

Abstract
This work is focused on the development of a plant virus-based carrier system for cargo delivery, specifically 30 nm-sized cowpea mosaic virus (CPMV). Whereas previous reports described the engineering of CPMV through genetic or chemical modification, we report a non-covalent infusion technique that facilitates efficient cargo loading. Infusion and retention of 130–155 fluorescent dye molecules per CPMV using DAPI (4’,6-diamidino-2-phenylindole dihydrochloride), propidium iodide (3,8-diamino-5-[3-(diethylmethylammonio)propyl]-6-phenylphenanthridinium diiodide), and acridine orange (3,6-bis(dimethylamino)acridinium chloride), as well as 140 copies of therapeutic payload proflavine (PF, acridine-3,6-diamine hydrochloride), is reported. Loading is achieved through interaction of the cargo with the CPMV’s encapsidated RNA molecules. The loading mechanism is specific; empty RNA-free eCPMV nanoparticles could not be loaded. Cargo-infused CPMV nanoparticles remain chemically active, and surface lysine residues were covalent modified with dyes leading to the development of dual-functional CPMV carrier systems. We demonstrate cargo-delivery to a panel of cancer cells (cervical, breast, and colon): CPMV nanoparticles enter cells via the surface marker vimentin, the nanoparticles target the endolysosome, where the carrier is degraded and the cargo released allowing imaging and/or cell killing. In conclusion, we demonstrate cargo-infusion and delivery to cells; the methods discussed provide a useful means for functionalization of CPMV toward its application as drug and/or contrast agent delivery vehicle.
Introduction
The application of nanomaterials as carrier systems to deliver imaging reagents and/or drugs has gained momentum in the medical field. Nanoparticles are advantageous because their large surface-area-to-volume ratio allows functionalization with multiple different payloads and ligands. Nanoparticles are used to partition cargos between diseased and healthy tissue, ideally avoiding healthy tissues or at least minimizing the accumulation of toxic substances in healthy organs. Disease targeting, such as cancer, is achieved making use of the unique biological features that distinguishes the tumor microenvironment from healthy cells. For example, based on their size, nanoparticles home to solid tumors based on leaky tumor blood vessels and the resulting enhanced permeability and retention effect [1,2]. Other targeting strategies include the use of receptor-specific ligands to direct the nanocarrier to receptors selectively over-expressed at the target disease site [3].
When it comes to cargo-loading and cargo-release, many different chemistries and mechanisms have been developed that control loading efficiency, affinity, and release rates; the choice of chemistry typically depends on the disease profile, cargo molecule, and carrier system of choice. Many different carrier systems are currently under investigation and development for drug delivery and tissue-specific imaging; each system has its advantages and disadvantages with regard to physiochemical properties, biodistribution and clearance, pharmacokinetic behavior, immunogenicity, and toxicity. Our research focuses on the development of bionanoparticles derived from plant viruses, also termed viral nanoparticles (VNPs).
The development and application of virus-based materials in medicine is a growing field with a strong potential impact [4–6]. There are many novel types of VNPs under development, with those based on bacteriophages and plant viruses favored because they are considered safer in humans than mammalian viruses [7]. Preclinical studies in mice have shown that plant viruses can be administered at doses of up to 100 mg (1016 VNPs) per kg body weight without signs of toxicity [8,9]. Like other protein-based nanomaterials they are immunogenic. However, strategies such as PEGylation can be used to overcome the immunogenicity of VNPs [10–15]. VNPs are genetically encoded and self-assemble into discrete and monodisperse structures with a precise shape and size. Many virus structures are understood at atomic resolution, allowing the development of protocols for high-precision VNP tailoring. This level of quality control cannot yet be achieved with synthetic nanoparticles. VNPs can be modified with targeting ligands and/or cargos using at least five approaches: genetic engineering, bioconjugate chemistry, self-assembly, mineralization, and infusion techniques [16].
In this work, we sought to develop the cowpea mosaic virus (CPMV) platform as a tool for cargo-delivery. CPMV is a plant picornavirus typically produced in black-eyed pea plants. CPMV capsids measure 30 nm in diameter and are comprised by 60 copies each of a small (S) and large (L) protein encapsulating a bipartite, single stranded, positive-sense RNA genome. CPMV has been extensively studied, developed, and tested for applications in the medical field. Bioconjugate chemistries on CPMV’s exterior and interior surfaces are well established [16–21] and its in vitro and in vivo properties are well understood. CPMV naturally is taken up by mammalian cells through interactions with surface-expressed vimentin [22]. This unique property can be used to target CPMV to endothelial cells for vascular imaging and tumor vessel mapping [23], targeting vimentin-expressing cancer cells in vitro or in vivo [24,25], as well as targeting and imaging sites of inflammation, such as atherosclerotic plaques or infections of the central nervous system [26,27]. Re-targeting of CPMV to receptors of interest can also be achieved through tailoring the surface chemistry with appropriate targeting ligands [28–31].
More recently, we turned toward the application of CPMV as a carrier for drug delivery and demonstrated cell toxicity of CPMV nanoparticles chemically modified with multiple copies of the chemotherapeutic drug doxorubicin [32]. Multistep chemical modification procedures, however, can be cumbersome, low yielding, and costly. We therefore sought to explore non-covalent cargo-loading strategies making use of the natural cargo, the nucleic acids. We tested the hypothesis that the encapsidated nucleic acids could act as a “sponge” to load imaging agents and drugs based on electrostatic interactions and/or affinity (Scheme 1). We discuss the loading of several fluorophores and therapeutic molecules. Furthermore, we report cargo-delivery in tissue culture and demonstrate imaging and treatment using a panel of cancer cell lines.
Scheme 1.
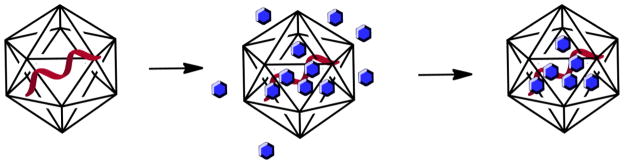
Cartoon of CPMV with its single stranded RNA molecule (in red), CPMV is exposed to a bathing solution containing the cargo of interest (in blue), washing and dialysis is used to remove excess cargo yielding intact CPMV with infused cargo.
Materials and Methods
CPMV propagation and purification
Black-eyed peas #5 (Vigna unguiculata) were inoculated with 20 ng/μ1 CPMV in 0.1 M potassium M phosphate buffer (pH 7.0) and propagated for 18–20 days using established procedures [33]. Virus concentration in plant extracts was determined by UV/visible spectroscopy and virus integrity was determined by size exclusion chromatography and UV/visible spectroscopy. A pure CPMV preparation has an absorbance ratio of A260 nm:A280 nm of 1.7±0.1. Empty CPMV (eCPMV) were provided by courtesy of Prof. Lomonossoff, John Innes Centre, Norwich, UK [34].
Cargo-loading via infusion
A solution of CPMV (at 1 mg mL−1 in 0.1 M potassium phosphate buffer pH 7.4, in the following referred to as KP buffer) was mixed with a 10,000-fold molar excess of the desired cargo molecule (see below) for 1 hour at room temperature in the dark. (The molecular weight of CPMV is 5.6x106 g mol−1.) Concentration curves were evaluated to determine the optimal excess to achieve efficient loading; CPMV was incubated with a molar excess 1,000, 2,000, 5,000, 10,000, and 50,000 cargo molecules per one CPMV. Time course studies were also conducted and it was found the loading does not improve after one hour of incubation. The following cargo molecules were studied: DAPI (4’,6-diamidino-2-phenylindole dihydrochloride, MP Biomedicals). Propidium iodide (3,8-diamino-5-[3-(diethylmethylammonio)propyl]-6-phenylphenanthridinium diiodide, Sigma Aldrich), acridine Orange (3,6-bis(dimethylamino)acridinium chloride, MP Biomedicals), and proflavine (PF, acridine-3,6-diamine hydrochloride, Sigma Aldrich). The reaction was then purified to remove cargo-loaded CPMV from excess reagents through extensive dialysis (Spectra/Por2, MWCO 12–14 KDa, Spectrum Laboratories) and multiple rounds of centrifuge filtration using spin columns (Amicon, MWCO 10 KDa). The cargo-loaded CPMV product was characterized using a combination of SEC, UV/Visible spectroscopy, and native and denaturing gel electrophoresis.
