

Abstract
HIV-1 protease is an important target for the development of antiviral inhibitors to treat AIDS. A room-temperature joint X-ray/neutron structure of the protease in complex with clinical drug amprenavir has been determined at 2.0 Å resolution. The structure provides direct determination of hydrogen atom positions in the enzyme active site. Analysis of the enzyme-drug interactions suggests that some hydrogen bonds may be weaker than deduced from the non-hydrogen interatomic distances. This information may be valuable for the design of improved protease inhibitors.
Keywords: neutron diffraction, HIV-1 protease, enzyme-drug complex, amprenavir, drug design
INTRODUCTION
Human immunodeficiency virus type-1 (HIV-1) protease (PR) is an essential enzyme for the viral life-cycle, aiding in the production of mature virions through hydrolysis of viral polyproteins Gag and Gag-Pol into structural and functional proteins. When the enzymatic activity of PR is inhibited immature noninfectious viral particles are generated.1,2 Therefore, PR is a valuable target for the design of small-molecule Protease Inhibitors (PIs). Since the first X-ray structures of PR were published3–5 in 1989, the design of PIs has been guided by the information gained from enzyme-inhibitor structures. The crystal structures of such PR-PI complexes can reveal unfilled spaces in the binding site for incorporation of hydrophobic groups and potential hydrogen (H) bond donors or acceptors to increase the binding affinity of modified inhibitors. The power of this structure-assisted approach to drug design is demonstrated by its contribution to the development of potent clinical PIs.6–9
Active PR is a homodimer of two identical subunits, with residues numbered 1–99 and 1'–99'. Each subunit provides a conserved triad Asp25-Thr26-Gly27 to form the catalytic site with the two aspartates positioned adjacent to each other, a characteristic feature in aspartic proteases. PIs and substrates bind to the cylindrically shaped active-site cavity between the catalytic aspartates and the flexible flaps comprising residues 45–55 and 45'–55'.10,11 Although early PIs mimicked the chemical structure and binding of substrates, later inhibitors, such as amprenavir (APV, Figure 1), incorporated less peptidic character and included a sulfonamide group to increase water solubility.6,12,13 Similar to more recent PIs, APV has a hydroxyethyleneamine core, which mimics the gem-diol tetrahedral intermediate of the PR-catalyzed hydrolysis reaction. APV was designed to maximize hydrophilic interactions with the active-site groups, and direct and water-mediated putative H bonding interactions have been reported in several X-ray structures of wild-type and mutant PR-APV complexes.14–16
Figure 1.
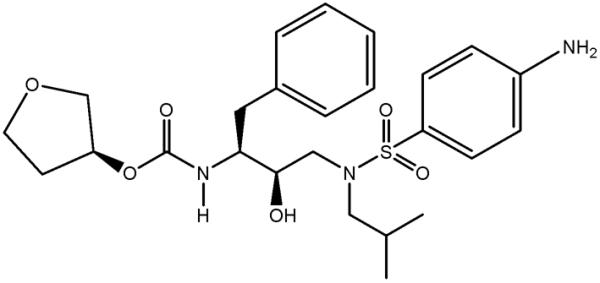
Chemical diagram of APV. Tetrahydrofuran group is termed P2, and aniline substituent is P2'.
X-ray crystallography is the most prevalent experimental method employed in structure-assisted drug design. Structure solution and refinement is increasingly automated, while new-generation synchrotron sources allow diffraction data to be collected rapidly and to high resolution from small crystal samples. A critical limitation of X-rays is, however, their weak sensitivity to H atoms. X-rays are scattered by electrons, and with only one electron H is difficult to observe in electron density maps. Even at ultra-high resolutions of < 1 Å many H atoms remain invisible due to thermal motions and atom-H covalent bond polarization that diminishes electron density on H. In fact, we could not detect any H atoms in the active site of the V32I mutant PR variant bound with the clinical drug darunavir even at the ultra-high resolution of 0.84 Å.17 This severe limitation precludes the direct observation of H atoms in the complex of a protein with bound drug and solvent molecules. Hence it is not possible to assign protonation states and interpret H bonding interactions, information which could be extremely valuable in designing better inhibitors with stronger drug-enzyme interactions.
