

Summary
An esterase which is encoded within a Thermotoga maritima chromosomal gene cluster for xylan degradation and utilization was characterized after heterologous expression of the corresponding gene in Escherichia coli and purification of the enzyme. The enzyme, designated AxeA, shares amino acid sequence similarity and its broad substrate specificity with the acetyl xylan esterase from Bacillus pumilus, the cephalosporin C deacetylase from Bacillus subtilis, and other (putative) esterases, allowing its classification as a member of carbohydrate esterase family 7. The recombinant enzyme displayed activity with p‐nitrophenyl‐acetate as well as with various acetylated sugar substrates such as glucose penta‐acetate, acetylated oat spelts xylan and DMSO (dimethyl sulfoxide)‐extracted beechwood xylan, and with cephalosporin C. Thermotoga maritimaAxeA represents the most thermostable acetyl xylan esterase known to date. In a 10 min assay at its optimum pH of 6.5 the enzyme's activity peaked at 90°C. The inactivation half‐life of AxeA at a protein concentration of 0.3 µg µl−1 in the absence of substrate was about 13 h at 98°C and about 67 h at 90°C. Differential scanning calorimetry analysis of the thermal stability of AxeA corroborated its extreme heat resistance. A multi‐phasic unfolding behaviour was found, with two apparent exothermic peaks at approximately 100–104°C and 107.5°C. In accordance with the crystal structure, gel filtration analysis at ambient temperature revealed that the enzyme has as a homohexameric oligomerization state, but a dimeric form was also found.
Introduction
Thermotoga maritima represents one of the few hyperthermophilic species of the domain of Bacteria, i.e. those with an optimum growth temperature of at least 80°C (see Huber and Stetter, 1992; Huber and Hannig, 2006). The obligate anaerobe, T. maritima, a model organism for hyperthermophilic bacteria, has a versatile organotrophic metabolism and is able to utilize various proteinaceous and carbohydrate substrates. This nutrition lifestyle is reflected in the T. maritima genome by one of the highest numbers of genes for carbohydrate‐active enzymes per megabasepair of all organisms, making T. maritima well suited for the analysis of mono‐, oligo‐ and polysaccharide utilization in hyperthermophilic bacteria.
Xylans represent hemicellulose components of plant cell walls which are usually associated with cellulose and lignin and consist of a backbone chain of 1,4‐linked β‐D‐xylopyranosyl residues which can carry various substituents, i.e. l‐arabinosyl, 4‐O‐methylglucuronyl, acetyl, feruloyl and p‐coumaroyl residues (see Kulkarni et al., 1999). The substitution pattern and degree of substitution in xylans depend on the source of plant material. For example, most hardwood xylans are glucuronoxylans with α‐(1→2)‐4‐O‐methyl‐d‐glucuronic acid or α‐(1→2)‐4‐O‐methyl‐d‐glucuronic acid groups on about 10% of the xylopyranosyl residues and are acetylated through ester linkages at position 2 or 3 of about 60–70% of the xylopyranosyl residues. The backbone of softwood xylans, on the other hand, is decorated with α‐(1→3)‐l‐arabinofuranosyl substituents but is not acetylated. The degradation of xylans requires the concerted action of main chain‐cleaving enzymes (endo‐1,4‐β‐xylanase, EC 3.2.1.8; β‐xylosidase, EC 3.2.1.37) and accessory, side‐group‐removing enzymes (α‐glucuronidase, EC 3.2.1.139; α‐arabinofuranosidase, EC 3.2.1.55; acetylesterase, EC 3.1.1.72). Since the backbone substituents can inhibit the main chain‐cleaving enzymes, the concerted and synergistic action of the accessory and main chain‐cleaving enzymes is important for efficient and complete xylan breakdown (Biely et al., 1986; Kormelink and Voragen, 1993; Castanares et al., 1995).
Plant xylans represent good growth substrates for T. maritimastrain MSB8 (DSM3109). Several enzymes of the T. maritima xylan utilization system have been investigated, including the two endo‐xylanases XynA and XynB (Winterhalter and Liebl, 1995; Winterhalter et al., 1995; Kleine and Liebl, 2006;Liebl et al., 2008), the β‐xylosidase XylA (BxlA) (E. Ossko and W. Liebl, unpubl. data) and the α‐glucuronidase AguA (Ruile et al., 1997).
In this report we describe the cloning and expression in Escherichia coli of a putative esterase gene identified within a xylan utilization gene cluster on the T. maritima MSB8 genome, and the characterization of the heterologously produced enzyme as the most thermoresistant acetyl xylan esterase currently known.
Results
In silico analysis of AxeA
The T. maritima axeA gene is located in an about 30 kb large gene cluster (TM0055–TM0077) whose function is proposed to be the breakdown and utilization of complex xylans (see Discussion). AxeA can be classified into the carbohydrate esterase family 7 according to the classification provided by the Cabohydrate‐Active enZymes Server (Coutinho and Henrissat, 1999) (CAZy; available at URL http://afmb.cnrs‐mrs.fr/CAZY/index.html). The amino acid sequence of AxeA was found to bear the highest similarity to sequences derived from putative genes for acetyl xylan esterases from Thermotogasp. RQ2 (97% identity/98% similarity), Thermotoga petrophila RKU‐1 and Marinitoga piezophila KA3 (96%/98%), Thermotoga neapolitana (90%/95%), a second putative orthologue from Thermotoga sp. RQ2 (72%/86%), Thermotoga lettingae TMO (67%/84%), Thermobaculum terrenum ATCC BAA‐798 (64%/77%), Roseiflexussp. RS‐1 (63%/76%), Herpetosiphon aurantiacusATCC 23779 (60%/75%), Streptomyces avermitilisMA‐4680 (56%/68%), or for cephalosporin C‐deacetylases from Acidothermus cellulolyticus11B (54%/66%) and Bacillus subtilis (42%/58%). Noteworthy, the most closely related sequences are all from other thermophiles, mostly from the Thermotogales group.
