1b–3b.
Results and discussion
Identification of metagenome‐derived hydrolases active towards the phenylalkyl carboxylic acid ethyl esters
In the first step, a library of 83 esterases from metagenomic origin was screened for activity towards the ethyl esters 1b–3b using a microtiter plate‐based method (Baumann et al., 2000) with bromothymol blue serving as pH indicator. This led to the identification of 20 enzymes, which were then subjected to analytical scale kinetic resolutions for the determination of enantioselectivity and ‐preference by gas chromatographic analysis. The best hits are shown in Fig. 2. Next, the reaction conditions were further optimized by variation of reaction temperature (from 30°C to 20°C) and the addition of the cosolvent DMSO. Thus, at least two enzymes were found for each substrate exhibiting high enantioselectivity (E‐values > 50) and complementary enantiopreference (Table 1).
Figure 2.
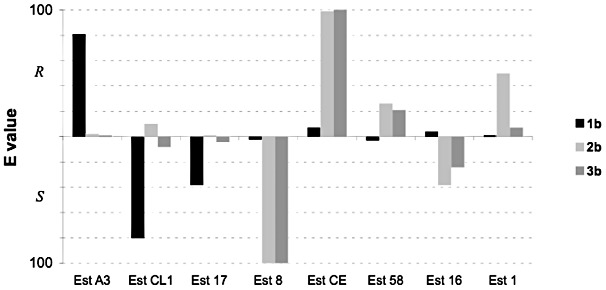
Esterases showing the highest enantioselectivity and opposite enantiopreference in the kinetic resolutions of substrates 1b–3b.
Table 1.
Results of kinetic resolutions of substrates 1b–3b in analytical scale.
| Substrate | Esterase | Ua | T (°C) | t (h) | eeSd (%) | eePe (%) | Cf (%) | Ef | Configuration |
|---|---|---|---|---|---|---|---|---|---|
| 1b | CL1 | 123b | 20g | 3 | 93 | 94 | 49 | > 100 | (−), R |
| 1b | A3 | 17b | 20g | 1.5 | 93 | 93 | 50 | 96 | (+), S |
| 1b | 17 | 0.1b | 30 | 1.5 | 88 | 80 | 48 | 38 | (−), R |
| 2b | 8 | 21b | 20g | 1.5 | 93 | 91 | 51 | 80 | (−), R |
| 2b | CE | 10b | 30g | 8 | 92 | 89 | 51 | 61 | (+), S |
| 2b | 58 | 0.07b | 30 | 1.5 | 82 | 81 | 49 | 26 | (+), S |
| 2b | 16 | 11b | 20 | 1.5 | 99 | 2 | 2 | n.d. | (−), R |
| 2b | 1 | 0.42b | 20 | 1.5 | 95 | 14 | 13 | 49 | (+), S |
| 3b | 8 | 21c | 30g | 8 | 98 | 97 | 50 | > 200 | (−), R |
| 3b | CE | 10c | 30g | 8 | 88 | 99 | 47 | > 200 | (+), S |
| 3b | 58 | 0.07c | 20 | 8 | 91 | 3 | 3 | n.d. | (+), S |
| 3b | 16 | 11c | 20 | 8 | 88 | 40 | 31 | 24 | (−), R |
Determined using p‐nitrophenyl acetate.
Substrate (20 mM).
Substrate (40 mM).
Determined by GC analysis.
Determined by GC analysis after derivatization with TMS diazomethane.
Calculated according to Chen and colleagues (1982).
In the presence of 10% DMSO.
n.d., not determined due to very low conversion.
Two enzymes, esterases CL1 and A3, showed high selectivity towards ethyl‐3‐phenyl butanoate 1b, whereas esterases CE and Est8 were most useful for the kinetic resolution of ethyl‐2‐phenyl propanoate 2b and the butanoate 3b. Surprisingly, in the case of esterases CE and Est8, the presence of DMSO increased the enzyme activity, without affecting significantly the enantioselectivity, probably due to the better solubility of the substrates in the reaction media. Finally, the kinetic resolutions were performed in preparative (1 g) scale (Table 2).
Table 2.