Covalent bioconjugation of CPMV
CPMV was labeled at surface-exposed lysine residues using N-hydroxysuccinimide (NHS) active AlexaFluor 555 (A555, Invitrogen) or NHS-activated Oregon Green 488 (O488, Invitrogen). Chemical modification was performed as a subsequent step, after cargo infusion. NHS-A555 or O488 in DMSO was added to CPMV (at 2 mg mL−1 in KP buffer) at a molar excess of 2000 NHS-A555/O488: 1 CPMV; the final DMSO concentration was adjusted to 10% by volume, the protein concentration was kept at 1 mg mL−1. The mix was reacted for two hours at room temperature with agitation in the dark. The reaction mix was purified through dialysis and spin filters as described above.
Size exclusion chromatography (SEC)
All CPMV nanoparticle preparations were analyzed by SEC using a Superose6 column on the ÄKTA Explorer chromatography system (GE Healthcare). Samples (100 μl of 1 mg mL−1) were analyzed at a flow rate of 0.5 mL min−1, using 0.1 M potassium phosphate buffer (pH 7.4).
UV/visible spectroscopy
A NanoDrop Spectrophotometer was used to measure the UV/visible spectra of native and modified CPMV nanoparticles. The degree of dye-loading was determined based on the concentration of dye:CPMV making use of Beer Lambert law and the dye and CPMV-specific extinction coefficients: CPMV: ε(260 nm)=8.1mL mg−1 cm−1, molecular weight of CPMV = 5.6x106 g mol−1, DAPI: ε(358 nm)=24,000 M−1cm−1, PI: ε(493 nm)= 5,900 M−1cm−1, AO: ε(470 nm)=43,000 M−1cm−, PF: ε(445 nm)=40,000 M−1cm−1, A555: ε(555 nm)=155,000 M−1cm−1, O488: ε(496 nm)=75,000 M−1cm−1. (It should be noted that the extinction coefficients may change in different chemical environments; degree of dye-loading is thus an approximation.)
Native and denaturing gel electrophoresis
CPMV nanoparticles were analyzed on native and denaturing gels. 5–10 μg sample was analyzed on 1.2% agarose gel in 1x TBE buffer, running buffer was 1x TBE. TBE = 45mM Tris, 45mM boric acid, 1.25 mM EDTA in MilliQ water. Protein subunits were analyzed on denaturing 4–12% NuPAGE gels (Invitrogen) using 1x MOPS buffer (Invitrogen). 10 μg sample (added SDS loading buffer; Invitrogen) was analyzed. If indicated, ethidium bromide was also used in gel staining for native gel samples. Otherwise, gels were photographed before and after staining with Coomassie Blue using AlphaImager (Biosciences) imaging system and UV or white light.
Tissue Culture
HeLa cells (cervical cancer) were obtained from ATCC®, and cultured and maintained in Minimum Essential Media (MeM) supplemented with 10% (v/v) FBS, 1% (w/v) penicillin-streptomycin, 1% (w/v) glutamine at 37°C and 5% CO2. PC-3 cell line (prostate cancer) was obtained from ATCC® and maintained in Dulbecco’s modified Eagle medium-F12 (DMEM/F12) that contained 10% (v/v) FBS, 1% (w/v) penicillin-streptomycin, 1% (w/v) glutamine at 37°C and 5% CO2. HT-29 cells (colon cancer) were obtained from ATCC®, and cultured and maintained in RPMI 1640 medium supplemented with 10% (v/v) FBS, 1% (w/v) penicillin-streptomycin, 1% (w/v) glutamine at 37°C and 5% CO2. All culture media reagents were purchased from Invitrogen.
Confocal Microscopy
Cellular uptake of CPMV
HeLa, PC-3, or HT-29 cells (25,000 cells/well) were grown for 24 hours on glass coverslips placed in an untreated 24-well plate in 200 μL media (see above) at 37°C, 5% CO2. Cells were washed and O488-CPMV, O488-CPMV-PF, O488-CPMV-CP (at 10 μg/well) were introduced in 100 μL of corresponding media, incubated for three hours, and then washed with saline to remove any unbound particles. Cells were fixed for five min at room temperature using DPBS containing 4% (v/v) paraformaldehyde and 0.3% (v/v) glutaraldehyde. Cell membranes were stained using 1 μg/mL wheat germ agglutinin (WGA) conjugated with AlexaFluor-555 (WGA-A555; Invitrogen) in 5% (w/v) goat serum (GS) for 45 min at room temperature in dark followed by subsequent washing with DPBS (Invitrogen). Nuclei were stained with DAPI (MP Biomedicals, 1:7500) for five min. Cells were washed with DPBS in between each staining step. Coverslips were then mounted onto glass slides using mounting media (Permount, Fisher Chemicals) and sealed using nail polish. Confocal images were captured on Olympus FluoView™ FV1000 LSCM and data processed using Image J 1.44o software (http://imagej.nih.gov/ij).
Co-localization of CPMV with endolysosomes
Native CPMV was used and stained using CPMV-specific antibodies. After incubation of HeLa cells with CPMV (as described above), cells were incubated with anti-CPMV antibodies (rabbit IgG, Pacific Immunology) at 1:200 dilution for 60 min at room temperature. Endolysosomes were stained using a mouse anti-human LAMP-1 antibody (Biolegend, 1:200; 5% GS) for 60 min. Secondary antibodies, goat anti-mouse-AlexaFluor 488 (secondary to LAMP-1 Ab) and goat anti-rabbit-AlexaFluor 555 (secondary to anti-CPMV Ab) at 1:500 dilutions (mixed together) were then used to label the primary antibodies for 60 min. DAPI staining and imaging was as described above.
Cellular uptake of CPMV-DAPI
HeLa cells (25,000 cells/well) were grown for 24 hours on glass coverslips placed on an untreated 24-well plate in 200 μL medium (see above) at 37°C, 5% CO2. Cells were washed and (A555)-CPMV-DAPI (1.7 nM CPMV, 0.233 μM DAPI /well) introduced in 100 μL medium, and cells were incubated for one to three hours at 37°C or 4°C, and then washed to remove any unbound CPMV particle with saline. Cells were fixed and stained with WGA-A488 (Invitrogen) as described above. CPMV was visualized either based on covalently-attached A555 dye or stained using anti-CPMV specific antibodies (see above). Confocal images were captured and analyzed as described above.
Fluorescence activated cell sorting (FACS)
HeLa and HT29 cells were grown to confluency, and collected using enzyme-free Hank′s based Cell Dissociation Buffer, and distributed in 200 μL aliquots at a concentration of 5 x 105 cell/mL in V-bottom 96-well plates. Cargo-loaded and dye-labeled CPMV samples (3 μg and 15 μg/ per well) were added to cells and incubated for 3 h at 37°C, 5% CO2. The cells were washed two times in FACS buffer (PBS solution of 1 mM EDTA, 25 mM HEPES at pH 7, 1 % FBS (v/v)) and fixed in 2% (v/v) formaldehyde in FACS buffer for 10 minutes at room temperature. Cells were washed and resuspended in FACS buffer and analyzed using a BD LSR II flow cytometer. At least 10,000 events (gated for live cells) were recorded. Experiments were repeated at least twice and triplicates of each sample were measured. Data were analyzed using FlowJo 8.6.3 software.
Cell viability assay
XTT Cell Proliferation Assay Kit (ATCC®) was used to determine cell viability. For XTT assay, HeLa, HT-29, and PC-3 cells were seeded on a 96-well plate (25,000 cells/well; 100 μl MEM/well), incubated for 24 h at 37°C, 5% CO2, washed twice, and then incubated in 100 μl MEM containing varying concentrations of cargo-loaded CPMV samples and respective controls (free CPMV and free drug). Time course studies were conducted: cells were treated with candidate formulation for 1 day, washed with saline to remove any unbound particles and drug, and placed in fresh medium for further incubation for 24 hours, 72 hours, and five days, prior to measuring cell viability. At the end of each incubation period, 50 μl of XTT reagent (reconstituted as per instructions in the kit) was added to each well and the plates were incubated for another 2–3 h for color development. The absorbance at 450 nm and 650 nm was then recorded on TECAN Infinite® 200 PRO multimode plate reader; data were analyzed as recommended by the supplier. All assays were analyzed in triplicates and repeated at least twice, data were analyzed using Microsoft Excel software.