In addition to the AxeA‐encoding gene studied here (ORF TM0077), T. maritima strain MSB8 has a second putative acetyl xylan esterase gene (76% identity/81% similarity). This gene (TM0435) is located on the genome adjacent to TM0434 which codes for an α‐glucuronidase of glycoside hydrolase family 4 within a cluster of genes (TM0430–TM0443) thought to be involved in pectin degradation (Chhabra et al., 2003).
Purification and properties of recombinant AxeA
Using the purification protocol described in Experimental procedures, AxeA from crude extract of the recombinant E. coli strain BL21(DE3)/pET24d‐axeA was purified with a yield of 22.5% to apparent gel electrophoretic homogeneity (Table 1, Fig. 1). Under optimized induction conditions the recombinant enzyme amounted to about 25% of the soluble proteins in the recombinant host. An amount of 27.7 mg pure AxeA was retrieved from 7.7 g wet cell mass. Using the standard assay for deacetylation of p‐nitrophenyl acetate (pNP‐acetate) in 50 mM sodium phosphate buffer pH 6.5 at 50°C, this enzyme preparation had a specific activity of 89 U mg−1. In 20 mM Hepes pH 6.5, its specific activity was 107 U mg−1.
Table 1.
Purification of AxeA from E. coli BL21 (DE3)/pET24d‐axeA.
| Purification step | Protein concentration (mg ml−1) | Total protein (mg) | Specific activity (U mg−1) | Total activity (U) | Purification factor | Yield (%) |
|---|---|---|---|---|---|---|
| Crude extract | 17.82 | 481.2 | 22.9 | 10 992 | 1 | 100 |
| Heat‐treated crude extract | 4.84 | 106.5 | 50.4 | 5 363 | 2.20 | 48.8 |
| Source 30 Q pooled fractions | 1.33 | 34.6 | 76.3 | 2 638 | 3.34 | 24.0 |
| Phenyl Sepharose pooled fractions | 0.66 | 27.7 | 89.1 | 2 469 | 3.90 | 22.5 |
The enzyme activities were determined at 50°C in 50 mM phosphate buffer pH 6.5 with p‐nitrophenyl acetate (pNP‐acetate) as the substrate. A slightly higher specific activity of the purified enzyme, i.e. 107 U mg−1, was measured in 20 mM Hepes buffer pH 6.5.
Figure 1.
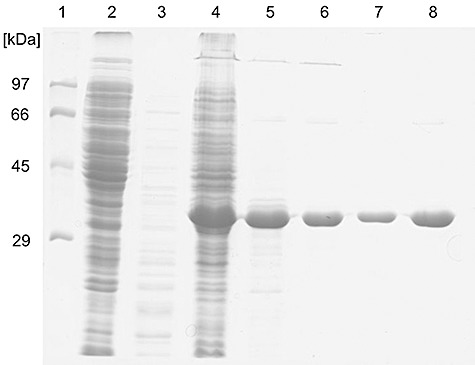
SDS‐PAGE analysis of AxeA purification steps. Lane 1, molecular mass standard proteins; lane 2, crude extract of E. coli BL21(DE3)/pET24d (50 µg); lane 3, crude extract of E. coli BL21(DE3)/pET24d after heat treatment (3.5 µg); lane 4, crude extract of E. coli BL21(DE3)/pET24d‐axeA (40 µg); lane 5, crude extract of E. coli BL21(DE3)/pET24d‐axeA after heat treatment (9 µg); lane 6, pooled active fractions after Source 30 Q ion exchange chromatography (4 µg); lane 7, pooled active fractions after Phenyl Sepharose HP chromatography (2.4 µg); lane 8, pooled active fractions after Phenyl Sepharose HP chromatography (5.8 µg).
The molecular mass of AxeA as determined by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) (Fig. 1) was in full accordance with the theoretical molecular mass calculated from the axeA‐derived sequence (37 kDa). Analytical gel filtration with a Superdex 200 prep grade column equilibrated with 50 mM TRIS‐HCl pH 8.0, 150 mM NaCl, yielded two symmetrical peaks corresponding to molecular masses of 229 and 74.4 kDa (Fig. 2). The protein from each of the peak fractions displayed similar specific esterase activity and yielded a single 37 kDa band after SDS‐PAGE analysis. These data indicate that active recombinant AxeA is found as a homohexamer and a homodimer respectively.
Figure 2.
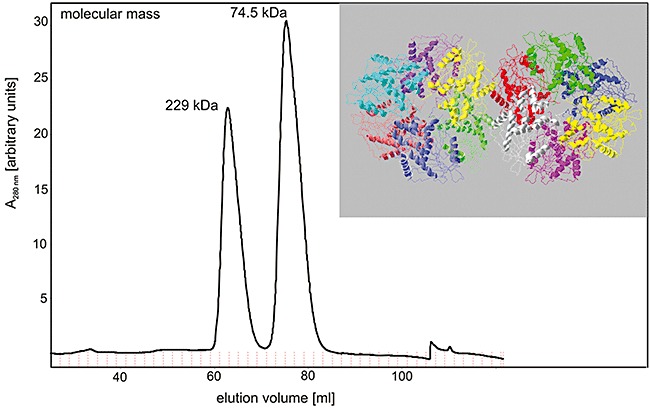
Size exclusion chromatography of purified recombinant AxeA, revealing active homodimeric and homohexameric forms of the enzyme. The elution volumes of the two symmetric peaks correspond to native molecular masses of 74.5 and 229.1 kDa respectively (for details see Experimental procedures). Insert: Oligomeric state of AxeA as derived from crystallographic data (PDB ID: 1vlq) which suggests 12 monomers arranged as two homohexamers in the asymmetric unit of the protein crystal.