Results of kinetic resolutions of substrates 1b–3b in preparative scale (in the presence of 10% DMSO).
| Substrate | Esterase | kUa | T (°C) | t (h) | eeSc (%) | eePd (%) | Ce (%) | Ee | Configuration |
|---|---|---|---|---|---|---|---|---|---|
| 1b | CL1 | 32b | 20 | 3 | 98 | 87 | 47 | > 200 | (−), R |
| 1b | A3 | 4b | 20 | 3 | 72 | 59 | 45 | 11 | (+), S |
| 2b | 8 | 6b | 20 | 1.5 | 92 | 56 | 62 | 11 | (−), R |
| 2b | CE | 3b | 30 | 8 | 82 | 97 | 46 | > 100 | (+), S |
| 3b | 8 | 3b | 30 | 8 | 97 | 95 | 50 | > 100 | (−), R |
| 3b | CE | 1b | 30 | 8 | 69 | > 99 | 41 | > 200 | (+), S |
Determined using p‐nitrophenyl acetate.
Substrate (40 mM).
Determined by GC analysis.
Determined by HPLC analysis.
Calculated according to Chen and colleagues (1982).
Esterases CE and A3 were thoroughly characterized before (Elend et al., 2006). Esterase Est8 was identified by an activity‐based screening of a metagenomic library using tributyrin as substrate. The library was set up from a soil sample collected from a vineyard near Weinheim, Germany. The protein comprises 247 amino acids giving a calculated molecular weight of 26.4 kDa. The primary structure shows a Gly–X–Ser–X–Gly pentapeptide typical of the structural motive in the immediate proximity of the active‐site serine of α/β‐hydrolases (Ollis et al., 1992). Furthermore, a Gly–Gly–Gly–Y motive was identified N‐terminal to the above‐mentioned pentapeptide, indicating that this enzyme is a member of the so‐called ‘GGG(A)X‐type’ esterases. This group of enzyme was shown to be capable of the conversion of esters of tertiary alcohols (Henke et al., 2002; Kourist et al., 2008). Est8 showed the highest identity of 65% to an esterase/lipase from an uncultured bacterium (AY833090.1).
In addition to the high enantioselectivity reported above, initial biochemical characterization of Est8 was also performed. Thus crude cell extracts of Escherichia coli expressing the gene encoding Est8 displayed the highest esterase activity at 50°C. Increasing the temperature further decreased the activity to about 10% at 70°C, 80°C and 90°C (Fig. 3).
Figure 3.
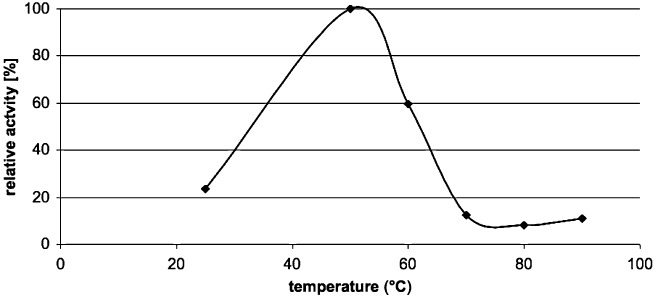
Activity profile of Est8. Crude cell extracts of E. coli cells expressing Est8 were incubated at the indicated temperatures and analysed spectrophotometrically for the conversion of p‐nitrophenylbutyrate at pH 8.0. The increase in absorbance was followed for 10 min and initial rates were determined. Each experiment was performed in duplicate. The highest activity determined was set to 100%.
Esterase CL1 was identified using the activity assay based on hydrolysis of tributyrin. This esterase was found in a metagenomic library isolated from biofilms in drinking water (Schmeisser et al., 2003); it comprises 318 amino acids resulting in its calculated molecular weight of 33 kDa. It reaches its maximum hydrolytic activity at pH 8, presenting the highest stability at pH 9–10. Further characterization and application of this protein are currently under investigation.
Conclusions
A library of esterases of metagenomic origin has been screened for enantioselectivity towards three phenylalkyl carboxylic acids. Among 83 candidates, efficient and highly enantioselective catalysts have been found for each substrate with complementary enantiopreference in all cases. This work demonstrates that the screening of environmental sources is a very promising tool for the identification of new biocatalysts with synthetically useful properties.