Unlike X-rays, neutrons are scattered by atomic nuclei and their scattering power is not directly proportional to atomic number. In particular, H and its heavier isotope deuterium, D, scatter neutrons as well as carbon, nitrogen and oxygen. Consequently, the positions of H and D atoms can be determined unambiguously in macromolecular structures refined against neutron diffraction data collected to medium resolutions of about 2.0–2.2 Å or better.18–20 Moreover, neutrons do not cause direct radiation damage and crystals do not need to be cryo-cooled but can be maintained at more physiologically relevant temperatures. On the other hand, due to the relatively weak fluxes of currently available neutron beamlines, a successful neutron crystallographic experiment requires crystals that are orders of magnitude larger than those utilized at synchrotrons. In addition, H possesses a large incoherent scattering cross section that contributes to background scattering and significantly reduces the signal-to-noise ratio. Replacing H by D (deuteration) can help overcome these problems, because D has a much lower incoherent scattering cross section than H. Deuteration can be partially achieved by soaking crystals in D2O solutions. Soaking replaces H2O by D2O and also labile and accessible H atoms on the protein and ligand by D atoms (e.g. OH, NH and SH groups), which typically make up ~15–20% of the solvent-accessible hydrogen content of the protein. Replacing all H atoms by D atoms, i.e. perdeuteration, can be achieved by expression of the protein from bacteria grown on deuterated media.21,22 Perdeuteration enhances data quality and permits the use of smaller crystal volumes by at least several-fold relative to D2O-soaked crystals23,24, without significantly affecting the protein structure.18,19
RESULTS AND DISCUSSION
Here, we report the neutron structure of wild-type PR in complex with the clinical inhibitor APV (PR-APV) determined at 2.0 Å resolution (Supporting Table 1). PR was perdeuterated at the level of 85% by expression in pure (100%) D2O with hydrogenous glucose as the sole carbon source (see Supporting Information); crystals of up to 0.2 mm3 were grown at pH 6.0 and then subjected to H/D exchange (Supporting Figure 1). Importantly, we learned that when the 100% perdeuterated PR was expressed using fully deuterated growth medium, the resulting PR-APV crystals were several-fold smaller. Both X-ray and neutron data were combined in joint X-ray/Neutron (XN) refinement.25 We saw no indication in the resulting structure of the inhibitor disorder (Figure 2a, 2b), which was observed in the previous atomic resolution X-ray structure of the complex at cryogenic temperature.16 We used hydrogenous APV for this study, therefore only H on aniline, hydroxyl and amide groups were exchanged by D, while the aliphatic and aromatic H atoms were visible as troughs in the negative 2FO-FC neutron scattering length density map, because H has a negative neutron scattering length (Figure 2c).
Figure 2.

Electron and neutron scattering length density maps for APV. (a) 2FO-FC electron density map contoured at 2.2σ level. (b) positive 2FO-FC neutron scattering length density map contoured at 1.5σ level. (c) negative 2FO-FC neutron scattering length density map contoured at 2.0σ level.
Previous solvent kinetic isotope effect and 13C NMR studies have established pKa values of ~3.5 and 6–6.5 for the two catalytic aspartates Asp25 and Asp25' indicating that only one Asp residue should be protonated in the 3–6.5 pH range.26–29 Our XN structure of the PR-APV complex confirms that the aspartate dyad is monoprotonated. We directly observed the location of the Asp25 D atom on the inner Oδ1 oxygen and the D atom on the hydroxyl oxygen of APV in the omit difference FO-FC neutron scattering length density map (Figure 3a). The protonated Asp25 donates the D in a short H bond (O…O separation is 2.5 Å) with the hydroxyl of APV, while the hydroxyl group of APV donates D in a similarly short H bond with the outer Oδ2 oxygen of Asp25'.30 Nonetheless, the former H bond is considerably more linear than the latter, and therefore may be stronger, with the O-D…O angles being 168° and 130°, respectively (Figure 3b). Despite the short O…O distances the positions of the D atoms clearly indicate that they are not low-barrier H bonds, which are signified by the symmetrical location of an H atom between the two heavier atoms, and therefore the pKa of the APV hydroxyl group is higher than those of the aspartates. Thus, these two H bonds likely possess asymmetric double-well potential energy profiles.31
Figure 3.