Thermotoga maritima AxeA displayed maximum deacetylation activity at pH 6.5, and revealed more than 50% of its maximum activity between pH 5.0 and pH 7.5. The temperature at which the highest deacetylation activity in a 10 min assay was recorded was 90°C. A sharp drop in its relative activity was observed above 90°C (Fig. 3).
Figure 3.
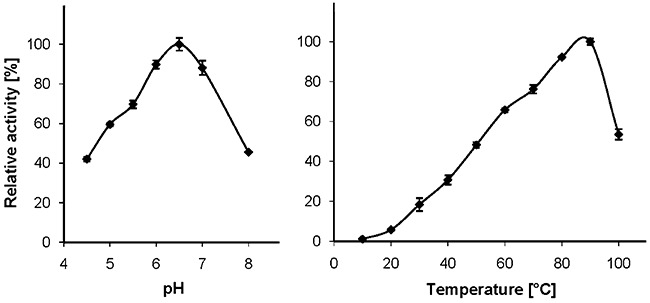
pH dependence (at 70°C) and temperature dependence (at pH 5.5) of AxeA activity, using a 10 min assay and chemically acetylated xylan as the substrate.
While some divalent cations (BaCl2, CaCl2, MgCl2, MnCl2) at a concentration of 3 mM stimulated the AxeA activity by about 40%, CdCl2 and ZnCl2 at 3 mM reduced the activity by 82% and 85% respectively.
Substrate specificity of AxeA
In addition to the hydrolysis of pNP‐acetate, the enzyme also was able to liberate acetate from glucose penta‐acetate. In 50 mM sodium phosphate buffer pH 6.5, at 1 mM substrate concentration, the specific activities with pNP‐acetate and glucose penta‐acetate were 89 and 40 U mg−1 respectively. With the latter substrate, an about eightfold higher activity (326 U mg−1) was determined at a glucose penta‐acetate concentration of 10 mM. No significant activity was detectable with 4‐methylumbelliferyl acetate and alpha‐naphthyl acetate, which was surprising because the related enzyme from Bacillus pumilus displayed high activity with alpha‐naphthyl acetate (Degrassi et al., 1998; 2000).
No acetate liberation could be measured with birchwood or beechwood xylan (up to a concentration of 4%) purchased from Sigma, probably due to the elimination of acetyl side‐groups during the preparation of these xylans. On the other hand, native beechwood xylan prepared by DMSO extraction after delignification of the wood with a sodium sulfite procedure (a kind gift by J. Puls, Hamburg) was deacetylated by AxeA with a specific activity of 5.2 U mg−1. Low activity (0.7 U mg−1) was measured with 1% oat spelts xylan purchased from Carl Roth GmbH. A much higher specific activity was achieved with chemically acetylated oat spelts xylan, i.e. 79 U mg−1 at a substrate concentration of 0.8% (w/v). At this concentration, substrate saturation of the enzyme was not yet reached, but higher concentrations could not be tested due to solubility limitations of the chemically acetylated oat spelts xylan.
Cephalosporin C deacetylase activity of AxeA
The amino acid sequence of T. maritimaAxeA bears 54% sequence identity to the cephalosporin C deacetylase from A. cellulolyticus. Therefore the β‐lactam cephalosporin C was tested as a potential substrate for esterase AxeA. Indeed, significant liberation of acetate from cephalosporin C was measured (Table 2).
Table 2.
Kinetic parameters of the hydrolysis of pNP‐acetate, glucose penta‐acetate and cephalosporin C by recombinant AxeA in 50 mM phosphate buffer pH 6.5.
| Substrate | Vmax(U mg−1) | KM(mM) | kcat (s−1) | kcat/KM (s−1 M−1) |
|---|---|---|---|---|
| pNP‐acetate | 113.5 ± 1.5 | 0.12 ± 0.08 | 69.9 | 5.83 × 105 |
| Glucose penta‐acetate | 366.8 ± 5.6 | 6.05 ± 0.26 | 226.2 | 3.74 × 104 |
| Cephalosporin C | 19.2 ± 1.4 | 4.18 ± 0.97 | 11.8 | 2.83 × 103 |
For glucose penta‐acetate these are apparent kinetic values.
Kinetics of pNP‐acetate, glucose penta‐acetate and cephalosporin C cleavage, and inhibition by acetate
The kinetic parameters of substrate cleavage were determined in 50 mM sodium phosphate buffer pH 6.5, using substrate concentrations between 0.1 and 25 mM. Next to pNP‐acetate, kinetic parameters were also measured for glucose penta‐acetate and cephalosporin C at 60°C. Classical Michaelis‐Menten‐like kinetics were observed. The kinetic values for pNP‐acetate and cephalosporin C, and the apparent kinetic values for glucose penta‐acetate are listed in Table 2. The substrate affinities were much lower for glucose penta‐acetate and cephalosporin C than for pNP‐acetate which was the best substrate in terms of catalytic efficiency.