Results
Loading CPMV nanoparticles with fluorescent dyes through infusion and nucleic acid-mediated retention
CPMV was propagated in Vigna unguiculata plants and purified using previously described procedures [35]. Typical yields were 100 mg of pure CPMV from 100 g of infected leaf material. The purity of CPMV preparation was assessed using size exclusion chromatography (SEC) and transmission electron microscopy (not shown). Samples were stored in 0.1 M potassium phosphate buffer pH 7.0 at 4°C.
To investigate the possibility and efficiency of dye-loading into the CPMV carrier system through infusion, we chose the following fluorophores: DAPI (4’,6-diamidino-2-phenylindole dihydrochloride), propidium iodide (PI, 3,8-diamino-5-[3-(diethylmethylammonio)propyl]-6-phenylphenanthridinium diiodide), and acridine orange (AO, 3,6-bis(dimethylamino)acridinium chloride), all of which are cationic, nucleic acid intercalating, fluorescent stains.
Intact CPMV nanoparticles were incubated in a bathing solution containing the fluorophores (DAPI, PI, or AO, Figure 1A) at various molar excesses (1,000, 2,000, 5,000, 10,000, and 50,000 dyes per 1 CPMV), incubation times were varied between one hour to overnight reactions. After completion, the reaction mix was extensively purified through several rounds of dialysis and spin filter centrifugation to remove excess reagents and dyes were quantified based on UV/visible absorbance spectroscopy (see materials and methods). Overall, we found that an excess of 10,000 dyes:1 CPMV nanoparticle and incubation for one hour gave most reproducible results in terms of yield of recovered CPMV and dye-loading efficiency. Recovery of purified, dye-loaded CPMV was 50–70% of the starting material. Structural integrity and loading with dye was confirmed using SEC, UV/visible spectroscopy, and native gel electrophoresis (Figure 1B-D).
Figure 1.

A) Structure of DAPI (4’,6-diamidino-2-phenylindole dihydrochloride), propidium iodide (PI, 3,8-diamino-5-[3-(diethylmethylammonio)propyl]-6-phenylphenanthridinium diiodide), and acridine orange (AO, 3,6-bis(dimethylamino)acridinium chloride). B) Size exclusion chromatography of CPMV-DAPI, CPMV-PI, and CPMV-AO shows the typical elution profile of intact CPMV, co-elution of the dyes indicates loading. C) UV/visible spectra of CPMV-DAPI, CPMV-PI, and CPMV-AO showing the CPMV typical peak at 260 nm and the dye-specific absorbance peak at 358, 493, and 470 nm, respectively. D) Native agarose gel electrophoresis of CPMV and eCPMV after incubation with DAPI, PI, AO. The gels were visualized and documented under UV light and then stained with Coomassie blue and photographed under white light.
SEC using FPLC and a Superose6 column showed the typical elution profiles for intact CPMV nanoparticles: CPMV loaded with DAPI, PI, and AO elute at 17.9 min, 17.5 min, and 17.6 min (Figure 1B), respectively, which is in agreement with elution profiles for native CPMV (not shown). The ratio of A260 nm:A280 nm provides additional information of the integrity of CPMV preparations, the peak at 260 nm is from the absorbance of encapsulated nucleic acids and absorbance at 280 nm reflects the protein capsid. Pure and intact CPMV preparations have a A260 nm:A280 nm ratio of 1.7±0.1 [dpv.web.net]. CPMV-DAPI, CPMV-PI, and CPMV-AO, each show A260 nm:A280 nm ratio of 1.7. For CPMV-AO, SEC elution profiles indicate a shoulder at 15.7 min, indicating that some aggregation occurred. Although a small fraction of the CPMV-AO formulation appeared to aggregate, the main peak is indicative of non-aggregated CPMV-AO nanoparticles. The latter was also confirmed by native agarose gel electrophoresis (see below).
FPLC elution profiles indicate successful loading of dyes: co-elution of the DAPI, PI, and AO as measured at 358 nm, 493 nm, and 470 nm, respectively, indicates loading of the dyes into the CPMV nanocarrier (see also UV and native gel data below). PI absorbance is low, which is reflected by its low extinction coefficient with εPI(493 nm)= 5,900 M−1cm−1, compared to DAPI and AO, which have extinction coefficients with values of εDAPI(358 nm)=24,000 M−1cm−1 and εAO(470 nm)=43,000 M−1cm−1.
The degree of dye-loading was quantified using UV/visible spectroscopy and the concentration ratio of dye:CPMV (see materials and methods). We found that CPMV could be loaded with 130±10% DAPI or PI and 155±10% AO; the increased AO ratios may be due to an overestimate based on the aggregated fraction in the preparation. Longer incubation times or larger excess of dye:CPMV did not yield more efficient loading, thus indicating that CPMV is saturated with dyes at a loading capacity of 130–155 dyes per CPMV nanoparticle.
Loading of the fluorescent cargos inside the CPMV carrier was further confirmed using native gel electrophoresis. RNA-containing CPMV and RNA-free empty eCPMV [34] nanoparticles were incubated with dyes, purified to remove unbound dyes, and then analyzed using agarose gels under native conditions. After separation of the intact (e)CPMV dye complexes, gels were visualized under UV light or stained with Coomassie and imaged under white light (Figure 1D). CPMV nanoparticles appear as double-band on native agarose gels; this band pattern reflects a proteolytic cleavage of the small (S) coat protein: CPMV particles with cleaved S have a higher mobility in the gel compared to fractions that contain the full length S protein. Depending on the preparation, the double bands may be more or less profound on the gel [36]. The overall band pattern is consistent with intact (e)CPMV nanoparticles. Furthermore, native gel electrophoresis data indicate that dyes DAPI, PI, and AO were successfully loaded into the CPMV capsids. Uptake of dye into RNA-free eCPMV nanoparticles was not apparent, thus indicating that the loading is dependent on the RNA molecules (see also discussion).
Chemical reactivity of dye-loaded CPMV nanoparticles
Next we sought to investigate the chemical reactivity of the CPMV surface lysine side chains after cargo-loading. Bioconjugation and addressability of the exterior CPMV surface is well known. CPMV nanoparticles display 300 reactive Lys side chains; all of which can be labeled using N-hydroxysuccinimide (NHS) active chemical modifiers and forcing conditions (high excess and long incubation periods) [37]. Using standard labeling protocols, typical labeling efficiency lies between 60–120 labels per CPMV. Here we used a standard labeling protocol (see methods), a NHS active ester of the fluorophore AlexaFluor555 (A555), and DAPI-loaded CPMV or native CPMV. We found that native and DAPI-loaded CPMV nanoparticles showed similar reactivity resulting in covalent display of 80±10% A555 dyes per CPMV and CPMV-DAPI nanoparticle, respectively. The degree of labeling was determined using UV/visible spectroscopy and the A555 specific extinction coefficient (Figure S1).
Native and denaturing gel electrophoresis techniques were used to confirm that DAPI was non-covalently loaded into the interior cavity of CPMV, complexed with the nucleic acids, and that A555 was covalently linked to the CPMV coat proteins (Figure 2). Gels were visualized under UV light and under white light after Coomassie staining. In denaturing gels, CPMV coat proteins are separated and visualized. The process of denaturing releases the encapsulated cargo (here DAPI), which is, based on its small molecular weight (MW = 277.324 gmol−1), detectable in the buffer front at the bottom of the gel (Figure 2A, lanes 2 and 4). The fluorescent appearance of coat proteins for A555-CPMV and A555-CPMV-DAPI (Figure 2A, lanes 3 and 4) indicates covalent modification of both the small (S, 24 kDa) and large (L, 42 kDa) coat protein of CPMV.
Figure 2.