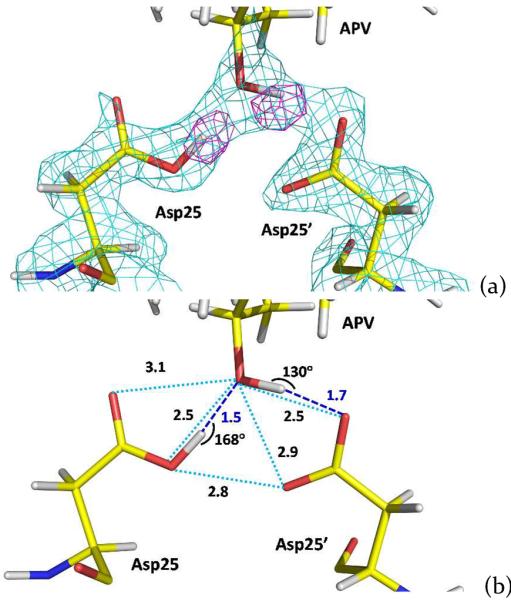
(a) 2FO-FC neutron scattering length density map contoured at 2.3σ (cyan) for the catalytic site showing residues Asp25, Asp25' and the hydroxyl group of amprenavir. Omit difference FO-FC neutron scattering length density map contoured at 4.5σ (magenta) shows the locations of D atoms on Asp25 and the drug's hydroxyl group. (b) H bonding interactions between the hydroxyl group of amprenavir and catalytic aspartic acid side chains. Distances are in Å.
The distribution of H bonds in the PR-APV complex differs from that found in an earlier neutron structure of wild-type PR in complex with a non-clinical inhibitor KNI-272.32 Unlike APV, KNI-272 contains a hydroxymethylcarbonyl isostere that binds to the catalytic site of the enzyme. In the PR-KNI-272 complex, the outer Oδ2 of one Asp residue was found to be protonated and donated its D in an H bond with the carbonyl group of the inhibitor, while the inner Oδ1 of the other Asp accepted a D in an H bond from the inhibitor's hydroxyl group. Importantly, comparison of the two neutron structures suggests that the exact location of the D on the protonated Asp and the nature of H bonding in the catalytic site are dependent on the chemical structure of the ligand's isostere. Further, both neutron structures are in agreement with previous proposals in which it was suggested that at different stages of the PR-catalyzed hydrolysis reaction the position of H on the protonated Asp may change between Oδ1 and Oδ2.33,34
Based on the atomic-resolution low-temperature X-ray structure of the PR-APV complex we had suggested a number of possible polar non-covalent interactions between the drug and the enzyme that might enhance binding.16 The lack of experimental positions for H atoms, however, precluded an analysis of which contacts might be interpreted as conventional or non-conventional H bonds. Unexpectedly, our new room-temperature XN structure indicates that we may have overestimated the importance of some of these interactions. In particular, the XN structure may have just two strong direct H bonds between APV and PR, both with the catalytic aspartates as discussed above. Other direct H bonds formed by the tetrahydrofuran (THF) and the amides of Asp29 and Asp30, and the water-mediated interactions that connect the drug molecule to the flaps and Asp30' have significantly longer distances (Figure 4). Another possible H bond interaction is formed between the main-chain amide N-D of Asp30' and the nitrogen of APV's aniline substituent. Interestingly, the terminal ND2 group of aniline is planar and rotated by 30° relative to the benzene plane, which probably strengthens the interaction, although the interatomic distances are long indicating a weak N-D⋯N interaction (Figure 4). Careful examination of the possible interaction between the APV carbamate's N-D and the carbonyl group of Gly27 reveals that the D⋯O=C angle is < 90°, suggesting very weak H bonding (Supporting Figure 2a). Similarly, the flap water is oriented to accept a D in an H bond with the main-chain amide of Ile50, but the main-chain amide of Ile50' faces the water's D atoms, perhaps substantially weakening the second interaction (Supporting Figure 2b). Thus, it appears the flap water forms three H bonds rather than four as was inferred from the X-ray structure, consistent with the occasional loss of one H-bond in mutant structures.35
Figure 4.
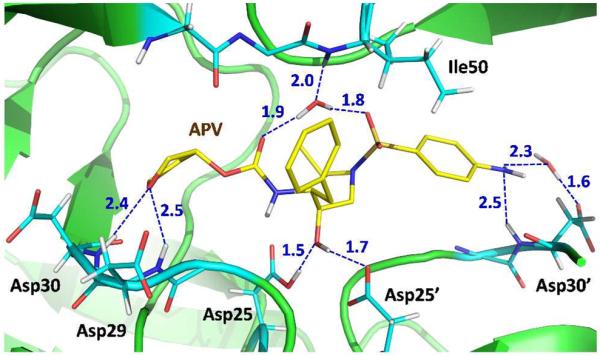
H bonds and water-mediated interactions formed by amprenavir with the active-site PR residues. Distances are in Å.