The influence of acetate on the hydrolysis of pNP‐acetate (1 mM final concentration) by AxeA was measured in 50 mM sodium phosphate buffer pH 6.5 and in 20 mM Hepes buffer pH 6.5 at 50°C at an enzyme concentration of 0.6 µg ml−1. At all acetate concentrations tested (0–15 mM) the enzyme displayed more than 95% of the activity measured without acetate addition. Since the acetate concentrations measured during deacetylation of acetyl xylan by AxeA were always below 10 mM, product inhibition by acetate thus does not appear to be a limiting factor of AxeA action.
Effect of temperature on activity and inactivation of AxeA
Using a 10 min assay in 100 mM sodium phosphate buffer at pH 5.5 and chemically acetylated xylan as the substrate (for each temperature a control reaction without enzyme was necessary to account for non‐enzymatic deacetylation; see Experimental procedures for details), the maximum activity of AxeA was measured at 90°C (Fig. 3). The influence of increasing temperature on AxeA inactivation, which was determined by incubation of pure AxeA at a concentration of 330 µg ml−1 in 50 mM sodium phosphate buffer pH 6.5 in the absence of substrate at various temperatures, withdrawing aliquots and measuring the residual activity with the pNP‐acetate standard assay, is depicted in Fig. 4.
Figure 4.
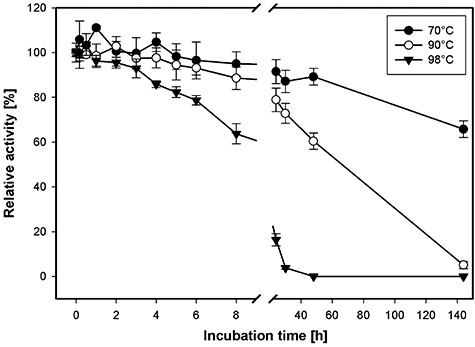
Temperature inactivation kinetics of recombinant AxeA at 70°C, 90°C and 98°C. The purified enzyme (at a concentration of 0.3 µg µl−1) was incubated in the absence of substrate at the respective temperatures, samples were withdrawn and the residual activity was determined with pNP‐acetate as described in Experimental procedures.
Differential scanning calorimetry
The thermal stability of AxeA was further investigated by differential scanning calorimetry (DSC). The observed unfolding transition was characterized by two apparent exothermic peaks, a broad one in the range between 100°C and 104°C, and a sharp one at 107.5°C (Fig. 5), which testifies to the extremely high thermostability of the enzyme. The deconvolution of the multi‐phasic unfolding behaviour yielded at least three intermediates, in accordance with the complex structure of AxeA (Fig. 2). However, an unambiguous assignment of the transitions to the denaturation or dissociation of the subunits of the oligomer was not possible. At higher temperatures an endothermic signal was observed, which is probably due to protein aggregation following denaturation. In accordance with this assumption, no signal was observed when reheating the sample after cooling.
Figure 5.
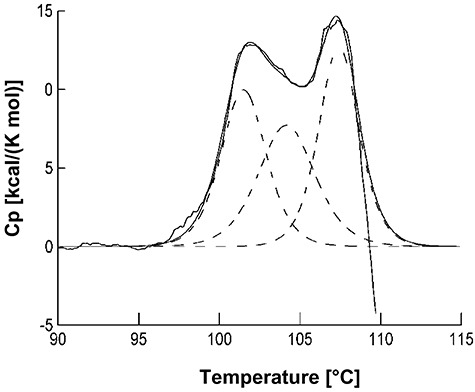
Differential scanning calorimetry (DSC) unfolding trace of AxeA. The protein (0.6 mg ml−1) was heated in 50 mM sodium phosphate, pH 7.5 with a rate of 1°C min−1. The experimentally observed transition (solid line) was deconvoluted (smoothed solid line), yielding three species with Tm values of 100°C, 104°C and 107.5°C (dashed lines).
Discussion
Purification and properties of AxeA
Due to the high expression level of the induced gene in the recombinant host a 3.9‐fold purification was sufficient to obtain a highly pure preparation of T. maritima AxeA. Gel filtration chromatography of the enzyme revealed protein peaks with similar specific esterase activities at elution volumes corresponding to 229 and 74 kDa. Considering the monomeric molecular mass (37 154 Da) predicted from the 325 residue polypeptide sequence which is in full accordance with the size determined by denaturing SDS‐PAGE analysis of purified recombinant AxeA (37 kDa), the gel filtration data indicate that AxeA exists as homohexameric and homodimeric forms respectively (Fig. 2). These results are in agreement with crystal structural data on AxeA (PDB ID: 1vlq; from a structural genomics project at the Joint Center for Structural Genomics, San Diego, CA, USA), which revealed homohexamers of the polypeptide, arranged as two homohexameric rings in the asymmetric unit of the protein crystal (see Fig. 2). However, our in vitro results did not indicate the presence of a dodecameric form of the enzyme. Crystallography has previously shown that the related multifunctional xylooligosaccharide/cephalosporin C deacetylase from B. subtilis also has a hexameric quaternary structure, formed by a trimer of dimers, with the active centres pointing towards the centre of the hexameric ring thereby being sequestered away from the cytoplasm (Vincent et al., 2003). In the case of the B. subtilis enzyme however, a dimeric form was not found experimentally, in contrast to T. maritima AxeA. The AxeA monomer is structurally similar to cephalosporin C deacetylases (PDB IDs: 1ods, 1l7a, 1odt) and we could also show that AxeA deacetylated cephalosporin C.
Taken together, our results concerning the comparative amino acid sequence analysis, but also the oligomerization state (hexameric and dimeric) and substrate specificity show that T. maritima AxeA can be classified as a typical member of the hexameric enzymes from carbohydrate esterase family 7, which share an unusual specificity for both acetylated xylan and cephalosporin C and an unusually high oligomeric state.