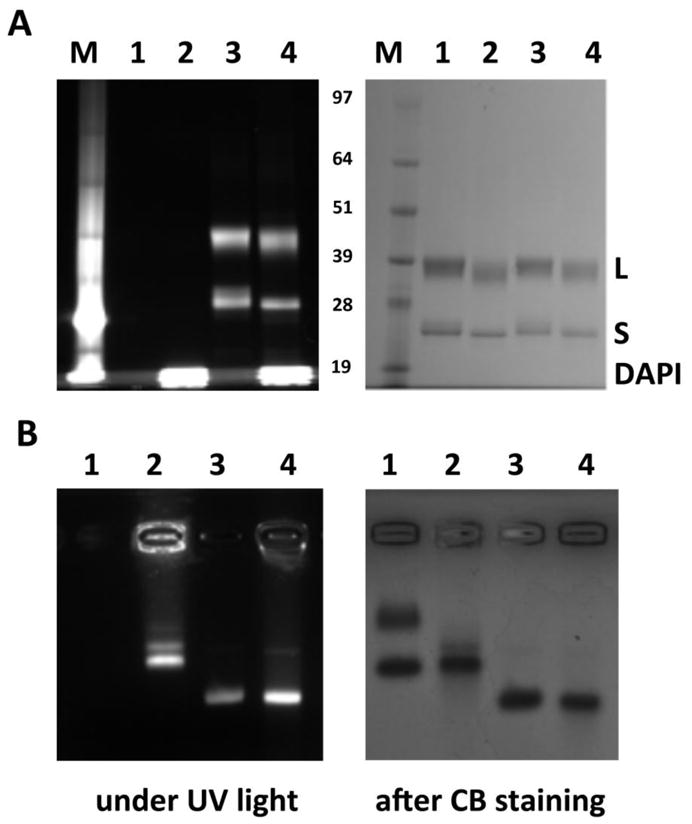
Electrophoretic separation of CPMV-DAPI and their coat proteins. A) Denaturing gel electrophoresis using a NuPAGE gel and B) native gel using an agarose gel. 1 = CPMV, 2 = CPMV-DAPI, 3 = A555-CPMV, 4 = A555-CPMV-DAPI, M = molecular weight standard; the bands are labeled in the center of the gels (in kDa). Gels were visualized under UV light and under white light after Coomassie blue (CB) staining.
In native agarose gels, intact CPMV nanoparticles are analyzed. DAPI-loaded and A555-labeled CPMV formulations appear fluorescent under UV light; free dye is not detected for any of the preparations; indicating that DAPI is stably encapsulated and not released during migration in the gel matrix (Figure 2B). The migration pattern toward the anode differs for the DAPI-loaded versus A555-labeled CPMV: DAPI is encapsulated on the interior of the CPMV particles, and alters the electrophoretic mobility only minimally. In contrast, A555, a non-charged molecule, is covalently attached to surface lysines. The A555-CPMV formulation displays fewer positive charges on its surface compared to native CPMV, and thus has enhanced mobility toward the anode.
CPMV particles have two electrophoretic forms; this is due to cleavage of the highly charged C-terminus of the S protein [36,38]. In denaturing gels this can be observed by the double band that appears for the S protein (Figure 2A). In the native gel both electrophoretic forms are detected for the native CPMV preparation (Figure 2B, lane 1). For DAPI-loaded and chemically-modified A555-labeled CPMV preparations, only the fast electrophoretic form appears (Figure 2B). We have observed this phenomenon previously; it is possible that labeling and purification conditions, further promote cleavage of the S protein.
Overall, data indicate that the chemical addressability for cargo-loaded CPMV nanoparticles is similar to that of native CPMV, allowing for the production of dual-modified CPMV carrier systems.
Cargo-delivery to cells
For a proof-of-concept study, we chose DAPI-loaded CPMV nanoparticles to study their fate in vitro and evaluate cargo delivery to cells. DAPI is a dye commonly used in tissue culture to stain the cell nuclei. The molecule is cell membrane permeable; it diffuses into the nucleus where it intercalates into the DNA. When bound to DNA, DAPI produces a blue fluorescence with excitation at about 360 nm and emission at 460 nm [39]. We hypothesized that CPMV carrying DAPI would bind and internalize into cells via endocytosis to localize within the endolysosomal compartment, where the CPMV carrier is degraded, and DAPI released to target the nucleus.
For our studies, the human cervical cancer cell line HeLa was used. CPMV-HeLa cell interactions are well characterized: We and others have previously reported that CPMV nanoparticles interact with mammalian cells via interaction with surface-expressed vimentin [22,40]. This property can be utilized to target cancer cells, e.g. cervical, colon, and prostate cancer cells [24,25]. (It should be noted that in addition to vimentin-mediated internalization, other endocytotic pathways also could play a role in CPMV-cell interactions). CPMV binds and internalizes into cells via energy-dependent endocytosis and translocates into the endolysosomal compartment [21,32,41].
Time and temperature-dependent cargo-delivery studies were performed: CPMV nanoparticles loaded with DAPI and covalently-labeled with A555 were incubated with HeLa for 10 min versus 60 min and at 4°C versus 37°C. CPMV uptake was not apparent at 4°C (Figure 3, panel E-H); this is consistent with previous studies reporting that CPMV uptake is an energy-dependent process [21,41]. At 37°C CPMV uptake was detectable after 60 min incubation with HeLa cells and accompanied by DAPI fluorescence from the nucleus (Figure 3, panel A-D). DAPI-fluorescence from the CPMV carriers is not detectable, which can be explained by the fact that the DAPI is only weakly fluorescent when incorporated into RNA structures [39]. Fluorescence from the nuclei indicates that DAPI is released from the CPMV carrier inside the cells allowing DAPI to diffuse into the nucleus, where it intercalates into the genomic DNA.
Figure 3.

The fate of CPMV-DAPI in HeLa cells. Time and temperature-dependency: CPMV-DAPI was incubated with HeLa cells at 4°C versus 37°C for 10 min versus 60 min. A, C, E, G = CPMV channel (in green), B, D, F, H = overlay of CPMV channel (in green), cell membrane stain (WGA, in red), and DAPI in blue. Concentration dependence: I-L showing cells after incubation with different concentrations of free DAPI versus CPMV-DAPI, cell membrane in red, DAPI in blue (no CPMV staining). Colocalization: M = overlay of DAPI in blue (note in this experiment cells were stained using free DAPI, and not DAPI delivered through CPMV), endolysosomes (Lamp-1 marker) in red, and CPMV in green, N = Lamp-1 only in red, O = CPMV only in green, P = colocalization analysis using ImageJ and colocalization highlighter plug-in, colocalized signals are shown in white. The scale bars are 50 μm in length.
To confirm that DAPI is indeed released inside the cells as opposed to leaking out of the CPMV carrier in medium during the 60 min incubation time; we conducted a concentration-dependent study: a typical cell nuclei staining protocol makes use of DAPI at 20 mM concentration or higher (Figure 3, panel J). For delivery studies, the DAPI concentration was five magnitudes lower measuring only 0.2 μM DAPI (see methods). Cells incubated with free DAPI at 0.2 μM do not show any apparent fluorescent signals from the nuclei. In stark contrast, 0.2 μM DAPI delivered to cells via the CPMV carrier shows fluorescent signals from the nuclei within 60 min of incubation (Figure 3, panel I-L). This indicates, that even though DAPI is a cell permeable dye, it enters cells more efficiently when delivered through the CPMV nanocarrier. Colocalization studies confirmed intracellular localization and translocation of CPMV into the endolysosomal compartment; this is indicated by co-localization with Lamp-1 marker (Figure 3, panel M-P).
Overall, this proof-of-concept study indicates that cargo infused into CPMV, bound to the viral nucleic acid, can be efficiently delivered into cells. We hypothesize that structural changes and degradation of the CPMV carrier within the endolysosomes triggers release of the cargo allowing for endolysosomal escape and targeting of the nucleus. These studies thus laid the foundation for drug delivery (see below).
Loading of drug molecules via infusion and nucleic acid retention
Next, we sought to investigate drug loading into CPMV followed by drug delivery to cancer cells. We chose proflavine (PF, acridine-3,6-diamine hydrochloride) (Figure 4) for a proof-of-concept study. Proflavine is mostly known as a bacteriostatic with applications as topical antiseptic. Cytotoxic activity of proflavine and its derivatives, e.g. proflavine diureas, in cancer cells and tumors has also been reported: the antiproliferative activity has been related to proflavine intercalation into DNA [42–45]. Although the use of proflavine, as well as other acridine derivatives, for modern chemotherapy may be controversial based on their inherent mutagenic properties [46], we reasoned that proflavine would be a reasonable guest molecule for our studies.
Figure 4.