The assignments of C-H⋯O non-conventional H bonds in the previous X-ray and current XN structures are in full agreement. However, a water-mediated O-H⋯π interaction seems to be missing. A water molecule located near the aromatic aniline moiety of APV is H bonded to the main-chain atoms of Gly27' and Asp29'. The D2O was found to be oriented almost parallel to the benzene ring (Supporting Figure 2c), casting doubt on the presence of this non-conventional contact, although the water is conserved in PR complexes with inhibitors based on the same chemical scaffold.36,37 This D2O molecule is however not rigid; its B-factor is about 40 Å2, which is twice as high as the B-factor of the flap water. Hence, it is plausible that the O-H⋯π interaction is dynamic, constantly forming and breaking.
Current drug design for HIV-1 PR is focused on improving the inhibitors' affinity for resistant variants of the enzyme. The comparison of drug resistant PR structures in complex with various PIs has demonstrated virtual invariance of the main-chain positions. Thus, one fruitful strategy has been to introduce more H bonding interactions with the main-chain atoms of the PR active site.9 Success of this approach has been demonstrated by the design of the clinical drug darunavir, whose chemical structure is similar to that of APV, except for the substitution of the THF moiety with a bis-THF. In our XN structure we observed that the THF (P2 substituent) group makes only weak O-H⋯O H bonds with the main-chain amides of Asp29 and Asp30 of the S2 binding subsite, the D⋯O distances are 2.4 and 2.5 Å, respectively. On the other side of APV, the aniline moiety (P2' substituent) makes a weak N-H⋯N contact with the main-chain amide of Asp30' of the S2' binding subsite, the D⋯O distance is similarly 2.5 Å (Figure 4). Analogous interactions occur in the PR-darunavir structures.17 Recently, we obtained low-temperature X-ray structures of the wild-type and mutant PR complexes with the potent antiviral PI that contains tris-THF substituent replacing the bis-THF of darunavir.38 The tris-THF PI showed the ability to make new, but weak, interactions, which are not observed in our current PR-APV structure, with the side chain of Asp30 in the wild-type enzyme and Asn30 in the mutant variant. Thus, these interactions should be further improved in the design of new inhibitors. The strongest H bonds are formed between the central hydroxyl of APV and the carboxylic groups of Asp25 and Asp25'. These H bonds are asymmetric because of the pKa mismatch. We hypothesize that these contacts may be strengthened if the pKa of APV's hydroxyl is lowered through introduction of a strong electron accepting atom (for instance, fluorine) on the CH2 of hydroxyethyleneamine isostere. Closer pKa values of the groups may trigger the formation of low-barrier H bonds, which are much stronger than those observed. Displacement of water molecules that are still present in the inhibitor-bound PR active site would improve the entropy of binding. Moreover, if these water-mediated drug-enzyme interactions are replaced with direct H bonds it would also improve the enthalpy of binding. We propose that it could be advantageous to replace a water-mediated interaction of aniline with the side chain of Asp30' and the weak N-H⋯N contact with the main chain of Asp30' by a stronger direct H bond. This D2O molecule has a relatively high B factor (~50 Å2) and is probably quite dynamic, diminishing the interaction strength. We have previously attempted to explore this idea in the design of PI GRL-06579A based only on the X-ray structures.39 In GRL-06579A the aniline of APV is replaced with the benzyl alcohol group (-C6H4-CH2-OH). The terminal hydroxyl of benzyl alcohol made similar H bond and water-mediated interactions with Asp30' as does APV; however, these O-H⋯O and N-H⋯O H bonds made by benzyl alcohol may be somewhat stronger than the N-H⋯O and N-H⋯N interactions made by aniline. Even so, it is clear that the interactions between P2, P2' and central hydroxyl of a PI and the PR atoms can be further redesigned to increase the binding efficiency, especially with the main chain groups.