Experimental procedures
Materials
All chemicals were purchased from Sigma‐Aldrich (Steinheim, Germany). Chiral gas chromatography (GC) analyses were performed by using a Heptakis‐(2,3‐di‐O‐acetyl‐6‐O‐t‐butyldimethylsilyl)‐β‐cyclodextrin‐column (Macherey‐Nagel, Düren, Germany) on a GC‐14A gas chromatograph (Shimadzu, Tokyo, Japan) and by using a Heptakis‐(2,6‐O‐di‐O‐methyl‐3‐O‐pentyl)‐β‐cyclodextrin‐column (Macherey‐Nagel, Düren, Germany) on a GC‐2010 gas chromatograph (Shimadzu, Tokyo, Japan) equipped with an AOC‐20i Autoinjector (Shimadzu, Tokyo, Japan). Chiral HPLC analyses were performed by using the column OD‐H (Daicel Chemical Industries, Osaka, Japan) with n‐hexane and 2‐propanol as solvent on a Hitachi Elite LaChrom device (Hitachi High Technologies America, San Jose, USA). All metagenomic esterases were produced and donated by B.R.A.I.N. AG (Zwingenberg, Germany) and used as glycerol‐stabilized crude cell extracts or as lyophilisates (Brüsehaber et al., 2008). Plasmids containing the genes for the esterases A3 (Elend et al., 2006), CE (Elend et al., 2006) and CL1 (Schmeisser et al., 2003) were cloned as described.
General procedure for the synthesis of the phenylalkyl carboxylic acid ethylesters
All ethyl esters were prepared by azeotropic esterification. One equivalent (18 mmol of 1a or 3a, and 20 mmol of 2a) of the phenylalkyl carboxylic acid was stirred with 3 eq. of ethanol at 110°C in toluene as solvent with some drops of concentrated sulfuric acid as catalyst. Water formed in the esterification reaction was removed by a water trap until completion. Next, the solvent was evaporated and the product was purified by silica gel column chromatography (n‐pentane/ethyl acetate, 10:1). Using this procedure, 1b was obtained as light yellow oil (1.8 g, 9.3 mmol, 52% yield), 2b was obtained as colourless oil (2.6 g, 14.6 mmol, 73% yield) and 3b was obtained as colourless oil (2.5 g, 13 mmol, 72% yield). The structures were confirmed by NMR spectroscopy and matched literature data (Yang et al., 1999).
High‐throughput screening
The test for hydrolytic activity towards the target substrates was based on a pH assay (Baumann et al., 2000). Wells were filled with 20 µl of a substrate stock solution (200 mM in DMSO), 5 µl of a solution of the pH indicator bromothymol blue (BTB, 16 mM in 16% ethanol and 84% water), 30 µl of a glycerol stock solution of the different esterases and 145 µl of sodium phosphate buffer (5 mM, pH 7.4) to get a final volume of 200 µl. Both positive and negative controls were present in each MTP. Positive controls contained 5 µl of BTB solution, 175 µl of sodium phosphate buffer (5 mM, pH 7.4) and 20 µl of DMSO containing the correspondent acids 1a–3a at final concentrations of 1–20 mM. Negative controls were identical, but without substrates. Each MTP was then sealed with adhesive foil, incubated at 37°C for 72 h and stirred at 400 r.p.m. in a thermoshaker (Eppendorf, Hamburg, Germany). Active esterases were then identified by visible colour changes and used in analytical scale experiments.