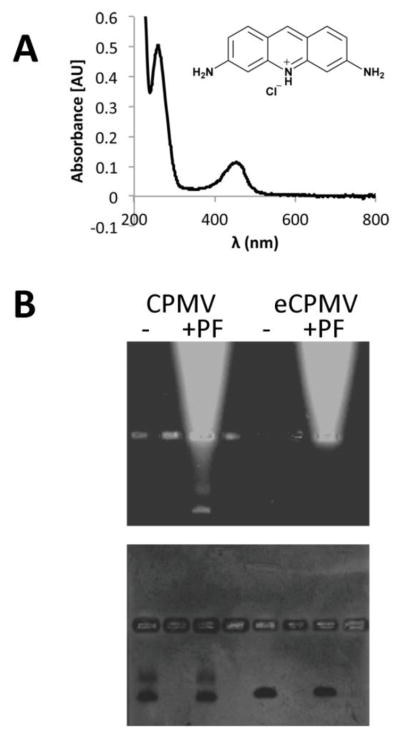
Characterization of drug-loaded CPMV. A) UV/visible spectroscopy of CPMV-PF showing the CPMV and proflavine-specific absorbance maxima at 260 nm and 450 nm. B) Native gel electrophoresis of CPMV and eCPMV with and without proflavine (PF) (it should be noted that non-purified samples were analyzed on the gel to show the migration pattern of free PF versus (e)CPMV); gels were documented under UV light, and then stained with Coomassie blue and photographed under white light. The bright bands in proflavine-positive samples indicate free dye that migrates towards the cathode in the electrophoretic field (on top).
First, loading of proflavine into RNA-containing CPMV and RNA-free eCPMV nanoparticles was studied: intact (e)CPMV nanoparticles were incubated in a bathing solution containing the drugs at various molar excesses ranging between 1,000–50,000 drugs per 1 CPMV (Supporting Figure S2). After completion, the reaction mix was extensively purified through several rounds of dialysis and spin filter centrifugation to remove excess reagents. Proflavine loading was quantified based on UV/visible absorbance spectroscopy (Figure 4A). According to results obtained from dye-loading, we found that an excess of 10,000 proflavine:1 CPMV nanoparticle gave most reproducible results in terms of yield of recovered CPMV (50–70% of starting materials) and drug-loading efficiency of 140±10% proflavine.
To confirm intactness of the preparation and analyze drug loading further, SEC and native gel electrophoresis was performed. Proflavine loading was studied by native gel electrophoresis and imaging gels under UV light (detection of the fluorescent proflavine compound) and under white light after Coomassie blue staining (detection of the protein-based viral nanoparticles). Loading of proflavine was only observed using RNA-containing CPMV nanoparticles (fluorescent bands on UV light, Figure 4B), non-specific uptake or interactions of proflavine with RNA-free eCPMV was not detectable by native gel electrophoresis (Figure 4B). Overall data indicate that proflavine diffuses inside the CPMV carrier where it is retained through interaction with the encapsulated nucleic acids.
Drug delivery, release, and cell killing
Next, we evaluated proflavine delivery to cancer cells. A panel of cancer cells was used for these studies: HeLa (cervical cancer cells), HT-29 (colon cancer cells), and PC-3 (prostate cancer cells). Drug delivery and cell killing was evaluated (Figure 5). CPMV-PF formulations show drug efficacy similar to that observed for free proflavine (Figure 5). In HeLa cells, free proflavine and CPMV-PF showed response with IC50 between 1.8 μM and 2.9 μM proflavine concentration. In HT-29 and PC-3 cells, the IC50 was determined at 6.13 μM for free and delivered drug. The CPMV carrier itself is not toxic to cells (Figure 5). Proflavine is an intercalating agent and this process is reversible; our data indicate that after the CPMV-PF complex enters the cells, the drug is released inducing cell toxicity (Figure 5).
Figure 5.
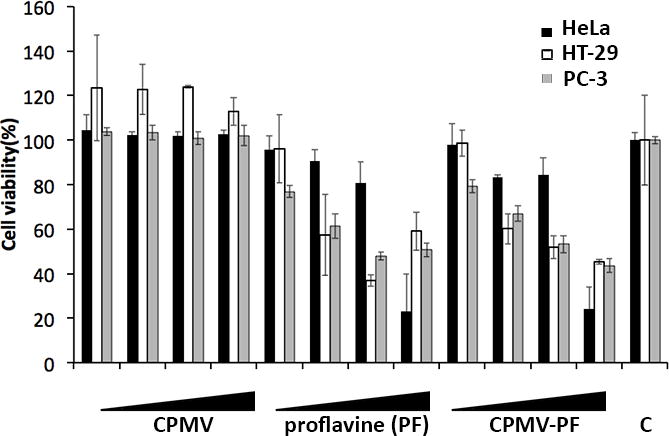
Cell viability assays. HeLa (in black) were exposed to proflavine and CPMV-PF at 0.3 μM, 0.6 μM, 1.8 μM, and 2.9 μM concentration of proflavine (equates to a CPMV concentration of 0.002 μM, 0.004 μM, 0.012 μM, and 0.02 μM) for 24 h, and washed, and incubated for further 24 h in tissue culture medium, prior to analysis of cell viability using XTT assay. C = untreated control cells. HT-29 (in white) and PC-3 (in grey) were exposed to proflavine and CPMV-PF at 1.46 μM, 3.07 μM, 6.13 μM, and 16.06 μM concentration of proflavine (equates to a CPMV concentration of 0.010 μM, 0.021 μM, 0.042 μM, and 0.11 μM) for 24 h, and washed, and incubated for further 24 h in tissue culture medium, prior to analysis of cell viability using XTT assay. C = untreated control cells.
Flow cytometry and confocal microscopy was used to confirm uptake of drug-loaded CPMV in HeLa, HT-29, and PC-3 cells. For these studies, dual-modified drug-loaded and dye-labeled CPMV nanoparticles were produced. First, the drug was loaded through infusion; second, A555 was covalently attached using an NHS ester and targeting lysine side chains. SEC, UV/visible spectroscopy, and native gels confirmed the integrity of dual-modified CPMV; 100±10% A555 were attached per CPMV-PF (not shown). Cell data confirmed binding (Figure 6A) and uptake of CPMV into HeLa, HT-29, and PC-3 cells (Figure 6B), this is consistent with previous reports: HeLa, HT-29, and PC-3 express surface vimentin, allowing CPMV to target, bind and get taken up into the cells [24,25]. In summary, we demonstrate that CPMV nanoparticles can be efficiently labeled with therapeutic cargos, and the natural CPMV-vimentin specificity enables targeting, uptake, and cargo delivery.
Figure 6.
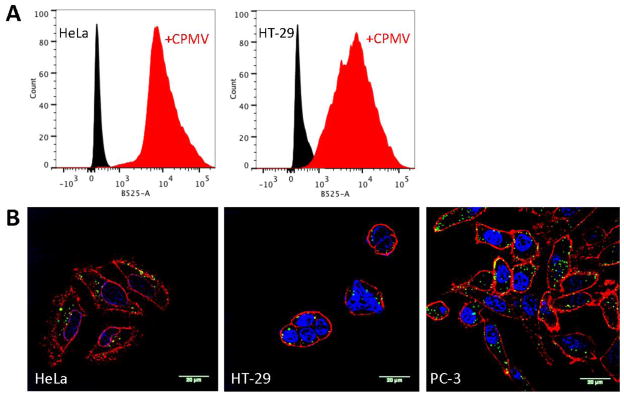
A) Cell binding of A555-CPMV-PF to HeLa and HT-29 cells after 60 min exposure. For these experiments cells were collected using non-enzymatic cell dissociation buffers to avoid the natural CPMV receptor being cleaved off the cell surface; we have not achieved collection of PC-3 cells using this method. B) Confocal microscopy images of HeLa, HT-29, and PC-3 cells after incubation with A555-CPMV-PF. Red = cell membrane (WGA staining), blue = nuclei (DAPI staining), and green = CPMV (from A555 dye). The scale bar is 20 μm.
Discussion
Nanoparticles in drug delivery
Nanoparticles are potentially useful for medical applications because they can be tailored to partition cargos between diseased and healthy cells and tissues. Diverse classes of materials are currently being considered; these include synthetic, man-made materials as well as natural nanomaterials, e.g. protein cages and capsids formed by viruses. Each class of nanomaterial offers distinct advantages and disadvantages. CPMV has many favorable properties for use as nanocarrier:
CPMV nanoparticles are non-pathogenic, non-toxic, and biodegradable in mammals at dosages of up to 100 mg (1016 CPMVs) per kg body weight [9].