Thermoactivity and thermostability of AxeA
Thermotoga maritima AxeA is by far the most thermoactive and most thermostable acetyl xylan esterase described to date. The temperature at which the highest deacetylation activity in a 10 min assay was recorded was 90°C (Fig. 3) which represents the maximum growth temperature of the authentic enzyme producer, T. maritima. Recombinant AxeA withstood long‐term incubation at 90°C, retaining 50% of its original activity after about 67 h at this temperature. Even at 98°C only a relatively slow rate of inactivation was observed. At this temperature, half of the initial activity was left after about 13 h (Fig. 4), which was unexpected because the enzyme's activity dropped sharply above 90°C (Fig. 3). The measurement of the thermal unfolding transition by DSC (Fig. 5) revealed two apparent exothermic peaks at 100–104°C and 107.5°C which underscores the extreme resistance of AxeA against heat denaturation. The precise basis of AxeA thermostabilization is unknown at this time, but the oligomerization state of AxeA, i.e. dimers and hexamers in vitro (see above, Fig. 2), may be important, since the association of polypeptides to oligomers is recognized as one of the factors that can increase protein stability (see Sterner and Liebl, 2001).
The extreme stability of AxeA renders this enzyme particularly suited for applications in biotechnology where robust, long‐lasting biocatalysts are needed. Possible applications include xylan deacetylation to improve efficacy of other xylan‐degradation enzymes during enzyme‐assisted lignocellulose degradation processes, the modification of cephalosporin antibiotics, but also the reversal of the hydrolysis reaction, i.e. the acetylation of xylan derivatives or similar polysaccharides for the introduction of acetyl functional groups into polysaccharides. Preliminary experiments in our group have shown that under conditions of reduced water concentration AxeA is capable of performing the reverse reaction, e.g. the acetylation of oat spelts xylan (data not shown). Current experiments are aimed at the determination of the efficiency and region specificity of this biosynthetic polymer‐modification reaction.
Genetic context of the axeA gene, and physiological role of AxeA
The axeA gene (TM0077) is located in an about 30‐kb‐large gene cluster which contains various genes for xylanolytic enzymes (see legend to Fig. 6), i.e. xylan backbone‐cleaving enzymes (endoxylanases XynA and XynB, and the β‐xylosidase BxlA) and accessory enzymes (α‐glucuronidase AguA and the acetyl xylan esterase AxeA). Also, this gene cluster contains genes for two ABC‐transport systems, which may be required for the uptake of substituted xylooligosaccharides, and a group of ORFs (TM0064–TM0069) which apparently code for a metabolic pathway for the utilization of glucuronic acid. Similarly, genes for glucuronate utilization are clustered in Bacillus stearothermophilus strain T‐6 (Shulami et al., 1999; 2007) but the gene order is different. According to DNA microarray experiments (Chhabra et al., 2003), TM0064–TM0069 are induced in T. maritima cells grown on xylan.
Figure 6.
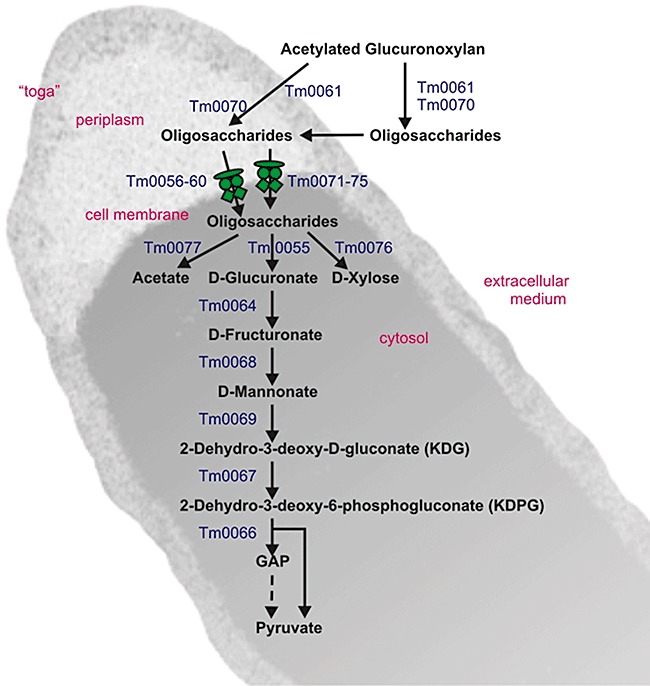
Working model showing the proposed metabolic functions encoded by the ORFs of the approximately 30 kb chromosomal gene cluster (TM0055–TM0077) for the breakdown and utilization of acetylated glucuronoxylan by T. maritima. The ORFs which are proposed to encode for the individual steps of the pathway are indicated next to the arrows. The proposed functional assignments of the ORFs are as follows (the relative orientation of the ORFs is indicated with ‘+’ and ‘−’ symbols): TM0055(+), α‐glucuronidase (aguA); TM0056(+), ABC‐transporter periplasmic binding protein; TM0057(+), ABC‐transporter ATP‐binding protein; TM0058(+), ABC‐transporter ATP‐binding protein; TM0059(+), ABC‐transporter permease protein; TM0060(+), ABC‐transporter permease protein; TM0061(−), endo‐1,4‐β‐xylanase [xynA, toga‐associated and partially released to the extracellular medium (Winterhalter and Liebl, 1995; Liebl et al. 2008)]; TM0062(−), hypothetical protein; TM0063(+), hypothetical protein; TM0064(+), glucuronate isomerase; TM0065(−), transcriptional regulator, IclR family; TM0066(−), 2‐dehydro‐3‐deoxyphosphogluconate aldolase/4‐hydroxy‐2‐oxoglutarate aldolase; TM0067(−), 2‐keto‐3‐deoxygluconate kinase; TM0068(−), d‐mannonate oxidoreductase; TM0069(−), mannonate dehydratase; TM0070(+), endo‐1,4‐β‐xylanase [xynB, XynB is thought to be present in the periplasm and also occurs extracellularly (Winterhalter and Liebl, 1995; Liebl et al., 2008)]; TM0071(−), ABC‐transporter periplasmic binding protein; TM0072(−), ABC‐transporter permease protein; TM0073(−), ABC‐transporter permease protein; TM0074(−), ABC‐transporter ATP‐binding protein; TM0075(−), ABC‐transporter ATP‐binding protein; TM0076(−), β‐xylosidase (bxlA); TM0077(−), acetyl xylan esterase (axeA). It is stressed that the proposed intracellular localization of AxeA has not been proven experimentally.