General procedure for esterase‐catalysed analytic scale kinetic resolutions
To a stirred solution of the ethyl esters 1b–3b (0.076 mmol of 1b or 2b, or 0.15 mmol of 3b) in 1.8 ml of sodium phosphate buffer (50 mM, pH 7.4) and 10% DMSO if necessary, 200 µl of esterase solution was added to a total volume of 2 ml. The reaction mixture was stirred at 1000 r.p.m. in a thermoshaker at 20°C or 30°C. Samples (400 µl) were taken after 2, 7 and 24 h. These samples were acidified with 20 µl of 1 N HCl and subsequently extracted three times with 400 µl diethyl ether. The combined organic layers were dried over anhydrous sodium sulfate and afterwards the organic solvent was evaporated in a Speed Vac (Thermo Fisher Scientific, Langenselbold, Germany). The mixture was then treated with TMS diazomethane (Hashimoto et al., 1981) in order to convert the carboxylic acid (hydrolysis product) in its methyl ester to allow analysis of substrate and product concentration and optical purity in one run by GC. Conversion and E values were then calculated from the enantiomeric excesses from substrate and product following the equations developed by Chen and colleagues (1982).
Esterase‐catalysed preparative‐scale kinetic resolution of 1b–3b
To a 40 mM solution of 1b–3b (∼1 g) in sodium phosphate buffer (50 mM, pH 7.4) containing 10% (v/v) DMSO, esterase (lyophilisate dissolved in buffer, units as given in Table 2) was added and hydrolysis was performed at 20°C or 30°C. After TLC analysis indicated 50% conversion, the reaction was stopped by addition volume of 1 N HCl, and substrate and product were extracted three times with diethyl ether. After drying the organic phase over anhydrous Na2SO4, the solvent was removed by evaporation under reduced pressure and the product was purified by column chromatography (n‐pentane/ethyl acetate). The purity of the biocatalysis products was confirmed by NMR spectroscopy and was in all cases > 99%.
Using this protocol, (R)(−)‐1b was obtained as a light yellow oil (411 mg, 21 mmol, 41.4% yield). Enantiomeric excess was determined after hydrolysis to 1a as 59%ee. (S)(+)‐1a was obtained as colourless oil (230 mg, 14 mmol, 27.7% yield, 72%ee); (S)(+)‐1b was obtained as light yellow oil (420 mg, 2.2 mmol, 42% yield). Enantiomeric excess was determined after hydrolysis to 1a as 87%ee; (R)(−)‐1a was obtained as light yellow oil (330 mg, 2 mmol, 38.5% yield, 98%ee); (R)(−)‐2b was obtained as colourless oil (164 mg, 9.2 mmol, 16.4% yield). Enantiomeric excess was determined after hydrolysis to 2a as 82%ee; (S)(+)‐2a was obtained as a white powder (181 mg, 12 mmol, 21.5% yield, 97%ee); (S)(+)‐2b was obtained as a colourless oil (218 mg, 12.2 mmol, 28.3% yield). Enantiomeric excess was determined after hydrolysis to 2a as 92%ee; (R)(−)‐2a was obtained as a white powder (180 mg, 12 mmol, 21.3%, 56%ee); (R)(−)‐3b was obtained as colourless oil (380 mg, 1.99 mmol, 38% yield). Enantiomeric excess was determined after hydrolysis to 3a as 69%ee; (S)(+)‐3a was obtained as yellow oil (360 mg, 2.18 mmol, 41.9% yield, > 99%ee); (S)(+)‐3b was obtained as colourless oil (150 mg, 0.78 mmol, 15% yield). Enantiomeric excess was determined after hydrolysis to 3a as 97%ee; (R)(−)‐3a was obtained after column chromatography (n‐pentane/ethyl acetate) as yellow oil (330 mg, 2 mmol, 39% yield, 95%ee).
Acknowledgments
We are grateful to the Deutsche Bundesstiftung Umwelt (Osnabrück, Germany) for financial support (AZ13198) and thank Anne Schätzchen and Julia Rick for their support in organic synthesis.