CPMV nanoparticles are 30 nm in size; this size regime is ideal for cell targeting and uptake [47]. Furthermore, based on their small size, CPMV has high likelihood to penetrate tissues more effectively compared to larger micelles or liposomes [2].
CPMV is monodisperse, and its structure known to atomic resolution. CPMV can be engineered with targeting ligands, drugs and/or imaging molecules at the exterior and interior surface using genetic engineering or bioconjugation protocols [20].
CPMV nanoparticles are stable under various solvent, pH, and temperature conditions.
We demonstrate in this work, that cargos are released efficiently upon targeting of the endolysosome; this is consistent with a previous study in which we delivered the chemotherapeutic molecule doxorubicin; in this case the drug was covalently introduced into the nanocarrier [32]. It appears that CPMV is metabolically cleared from cells within a few days [21,48]. The slow processing of the CPMV nanoparticles inside the endolysosome results in delayed drug release when the cargo is conjugated via a covalent mechanism [32]. In contrast, we report here, that cargos stably loaded via infusion technique were released quickly upon cell entry. For example, DAPI delivered by CPMV was detectable in the nucleus after 60 min exposure. It is possible that conformational changes in the capsid structure are induced upon entry into the acidic environment of the endolysosomal compartment, and thus inducing cargo release and eventual degradation of the carrier material.
Based on its biology and natural affinity to surface expressed vimentin, CPMV provides an interesting carrier system to deliver cargos to vimentin-positive (cancer) cells [22].
Besides all its advantages it should be noted, that a potential disadvantage of the protein-based carrier systems is that the repetitive coat proteins can induce immunogenicity, but this can be overcome by PEGylation [15].
Modification of virus-based materials
Based on the versatility of virus-based materials as carrier systems, we and others have reported various modification techniques to functionalize the carriers with cargos and/or targeting ligands. A majority of efforts have focused on genetic and chemical modification [16]. Non-covalent techniques such as infusion have several advantages:
While genetic engineering is only applicable to amino acid-based compounds, infusion-based cargo-loading is, at least theoretically, applicable to any material, including peptides, organic fluorophores, contrast agents, or chemotherapeutic drugs.
Infusion-based methods do not alter the composition or structure of the cargo; in contrast covalent modification can introduce alternations to the cargo rendering it less or non-active. Metabolic degradation and/or structural changes of the CPMV carrier within the endolysosomal compartment allow cargo-release without the need of introduction of release mechanisms, which could further hamper the functionality of the cargo.
Some genetic and/or chemical modifications can destabilize the protein structure. Modifications are not required for infusion-based cargo loading. Intact and native CPMV nanoparticles are used; which means that no structural changes are made to the virus-based carrier.
A few non-covalent VNP modification strategies have been developed and tested, for example cowpea chlorotic mottle virus (CCMV) was used to complex lanthanides at the interface of coat protein subunits. Under physiological conditions Ca2+ ions are bound to these sites. Ca2+ ions can be replaced with Gd3+ or Tb3+ cations; resulting in binding of 180 lanthanides [49,50]. These complexes could be potentially useful for magnetic resonance imaging applications. Similarly, the lanthanides Gd3+ and Tb3+ were infused and entrapped into CPMV particles making use of the encapsidated nucleic acids. Around 80 ± 20 Gd3+ and Tb3+ ions can be stably bound and trapped inside CPMV based on RNA interactions [9,51].
Using red clover necrotic mosaic virus (RCNMV), it was demonstrated that fluorescent dyes and doxorubicin could be infused making use of RCNMV’s pH- and metal ion-dependent reversible gating mechanism; at low pH the particles are in a compact conformation, upon increase of pH a structural transition leads to a swelling and pore-opening [52] In the swollen, open conformation and in the presence of RNA molecules, small positively charged molecules can freely diffuse into the interior cavity of the particles, where they bind to the negatively charged viral nucleic acids via electrostatic interactions. Lowering of the pH reverts the structural transitions; the particles appear in the compact, closed conformation and the infused molecules are trapped [53]. Making use of this gating mechanisms has also been shown a feasible approach to entrap negatively-charged polymers within the RCNMV nanoparticle [54].
Another approach was developed studying bacteriophage MS2: MS2 phages contain a translational repression (TR) operator that binds to a TR RNA stem loop. TR operator proteins can be chemically engineered and small drug molecules can be covalently attached. When intact MS2 particles are exposed to such TR operators the proteins diffuse inside the VNPs and bind stably to the 90 RNA stem loops. Therapeutic molecules such as plant toxin ricin A chain or 5-fluorouridine have been successfully incorporated into MS2 using these design principles. In vitro cell studies confirmed cargo delivery and successful cell killing of target cells [55,56].
Infusion of small guest molecules into CPMV, as we report here, presents a convenient means of loading cargos into RNA-containing CPMV nanoparticles. The requirement for the cargo is that is has positive charges and/or affinity toward nucleic acids. To enable release, the interaction with nucleic acids must be reversible (see Figure 5). Our data indicate that nucleic acid intercalating molecules such as DAPI and proflavine bind to CPMV carriers via a reversible mechanism and thus can be released inside cells (see Figures 3 and 5).
We demonstrated that cargo molecules were stably bound inside RNA-containing CPMV nanoparticles; non-specific loading into eCPMV nanoparticles was not observed (see Figures 2 and 4). The formulations remained structurally sound and the guest molecules were stably encapsulated for several weeks upon storage in the fridge in phosphate buffered saline solution at physiological pH. Cargo release in medium and during electrophoresis was not observed. Upon entry into the endolysosome, efficient release over relatively short time scales is triggered: it was indicated that DAPI was released within 60 min of exposure (see Figure 3). Further, the IC50 of CPMV-PF was comparable to that of free proflavine, further indicating efficient release (see Figure 5).
RNA-containing CPMV nanoparticles are non-infectious toward mammalian cells, and therefore can be considered as safe. From an agricultural point of view, of course, RNA-containing nanoparticles are infectious toward legumes, such as black-eyed peas. To produce cargo-loaded CPMV-based nanoparticles that are safe from an agricultural point of view, one could consider the following strategy: three forms of CPMV nanoparticles can be isolated from infectious leaves by isopycnic centrifugation on density gradients. The three components have identical protein composition but differ in their RNA contents [57–59]. The particles of the top (T) component are devoid of RNA, while the M and B components each contain a single RNA molecule, RNA-2 and RNA-1, respectively [60]. While RNA-1 encodes the replication machinery, RNA-2 encodes the coat proteins. The presence of both RNA molecules is required to yield an infection and production of intact CPMV particles in the plants. One could consider separating B and M components for downstream medical applications to avoid any potential agricultural safety issues.
It would be interesting to conduct future loading experiments on separated B and M components. RNA-1 is 5889 nucleotides long and thus 1.7 times longer compared to RNA-2 that consists of 3481 nucleotides [61,62]. It is possible that more efficient cargo loading could be achieved by using purified B components that contain the longer RNA-1 polymer, however, this will also largely depend on the secondary structure of the encapsidated RNA molecules. Generally, we observed reproducible labeling from batch-to-batch with some variation between different compounds tested, resulting in infusion of 130 and 155 dye/drug molecules per CPMV nanoparticle (see Figures 2+4).
The chemical reactivity of cargo-loaded CPMV nanoparticles appears to be non-altered (see Figure 2), which provides a foundation for the synthesis of dual-modified nanoparticles, e.g. encapsulating a therapeutic cargo while displaying contrast agents on the exterior surface toward the development of theranostic devices.