No signal peptide could be detected in the AxeA amino acid sequence with the software SignalP 3.0 (http://www.cbs.dtu.dk/services/SignalP). Degrassi and colleagues (2000) speculate that the similar acetyl esterase from B. pumilus is secreted due to an internal secretion signal. In contrast to this theory it seems more plausible that AxeA is not secreted but an intracellular enzyme, which was also proposed for the acetyl xylan esterase AxeI from Thermoanaerobacteriumsp. JW/SL (Lorenz and Wiegel, 1997). The physiological function in this case would be the deacetylation of acetylated xylooligosaccharides rather than complex xylan. In the case of T. maritima it could be that xylan is first degraded by the endoxylanases XynA or XynB (Winterhalter et al., 1995) followed by the internalization of xylooligosaccharides via the transporters encoded in the same xylan degradation gene cluster. The putative functions of the proteins encoded by this gene cluster are summarized in the metabolic scheme shown in Fig. 6. The scheme takes into account the peculiar cell morphology of T. maritima with the outer sheath (‘toga’) enclosing a large periplasmic space, as well as experimental evidence from previous studies concerning the localization and catalytic properties of the xylanases (XynA and XynB) and accessory enzymes (AguA, BxlA) studied in our group (Winterhalter and Liebl, 1995; Winterhalter et al., 1995; Ruile et al., 1997; E. Ossko and W. Liebl, unpubl. data). While the precise specificities of the transport systems cannot be deduced from the gene sequences and therefore must remain speculative, the similarity‐based assignment of functions to most of the other ORFs could be performed with a high level of confidence. In summary, the enzymatic properties of AxeA as well as the presence of its gene in a gene cluster together with genes for other xylanolytic enzymes indicate that AxeA and all or most of the other 22 proteins encoded by the genes of this cluster are involved in the utilization of xylans as a carbon and energy source by T. maritima.
Experimental procedures
Strain and growth conditions
Thermotoga maritima strain MSB8 (DSM 3109) was grown anaerobically at 80°C in Difco Marine Broth medium #2216 (Difco, Detroit, MI, USA) or a reduced medium with 0.25% soluble starch as described by Winterhalter and colleagues (1995). Escherichia coli strains were cultivated in Luria–Bertani medium (1% tryptone, 0.5% yeast extract, 0.5% sodium chloride) containing kanamycin at a concentration of 50 µg ml−1.
General DNA modification and DNA sequencing methods
Genomic DNA was isolated from 0.6 g of T. maritima MSB8 cells using the method described by Gabelsberger and colleagues (1993). Plasmid DNA isolation, transformation of E. coli, DNA modifications and sequence analysis were performed with standard techniques.
Amplification of DNA fragments encoding AxeA (T. maritimaORF TM0077, NP_227893.1) via the polymerase chain reaction (PCR) was performed using Pfu polymerase (Stratagene, Heidelberg, Germany) and the primers axeA_fwd (GGAGTGAAAccATGGCCTTCTTCGATTTAC) and axeA_rev (AAACGGAATTCGTGGTACTTGA). The PCR amplification was carried out with 30 cycles of 94°C/45 s, 50–65°C/45 s and 72°C/180 s. The axeA gene was cloned into pET24d (Novagen, Darmstadt, Germany) via restriction sites introduced by PCR and the resulting plasmid pET24d‐axeA was introduced into E. coli BL21 (DE3) cells.
Protein analytical methods and enzyme assays
The determination of protein concentrations and SDS‐PAGE were performed as described in Liebl and colleagues (1992). Size exclusion chromatography was carried out using a Superdex 200 HiLoad 16/60 column equilibrated with 50 mM TRIS (pH 8.0), 150 mM NaCl (column volume 120 ml). An enzyme sample (0.6 mg in 1 ml equilibration buffer) was applied to the column and eluted with the same buffer at a flow rate of 0.3 ml per minute. The column was calibrated with the standard proteins cytochrome c (12.5 kDa), albumin (68 kDa) and ferritin (450 kDa).
The pH of buffers was generally adjusted at the temperature of use. The routine assay used to measure AxeA activity was based on the liberation of p‐nitrophenol from pNP‐acetate as described by Degrassi and colleagues (1998). The reactions (2 min) were performed at 50°C and pH 6.5 in 50 mM phosphate or 20 mM Hepes buffer containing enzyme and 1 mM substrate. One unit was defined as the amount of enzyme that liberated 1 µmol pNP per minute. Due to the lability of pNP‐acetate at 60°C or higher, this pNP‐acetate assay was not used at temperatures above 50°C.