References
- Arnold F.H., Georgiou G. Humana Press; 2003. [Google Scholar]
- Baumann M., Hauer B.H., Bornscheuer U.T. Rapid screening of hydrolases for the enantioselective conversion of ‘difficult‐to‐resolve’ substrates. Tetrahedron: Asymmetry. 2000;11:4781–4790. [Google Scholar]
- Bornscheuer U.T. Methods to increase enantioselectivity of lipases and esterases. Curr Opin Biotechnol. 2002;13:543–547. doi: 10.1016/s0958-1669(02)00350-6. [DOI] [PubMed] [Google Scholar]
- Bornscheuer U.T., Kazlauskas R.J. Wiley‐VCH; 2006. [Google Scholar]
- Brüsehaber E., Böttcher D., Liebeton K., Eck J., Naumer C., Bornscheuer U.T. Asymmetric synthesis of cis‐3,5‐diacetoxycyclopent‐1‐ene using metagenome‐derived hydrolases. Tetrahedron: Asymmetry. 2008;19:730–732. [Google Scholar]
- Chavda S., Coumbarides G.S., Dingjan M., Eames J., Flinn A., Ghilagaber S. Synthesis of enantiomerically pure isotopomers of 2‐phenylpropionic acids. Chirality. 2007;19:366–373. doi: 10.1002/chir.20384. et al. [DOI] [PubMed] [Google Scholar]
- Chen C.S., Fujimoto Y., Girdaukas G., Sih C.J. Quantitative analyses of biochemical kinetic resolutions of enantiomers. J Am Chem Soc. 1982;104:7294–7299. [Google Scholar]
- Elend C., Schmeisser C., Leggewie C., Babiak P., Carballeira J.D., Steele H.L. Isolation and biochemical characterization of two novel metagenome‐derived esterases. Appl Environ Microbiol. 2006;72:3637–3645. doi: 10.1128/AEM.72.5.3637-3645.2006. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber K. Springer Verlag; 2004. [Google Scholar]
- Fuganti C., Serra S. Baker's yeast‐mediated enantioselective synthesis of the bisabolane sesquiterpenes (+)‐curcuphenol, (+)‐xanthorrhizol, (−)‐curcuquinone and (+)‐curcuhydroquinone. J Chem Soc, Perkin Trans 1. 2000:3758–3764. [Google Scholar]
- Fuganti C., Serra S., Dulio A. Baker's yeast mediated enantioselective synthesis of the bisabolane sesquiterpenes curcumene, turmerone, dehydrocurcumene and nuciferal. J Chem Soc, Perkin Trans 1. 1999:279–282. [Google Scholar]
- Gabor E., Liebeton K., Nielhaus F., Eck J., Lorenz P. Updating the metagenomics toolbox. Biotechnol J. 2007;2:201–206. doi: 10.1002/biot.200600250. [DOI] [PubMed] [Google Scholar]
- Hashimoto N., Aoyama T., Shioiri T. New methods and reagents in organic‐synthesis. 14. A simple efficient preparation of methyl esters with trimethylsilyldiazomethane (TMSCHN2) and its application to gas chromatographic analysis of fatty acids. Chem Pharm Bull. 1981;29:1475–1478. [Google Scholar]
- Henke E., Pleiss J., Bornscheuer U.T. Activity of lipases and esterases towards tertiary alcohols: insights into structure‐function relationships. Angew Chem Int Ed Engl. 2002;41:3211–3213. doi: 10.1002/1521-3773(20020902)41:17<3211::AID-ANIE3211>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Kamal A., Malik M.S., Shaik A.A., Azeeza S. Enantioselective synthesis of (R)‐ and (S)‐curcumene and curcuphenol: an efficient chemoenzymatic route. Tetrahedron: Asymmetry. 2007;18:2547–2553. [Google Scholar]
- Kogure T., Eliel E.L. A convergent asymmetric synthesis of (−)‐malyngolide and its three stereoisomers. J Org Chem. 1984;49:576–578. [Google Scholar]
- Kourist R., Krishna S.H., Patel J.S., Bartnek F., Hitchman T.S., Weiner D.P., Bornscheuer U.T. Identification of a metagenome‐derived esterase with high enantioselectivity in the kinetic resolution of arylaliphatic tertiary alcohols. Org Biomol Chem. 2007;5:3310–3313. doi: 10.1039/b709965g. [DOI] [PubMed] [Google Scholar]