Supplementary Material
Acknowledgments
This work was supported by NIH grants NIBIB R00 EB009105 (NFS), a NCI R25 CA148052 Cancer Pharmacology training grant (KLL), and Mt. Sinai Foundation (NFS). Pooja Saxena and Prof. George Lomonossoff (John Innes Centre, Norwich, UK) are thanked for providing eCPMV samples.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J Control Release. 2000;65(1–2):271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 2.Perrault SD, Walkey C, Jennings T, Fischer HC, Chan WC. Mediating tumor targeting efficiency of nanoparticles through design. Nano letters. 2009;9(5):1909–1915. doi: 10.1021/nl900031y. [DOI] [PubMed] [Google Scholar]
- 3.Ruoslahti E. Vascular zip codes in angiogenesis and metastasis. Biochem Soc Trans. 2004;32(Pt3):397–402. doi: 10.1042/BST0320397. [DOI] [PubMed] [Google Scholar]
- 4.Chitale R. Merck hopes to extend gardasil vaccine to men. J Natl Cancer Inst. 2009;101(4):222–223. doi: 10.1093/jnci/djp014. [DOI] [PubMed] [Google Scholar]
- 5.Liu TC, Galanis E, Kirn D. Clinical trial results with oncolytic virotherapy: A century of promise, a decade of progress. Nat Clin Pract Oncol. 2007;4(2):101–117. doi: 10.1038/ncponc0736. [DOI] [PubMed] [Google Scholar]
- 6.Shirakawa T. Clinical trial design for adenoviral gene therapy products. Drug News Perspect. 2009;22(3):140–145. doi: 10.1358/dnp.2009.22.3.1354090. [DOI] [PubMed] [Google Scholar]
- 7.Manchester M, Singh P. Virus-based nanoparticles (VNPs): Platform technologies for diagnostic imaging. Adv Drug Deliv Rev. 2006;58(14):1505–1522. doi: 10.1016/j.addr.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 8.Kaiser CR, Flenniken ML, Gillitzer E, Harmsen AL, Harmsen AG, Jutila MA, Douglas T, Young MJ. Biodistribution studies of protein cage nanoparticles demonstrate broad tissue distribution and rapid clearance in vivo. Int J Nanomedicine. 2007;2(4):715–733. [PMC free article] [PubMed] [Google Scholar]
- 9.Singh P, Prasuhn D, Yeh RM, Destito G, Rae CS, Osborn K, Finn MG, Manchester M. Bio-distribution, toxicity and pathology of cowpea mosaic virus nanoparticles in vivo. J Control Release. 2007;120(1–2):41–50. doi: 10.1016/j.jconrel.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou JG, Chen YM. Research on PEGylation of porcine prothrombin for improving biostability and reducing animal immunogenicity. Bioorg Med Chem Lett. 2011;21(11):3268–3272. doi: 10.1016/j.bmcl.2011.04.037. [DOI] [PubMed] [Google Scholar]
- 11.Park JB, Kwon YM, Lee TY, Brim R, Ko MC, Sunahara RK, Woods JH, Yang VC. PEGylation of bacterial cocaine esterase for protection against protease digestion and immunogenicity. J Control Release. 2010;142(2):174–179. doi: 10.1016/j.jconrel.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Basu A, Yang K, Wang M, Liu S, Chintala R, Palm T, Zhao H, Peng P, Wu D, Zhang Z, Hua J, et al. Structure-function engineering of interferon-beta-1b for improving stability, solubility, potency, immunogenicity, and pharmacokinetic properties by site-selective mono-PEGylation. Bioconjug Chem. 2006;17(3):618–630. doi: 10.1021/bc050322y. [DOI] [PubMed] [Google Scholar]
- 13.Yang Z, Wang J, Lu Q, Xu J, Kobayashi Y, Takakura T, Takimoto A, Yoshioka T, Lian C, Chen C, Zhang D, et al. PEGylation confers greatly extended half-life and attenuated immunogenicity to recombinant methioninase in primates. Cancer Res. 2004;64(18):6673–6678. doi: 10.1158/0008-5472.CAN-04-1822. [DOI] [PubMed] [Google Scholar]
- 14.O’Riordan CR, Lachapelle A, Delgado C, Parkes V, Wadsworth SC, Smith AE, Francis GE. PEGylation of adenovirus with retention of infectivity and protection from neutralizing antibody in vitro and in vivo. Hum Gene Ther. 1999;10(8):1349–1358. doi: 10.1089/10430349950018021. [DOI] [PubMed] [Google Scholar]
- 15.Raja KS, Wang Q, Gonzalez MJ, Manchester M, Johnson JE, Finn MG. Hybrid virus-polymer materials. 1. Synthesis and properties of PEG-decorated cowpea mosaic virus. 2003;3:472–476. doi: 10.1021/bm025740+. [DOI] [PubMed] [Google Scholar]
- 16.Pokorski JK, Steinmetz NF. The art of engineering viral nanoparticles. Mol Pharm. 2011;8(1):29–43. doi: 10.1021/mp100225y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chatterji A, Ochoa WF, Ueno T, Lin T, Johnson JE. A virus-based nanoblock with tunable electrostatic properties. Nano Lett. 2005;5(4):597–602. doi: 10.1021/nl048007s. [DOI] [PubMed] [Google Scholar]
- 18.Hong V, Presolski SI, Ma C, Finn MG. Analysis and optimization of copper-catalyzed azide-alkyne cycloaddition for bioconjugation. Angewandte Chemie (International ed. 2009;48(52):9879–9883. doi: 10.1002/anie.200905087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Q, Lin T, Johnson JE, Finn MG. Natural supramolecular building blocks: Cysteine-added mutants of cowpea mosaic virus. Chem Biol. 2002;9(7):813–819. doi: 10.1016/s1074-5521(02)00166-7. [DOI] [PubMed] [Google Scholar]
- 20.Wen AM, Shukla S, Saxena P, Aljabali AA, Yildiz I, Dey S, Mealy JE, Yang AC, Evans DJ, Lomonossoff GP, Steinmetz NF. Interior engineering of a viral nanoparticle and its tumor homing properties. Biomacromolecules. 2012;13(12):3990–4001. doi: 10.1021/bm301278f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu Z, Chen K, Yildiz I, Dirksen A, Fischer R, Dawson PE, Steinmetz NF. Development of viral nanoparticles for efficient intracellular delivery. Nanoscale. 2012;4(11):3567–3576. doi: 10.1039/c2nr30366c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koudelka KJ, Destito G, Plummer EM, Trauger SA, Siuzdak G, Manchester M. Endothelial targeting of cowpea mosaic virus (CPMV) via surface vimentin. PLoS Pathog. 2009;5(5):e1000417. doi: 10.1371/journal.ppat.1000417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lewis JD, Destito G, Zijlstra A, Gonzalez MJ, Quigley JP, Manchester M, Stuhlmann H. Viral nanoparticles as tools for intravital vascular imaging. Nature medicine. 2006;12(3):354–360. doi: 10.1038/nm1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Steinmetz NF, Cho CF, Ablack A, Lewis JD, Manchester M. Cowpea mosaic virus nanoparticles target surface vimentin on cancer cells. Nanomedicine (Lond) 2011;6(2):351–364. doi: 10.2217/nnm.10.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steinmetz NF, Mauger J, Sheng H, Bensussan A, Marcic I, Kumar V, Braciak T. Two domains of vimentin are expressed on the surface of lymph node, bone and brain metastatic prostate cancer lines along with the putative stem cell marker proteins CD44 and CD133. Cancers. 2011;3:2870–2885. doi: 10.3390/cancers3032870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Plummer EM, Thomas D, Destito G, Shriver LP, Manchester M. Interaction of cowpea mosaic virus nanoparticles with surface vimentin and inflammatory cells in atherosclerotic lesions. Nanomedicine. 2012;7(6):877–888. doi: 10.2217/nnm.11.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shriver LP, Koudelka KJ, Manchester M. Viral nanoparticles associate with regions of inflammation and blood brain barrier disruption during CNS infection. Journal of Neuroimmunology. 2009 doi: 10.1016/j.jneuroim.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brunel FM, Lewis JD, Destito G, Steinmetz NF, Manchester M, Stuhlmann H, Dawson PE. Hydrazone ligation strategy to assemble multifunctional viral nanoparticles for cell imaging and tumor targeting. Nano Lett. 2010;10(3):1093–1097. doi: 10.1021/nl1002526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Destito G, Yeh R, Rae CS, Finn MG, Manchester M. Folic acid-mediated targeting of cowpea mosaic virus particles to tumor cells. Chem Biol. 2007;14(10):1152–1162. doi: 10.1016/j.chembiol.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hovlid ML, Steinmetz NF, Laufer B, Lau JL, Kuzelka J, Wang Q, Hyypia T, Nemerow GR, Kessler H, Manchester M, Finn MG. Guiding plant virus particles to integrin-displaying cells. Nanoscale. 2012;4(12):3698–3705. doi: 10.1039/c2nr30571b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steinmetz NF, Ablack AL, Hickey JL, Ablack J, Manocha B, Mymryk JS, Luyt LG, Lewis JD. Intravital imaging of human prostate cancer using viral nanoparticles targeted to gastrin-releasing peptide receptors. Small. 2011;7(12):1664–1672. doi: 10.1002/smll.201000435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aljabali AA, Shukla S, Lomonossoff GP, Steinmetz NF, Evans DJ. CPMV-DOX delivers. Molecular Pharmaceutics. 2012 doi: 10.1021/mp3002057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wellink J. Comovirus isolation and rna extraction. Meth Mol Biol. 1998;81:205–209. doi: 10.1385/0-89603-385-6:205. [DOI] [PubMed] [Google Scholar]