The hydrolysis of acetyl ester linkages in glucose penta‐acetate was measured in 10 min assays at 55°C in reaction mixtures (500 µl) containing 50 mM phosphate buffer pH 6.5 and 1–10 mM glucose penta‐acetate. Acetic acid liberated was quantified using a commercially available kit (r‐biopharm, Darmstadt, Germany).
The deacetylation of xylan by AxeA was carried out in reaction mixtures (500 µl) containing 100 mM sodium phosphate buffer pH 6.5 (or alternatively 20 mM Hepes buffer pH 6.5), 0.2–2% xylan, and enzyme, at the temperatures specified in the text. Routinely, the reaction was stopped after 10 min by adding 10 µl of 1 N sulfuric acid. Particulate material was sedimented (8000 r.p.m., 3 min, 4°C in a bench‐top centrifuge) and acetic acid in the supernatant was quantified as described above.
The pH dependence of AxeA activity was determined at 70°C in 100 mM sodium phosphate buffer adjusted to the desired pH values, using chemically acetylated xylan (prepared from oat spelts xylan as described by Johnson et al., 1988) as the substrate. The influence of the temperature on AxeA activity on chemically acetylated xylan was determined in 10 min assays between 30°C and 100°C in 100 mM sodium phosphate buffer. Due to the significant spontaneous deacetylation of the substrate above 80°C at pH 6.5 these assays were conducted at pH 5.5. For each condition a control reaction without enzyme was carried out to account for non‐enzymatic deacetylation.
Xylanase activity was determined at 75°C in 250 mM NaCl, 50 mM BIS‐TRIS buffer pH 6.2 as described previously (Winterhalter and Liebl, 1995). One unit of xylanase activity liberates 1 µmol of reducing groups (as xylose equivalents) per minute.
Purification of recombinant AxeA
AxeA was purified from the recombinant E. coli strain BL21(DE3)/pET24d‐axeA grown aerobically at 37°C in LB medium with kanamycin (50 µg ml−1). At OD = 0.8, 0.1 mM IPTG was added and incubation was continued for 4 h before harvesting. The cells were washed with 50 mM sodium phosphate buffer pH 7.5, suspended in the washing buffer (0.25 g wet cell mass per ml) and lysed by passage through a French press cell at 6.9 MPa. Upon centrifugation the cleared crude extract which had a protein concentration of about 18 mg ml−1 was subjected to a heat treatment (70°C, 20 min) to precipitate the heat‐labile E. coli host proteins which were removed by centrifugation (Sorvall SS34 rotor, 10 000 r.p.m., 20 min, 4°C). The supernatant was dialysed at 4°C against 50 mM sodium phosphate buffer pH 8.0 and applied to a Source 30 Q HR 10/10 column. Proteins were eluted at a flow rate of 2 ml min−1 with a linear 0 M–1 M sodium chloride gradient in 50 mM sodium phosphate buffer pH 8.0. Fractions with activity were combined, dialysed at 4°C in 50 mM TRIS‐HCl buffer pH 8.0, 1 M ammonium sulfate, and applied to a Phenyl Sepharose HP XK 16/10 column. Elution was achieved with a linear 1 M–0 M ammonium sulfate gradient, and fractions containing acetyl xylan esterase activity were pooled, dialysed and stored in aliquots at −20°C.
Differential scanning calorimetry
Differential scanning calorimetry was performed with a CSC 6100 II differential scanning calorimeter [Calorimetry Sciences Corporation (CSC), Provo, UT, USA] using a AxeA concentration of 0.6 mg ml−1 in 50 mM sodium phosphate pH 7.5. Buffers and samples were degassed before calorimetric analysis, which was performed between 25°C and 125°C with a scan rate of 1°C min−1. Buffer baselines were measured under identical conditions. The data were evaluated with the CSC‐program CpCalc 2.1 to determine apparent melting temperatures (Tm), which are those temperatures where the specific heat capacity of the individual species peaked. The measurements were performed three times to ensure the reproducibility of the unfolding transition.
Acknowledgments
The authors wish to thank M. Bömeke for skilful technical assistance. We are indebted to J. Puls, Hamburg, Germany, for supplying DMSO‐extracted native xylan. Financial support by the German Federal Ministries of Education and Research and of Food, Agriculture and Consumer Protection is gratefully acknowledged.