- Kourist R., Nguyen G.S., Strübing D., Böttcher D., Liebeton K., Naumer C. Hydrolase‐catalyzed stereoselective preparation of protected α, α‐dialkyl‐α‐hydroxycarboxylic acids. Tetrahedron: Asymmetry. 2008;19:1839–1943. et al. [Google Scholar]
- Lorenz P., Eck J. Metagenomics and industrial applications. Nat Rev Microbiol. 2005;3:510–516. doi: 10.1038/nrmicro1161. [DOI] [PubMed] [Google Scholar]
- Lorenz P., Liebeton K., Niehaus F., Schleper C., Eck J. The impact of non‐cultivated biodiversity on enzyme discovery and evolution. Biocatal Biotransform. 2003;21:87–91. [Google Scholar]
- Mori K. Synthesis of (R)‐ar‐turmerone and its conversion to (R)‐ar‐himachalene, a pheromone component of the flea beetle: (R)‐ar‐himachalene is dextrorotatory in hexane, while levorotatory in chloroform. Tetrahedron: Asymmetry. 2005;16:1721–1721. [Google Scholar]
- Noorduin W.L., Kaptein B., Meekes H., Van Enckevort W.J., Kellogg R.M., Vlieg E. Fast attrition‐enhanced deracemization of naproxen by a gradual in situ feed. Angew Chem Int Ed Engl. 2009;48:4581–4583. doi: 10.1002/anie.200901386. [DOI] [PubMed] [Google Scholar]
- Ollis D.L., Cheah E., Cygler M.E., Eckstein E., Dijkstra B.W., Frolow F. The alpha/beta hydrolase fold. Protein Eng. 1992;5:197–211. doi: 10.1093/protein/5.3.197. et al. [DOI] [PubMed] [Google Scholar]
- Piccolo O., Azzena U., Melloni G., Delogu G., Valoti E. Stereospecific Friedel‐Crafts alkylation of aromatic compounds: synthesis of optically active 2‐and 3‐arylalkanoic esters. J Org Chem. 1991;56:183–187. [Google Scholar]
- Schmeisser C., Stockigt C., Raasch C., Wingender J., Timmis K.N., Wenderoth D.F. Metagenome survey of biofilms in drinking‐water networks. Appl Environ Microbiol. 2003;69:7298–7309. doi: 10.1128/AEM.69.12.7298-7309.2003. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonawane H.R., Bellur N.S., Ahuja J.R., Kulkarni D.G. Recent developments in the synthesis of optically active alpha‐arylpropanoic acids – an important class of nonsteroidal antiinflammatory agents. Tetrahedron: Asymmetry. 1992;3:163–192. [Google Scholar]
- Steele H.L., Jaeger K.E., Daniel R., Streit W.R. Advances in recovery of novel biocatalysts from metagenomes. J Mol Microbiol Biotechnol. 2009;16:25–37. doi: 10.1159/000142892. [DOI] [PubMed] [Google Scholar]
- Stephan E., Rocher R., Aubouet J., Pourcelot G., Cresson P. Preparation of chiral indanones and dihydrocoumarins – application to synthesis of (+)‐3‐(2,6‐dimethoxyphenyl) pentanoic acid. Tetrahedron: Asymmetry. 1994;5:41–44. [Google Scholar]
- Sun X.F., Zhou L., Wang C.J., Zhang X.M. ) Rh‐catalyzed highly enantioselective synthesis of 3‐arylbutanoic acids. Angew Chem Int Ed Engl. 2007;46:2623–2626. doi: 10.1002/anie.200604810. [DOI] [PubMed] [Google Scholar]
- Um P.J., Drueckhammer D.G. Dynamic enzymatic resolution of thioesters. J Am Chem Soc. 1998;120:5605–5610. [Google Scholar]
- Varadharaj G., Hazell K., Reeve C.D. An efficient preparative scale resolution of 3‐phenylbutyric acid by lipase from Burkholderia cepacia (Chirazyme L1) Tetrahedron: Asymmetry. 1998;9:1191–1195. [Google Scholar]
- Yang H., Henke E., Bornscheuer U.T. The use of vinyl esters significantly enhanced enantioselectivities and reaction rates in lipase‐catalyzed resolutions of arylaliphatic carboxylic acids. J Org Chem. 1999;64:1709–1712. doi: 10.1021/jo981780l. [DOI] [PubMed] [Google Scholar]