- 34.Saunders K, Sainsbury F, Lomonossoff GP. Efficient generation of cowpea mosaicvirus empty virus-like particles by the proteolytic processing of precursors in insect cells and plants. Virology. 2009 doi: 10.1016/j.virol.2009.08.023. [DOI] [PubMed] [Google Scholar]
- 35.Wen AM, Lee KL, Yildiz I, Bruckman MA, Shukla S, Steinmetz NF. Viral nanoparticles for in vivo tumor imaging. J Vis Exp. 2012:69. doi: 10.3791/4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Steinmetz NF, Evans DJ, Lomonossoff GP. Chemical introduction of reactive thiols into a viral nanoscaffold: A method that avoids virus aggregation. Chembiochem. 2007;8(10):1131–1136. doi: 10.1002/cbic.200700126. [DOI] [PubMed] [Google Scholar]
- 37.Chatterji A, Ochoa W, Paine M, Ratna BR, Johnson JE, Lin T. New addresses on an addressable virus nanoblock: Uniquely reactive lys residues on cowpea mosaic virus. Chem Biol. 2004;11(6):855–863. doi: 10.1016/j.chembiol.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 38.Sainsbury F, Saunders K, Aljabali AA, Evans DJ, Lomonossoff GP. Peptide-controlled access to the interior surface of empty virus nanoparticles. Chembiochem. 2011;12(16):2435–2440. doi: 10.1002/cbic.201100482. [DOI] [PubMed] [Google Scholar]
- 39.Kapuscinski J. DAPI: A DNA-specific fluorescent probe. Biotech Histochem. 1995;70(5):220–233. doi: 10.3109/10520299509108199. [DOI] [PubMed] [Google Scholar]
- 40.Koudelka KJ, Rae CS, Gonzalez MJ, Manchester M. Interaction between a 54-kilodalton mammalian cell surface protein and cowpea mosaic virus. J Virol. 2007;81 (4):1632–1640. doi: 10.1128/JVI.00960-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Plummer EM, Manchester M. Endocytic uptake pathways utilized by cpmv nanoparticles. Molecular Pharmaceutics. 2013;10(1):26–32. doi: 10.1021/mp300238w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kozurkova M, Sabolova D, Janovec L, Mikes J, Koval J, Ungvarsky J, Stefanisinova M, Fedorocko P, Kristian P, Imrich J. Cytotoxic activity of proflavine diureas: Synthesis, antitumor, evaluation and DNA binding properties of 1’,1”-(acridin-3,6-diyl)-3’,3”-dialkyldiureas. Bioorganic & medicinal chemistry. 2008;16(7):3976–3984. doi: 10.1016/j.bmc.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 43.Sasikala WD, Mukherjee A. Molecular mechanism of direct proflavine-DNA intercalation: Evidence for drug-induced minimum base-stacking penalty pathway. The journal of physical chemistry B. 2012;116(40):12208–12212. doi: 10.1021/jp307911r. [DOI] [PubMed] [Google Scholar]
- 44.Aggarwal AK, Neidle S. Nucleic acid binding drugs. Part xiii. Molecular motion in a drug-nucleic acid model system: Thermal motion analysis of a proflavine-dinucleoside crystal structure. Nucleic acids research. 1985;13(15):5671–5684. doi: 10.1093/nar/13.15.5671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Balda BR, Birkmayer GD. Drug effects in melanoma: Tumor-specific interactions of proflavine and ethidiumbromide. Yale J Biol Med. 1973;46(5):464–470. [PMC free article] [PubMed] [Google Scholar]
- 46.Wainwright M. Acridine-a neglected antibacterial chromophore. The Journal of antimicrobial chemotherapy. 2001;47(1):1–13. doi: 10.1093/jac/47.1.1. [DOI] [PubMed] [Google Scholar]
- 47.Gao H, Shi W, Freund LB. Mechanics of receptor-mediated endocytosis. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(27):9469–9474. doi: 10.1073/pnas.0503879102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rae CS, Khor IW, Wang Q, Destito G, Gonzalez MJ, Singh P, Thomas DM, Estrada MN, Powell E, Finn MG, Manchester M. Systemic trafficking of plant virus nanoparticles in mice via the oral route. Virology. 2005;343(2):224–235. doi: 10.1016/j.virol.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 49.Allen M, Bulte JW, Liepold L, Basu G, Zywicke HA, Frank JA, Young M, Douglas T. Paramagnetic viral nanoparticles as potential high-relaxivity magnetic resonance contrast agents. Magn Reson Med. 2005;54(4):807–812. doi: 10.1002/mrm.20614. [DOI] [PubMed] [Google Scholar]
- 50.Basu G, Allen M, Willits D, Young M, Douglas T. Metal binding to cowpea chlorotic mottle virus using terbium(III) fluorescence. J Biol Inorg Chem. 2003;8(7):721–725. doi: 10.1007/s00775-003-0470-7. [DOI] [PubMed] [Google Scholar]
- 51.Prasuhn DE, Jr, Yeh RM, Obenaus A, Manchester M, Finn MG. Viral MRI contrast agents: Coordination of Gd by native virions and attachment of Gd complexes by azide-alkyne cycloaddition. Chem Commun. 2007;12:1269–1271. doi: 10.1039/b615084e. [DOI] [PubMed] [Google Scholar]
- 52.Sherman MRHG, Tama F, Sit TL, Brooks CL, Mikhailov AMEVO, Baker TS, Lommel SA. J Virol. 2006;80:10395–10406. doi: 10.1128/JVI.01137-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Loo L, Guenther RH, Lommel SA, Franzen S. Infusion of dye molecules into red clover necrotic mosaic virus. Chemical communications (Cambridge, England) 2008;1:88–90. doi: 10.1039/b714748a. [DOI] [PubMed] [Google Scholar]
- 54.Douglas T, Young M. Host-guest encapsulation of materials by assembled virus protein cages. Nature. 1998;393:152–155. [Google Scholar]
- 55.Brown WL, Mastico RA, Wu M, Heal KG, Adams CJ, Murray JB, Simpson JC, Lord JM, Taylor-Robinson AW, Stockley PG. Rna bacteriophage capsid-mediated drug delivery and epitope presentation. Intervirology. 2002;45(4–6):371–380. doi: 10.1159/000067930. [DOI] [PubMed] [Google Scholar]
- 56.Wu M, Brown WL, Stockley PG. Cell-specific delivery of bacteriophage-encapsidated ricin a chain. Bioconjug Chem. 1995;6(5):587–595. doi: 10.1021/bc00035a013. [DOI] [PubMed] [Google Scholar]
- 57.Bancroft JB. Purification and properties of bean pod mottle virus and associated centrifugal and electrophoretic components. Virology. 1962;16:419–427. doi: 10.1016/0042-6822(62)90222-2. [DOI] [PubMed] [Google Scholar]
- 58.Bruening G, Agrawal HO. Infectivity of a mixture of cowpea mosaic virus ribonucleoprotein components. Virology. 1967;32(2):306–320. doi: 10.1016/0042-6822(67)90279-6. [DOI] [PubMed] [Google Scholar]
- 59.Wu GJ, Bruening G. Two proteins from cowpea mosaic virus. Virology. 1971;46(3):596–612. doi: 10.1016/0042-6822(71)90063-8. [DOI] [PubMed] [Google Scholar]
- 60.Lomonossoff GP, Johnson JE. The synthesis and structure of comovirus capsids. Prog Biophys Mol Biol. 1991;55(2):107–137. doi: 10.1016/0079-6107(91)90003-b. [DOI] [PubMed] [Google Scholar]
- 61.Goldbach RW, Rezelman G, van Kammen A. Nature. 1980;286:297–300. [Google Scholar]
- 62.Pelham HR. Synthesis and proteolytic processing of cowpea mosaic virus proteins in reticulocyte lysates. Virology. 1979;96(2):463–477. doi: 10.1016/0042-6822(79)90104-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.