References
- Biely P., MacKenzie C.R., Puls J., Schneider H. Bio/Technology. 1986;4:731–733. [Google Scholar]
- Castanares A., McCrae S.I., Wood T.M. d‐Xylan‐degrading enzyme system from the fungus Phanerochaete chrysosporium: isolation and partial characterisation of an α‐(4‐O‐methyl)‐d‐glucuronidase. J Biotechnol. 1995;43:183–194. doi: 10.1016/0168-1656(95)00128-x. [DOI] [PubMed] [Google Scholar]
- Chhabra S.R., Shockley K.R., Conners S.B., Scott K.L., Wolfinger R.D., Kelly R.M. Carbohydrate‐induced differential gene expression patterns in the hyperthermophilic bacterium Thermotoga maritima. J Biol Chem. 2003;278:7540–7552. doi: 10.1074/jbc.M211748200. [DOI] [PubMed] [Google Scholar]
- Coutinho P.M., Henrissat B. Carbohydrate‐active enzymes: an integrated database approach. In: Gilbert H.J., Davies G., Henrissat B., Svensson B., editors. The Royal Society of Chemistry; 1999. pp. 3–12. , and . In Recent Advances in Carbohydrate Bioengineering. , and (eds). Cambridge, UK: , pp. [WWW document]. URL http://www.cazy.org/ [Google Scholar]
- Degrassi G., Okeke B.C., Bruschi C.V., Venturi V. Purification and characterization of an acetyl xylan esterase from Bacillus pumilus. Appl Environ Microbiol. 1998;64:789–792. doi: 10.1128/aem.64.2.789-792.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degrassi G., Kojic M., Ljubijankic G., Venturi V. The acetyl xylan esterase of Bacillus pumilus belongs to a family of esterases with broad substrate specificity. Microbiology. 2000;146:1585–1591. doi: 10.1099/00221287-146-7-1585. [DOI] [PubMed] [Google Scholar]
- Gabelsberger J., Liebl W., Schleifer K.H. Cloning and characterization of β‐galactoside and β‐glucoside hydrolysing enzymes of Thermotoga maritima. FEMS Microbiol Lett. 1993;109:131–138. [Google Scholar]
- Huber R., Hannig M. Thermotogales. In: Dworkin M., Falkow S., Rosenberg E., Schleifer K.‐H., Stackebrandt E., editors. 3rd. Springer‐Verlag; 2006. pp. 899–922. [Google Scholar]
- Huber R., Stetter K.O. The order Thermotogales. In: Balows A., Trüper H.G., Dworkin M., Harder W., Schleifer K.H., editors. 2nd. Springer‐Verlag; 1992. pp. 3809–3815. [Google Scholar]
- Johnson K.G., Fontana D., MacKenzie C.R. 1988;160:551–560. , and ) Measurement of acetylxylan esterase in Streptomyces Meth Enzymol. [Google Scholar]
- Kleine J., Liebl W. Comparative characterization of deletion derivatives of the modular xylanase XynA of Thermotoga maritima. Extremophiles. 2006;10:378–381. doi: 10.1007/s00792-006-0509-0. [DOI] [PubMed] [Google Scholar]
- Kormelink F.J.M., Voragen A.G.J. Degradation of different [(glucurono)arabino]xylans by combination of purified xylan‐degrading enzymes. Appl Microbiol Biotechnol. 1993;38:688–695. [Google Scholar]
- Kulkarni N., Shendye A., Rao M. Molecular and biotechnological aspects of xylanases. FEMS Microbiol Rev. 1999;23:411–456. doi: 10.1111/j.1574-6976.1999.tb00407.x. [DOI] [PubMed] [Google Scholar]
- Liebl W., Feil R., Gabelsberger J., Kellermann J., Schleifer K.H. Purification and characterization of a novel thermostable 4‐α‐glucanotransferase of Thermotoga maritima cloned in Escherichia coli. Eur J Biochem. 1992;207:81–88. doi: 10.1111/j.1432-1033.1992.tb17023.x. [DOI] [PubMed] [Google Scholar]
- Liebl W., Winterhalter C., Baumeister W., Armbrecht M., Valdez M. Xylanase attachment to the cell wall of the hyperthermophilic bacterium Thermotoga maritima. J Bacteriol. 2008;190:1350–1358. doi: 10.1128/JB.01149-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz W.W., Wiegel J. Isolation, analysis, and expression of two genes from Thermoanaerobacterium sp. strain JW/SL YS485: a beta‐xylosidase and a novel acetyl xylan esterase with cephalosporin C deacetylase activity. Appl Environ Microbiol. 1997;179:5436–5441. doi: 10.1128/jb.179.17.5436-5441.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruile P., Winterhalter C., Liebl W. Isolation and analysis of a gene encoding α‐glucuronidase, an enzyme with a novel primary structure involved in the breakdown of xylan. Mol Microbiol. 1997;23:267–279. doi: 10.1046/j.1365-2958.1997.2011568.x. [DOI] [PubMed] [Google Scholar]
- Shulami S., Gat O., Sonenshein A.L., Shoham Y. The glucuronic acid utilization gene cluster from Bacillus stearothermophilus T‐6. J Bacteriol. 1999;181:3695–3704. doi: 10.1128/jb.181.12.3695-3704.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulami S., Zaide G., Zolotnitsky G., Langut Y., Feld G., Sonenshein A.L., Shoham Y. A two‐component system regulates the expression of an ABC transporter for xylo‐oligosaccharides in Geobacillus stearothermophilus. Appl Environ Microbiol. 2007;73:874–884. doi: 10.1128/AEM.02367-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterner R., Liebl W. Thermophilic adaptation of proteins. Crit Rev Biochem Mol Biol. 2001;36:39–106. doi: 10.1080/20014091074174. [DOI] [PubMed] [Google Scholar]
- Vincent F., Charnock S.J., Verschueren K.H.G., Turkenburg J.P., Scott D.J., Offen W.A. Multifunctional xylooligosaccharide/cephalosporin C deacetylase revealed by the hexameric structure of the Bacillus subtilis enzyme at 1.9 A resolution. J Mol Biol. 2003;330:593–606. doi: 10.1016/s0022-2836(03)00632-6. et al. [DOI] [PubMed] [Google Scholar]
- Winterhalter C., Liebl W. Two extremely thermostable xylanases of the hyperthermophilic bacterium Thermotoga maritima strain MSB8. Appl Environ Microbiol. 1995;61:1810–1815. doi: 10.1128/aem.61.5.1810-1815.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterhalter C., Heinrich P., Candussio A., Wich G., Liebl W. Identification of a novel cellulose‐binding domain within the multi‐domain 120 kDa xylanase XynA of the hyperthermophilic bacterium Thermotoga maritima. Mol Microbiol. 1995;15:431–444. doi: 10.1111/j.1365-2958.1995.tb02257.x. [DOI] [PubMed] [Google Scholar]