CONCLUSIONS
2.0 Å joint XN structure of PR-APV has unambiguously revealed the protonation of the inner Oq1 oxygen atom of Asp25, location of D atom on the APV's hydroxyl group and orientations of the aniline's ND2 group and water molecules. The monoprotonation of the catalytic aspartic dyad is in agreement with the earlier neutron structure of PR-KNI-272 complex. The comparison of the two neutron structures indicates that the exact location of the H atom and the H bond arrangement depend on the nature of the chemical groups bound to the catalytic site. These observations are also in accord with the proposed mechanism of the hydrolysis reaction catalyzed by PR. Examination of the drug-enzyme interactions in PR-APV based on the determined positions of D atoms has shown that geometrical parameters for some contacts are significantly distorted from the accepted values for H bonds. This neutron structure suggests that H bonding of the inhibitor to PR contributes less to its affinity than was inferred from the X-ray structures, which may be valuable information for the design of new PIs with improved H bonds.
Supplementary Material
ACKNOWLEDGMENT
MJW was partly supported by a DOE-OBER grant to the neutron Protein Crystallography Station at LANSCE. AYK, PL and MM were partly supported by DOE-BES. PL was partly supported by an NIH-NIGMS-funded consortium (1R01GM071939-01) between ORNL and LBNL. IW was partly supported by NIH grant R01GM02920. We thank Dr. Srinivas Iyer of the Bioscience Division of Los Alamos National Laboratory for performing mass spectrometry measurements.
ABBREVIATIONS USED
- PR
HIV-1 protease
- PI
HIV-1 protease inhibitor
- APV
amprenavir
- THF
tetrahydrofuran
Footnotes
Supporting Information. Protein Data Bank code 4JEC. Experimental section, Supporting Table 1, Supporting Figures 1 and 2. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Gottlinger HG, Sodroski JG, Haseltine WA. Role of capsid precursor processing and myristoylation in morphogenesis and infectivity of human imuunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA. 1989;86:5781–5785. doi: 10.1073/pnas.86.15.5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Louis JM, Ishima R, Torchia DA, Weber IT. HIV-1 protease: structure, dynamics, and inhibition. Adv. Pharmacol. 2007;55:261–298. doi: 10.1016/S1054-3589(07)55008-8. [DOI] [PubMed] [Google Scholar]
- (3).Navia MA, Fitzgerald PMD, McKeever BM, Leu C-T, Heimbach JC, Herber WK, Sigal IS, Darke PL, Springer JP. Three-dimensional structure of aspartyl protease from human immunodeficiency virus HIV-1. Nature. 1989;337:615–620. doi: 10.1038/337615a0. [DOI] [PubMed] [Google Scholar]
- (4).Wlodawer A, Miller M, Jaskolski M, Sathyanarayana BK, Baldwin E, Weber IT, Selk LM, Clawson L, Schneider J, Kent SB. Conserved folding in retroviral proteases: crystal structure of a synthetic HIV-1 protease. Science. 1989;245:616–621. doi: 10.1126/science.2548279. [DOI] [PubMed] [Google Scholar]
- (5).Miller M, Schneider J, Sathyanarayana BK, Toth MV, Marshall GR, Clawson L, Selk LM, Kent SB, Wlodawer A. Structure of complex of synthetic HIV-1 protease with a substrate-based inhibitor at 2.3Å resolution. Science. 1989;246:1149–1152. doi: 10.1126/science.2686029. [DOI] [PubMed] [Google Scholar]
- (6).Wlodawer A, Vondrasek J. Inhibitors of HIV-1 protease: a major success of structure-assisted drug design. Annu. Rev. Biophys. Biomol. Struct. 1998;27:249–284. doi: 10.1146/annurev.biophys.27.1.249. [DOI] [PubMed] [Google Scholar]
- (7).Mastrolorenzo A, Rusconi S, Scozzafava A, Supuran CT. Inhibitors of HIV-1 protease: 10 years after. Expert Opin. Ther. Patents. 2006;16:1067–1091. [Google Scholar]
- (8).Mitsuya H, Maeda K, Das D, Ghosh AK. Development of protease inhibitors and the fight with drug-resistant HIV-1 variants. Adv. Pharmacol. 2008;56:169–197. doi: 10.1016/S1054-3589(07)56006-0. [DOI] [PubMed] [Google Scholar]
- (9).Ghosh AK, Anderson DD, Weber IT, Mitsuya H. Enhancing protein backbone binding – a fruitful concept for combating drug-resistant HIV. Angew. Chem. Int. Ed. 2012;51:1778–1802. doi: 10.1002/anie.201102762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Weber IT, Kovalevsky AY, Harrison RW. Frontiers in Drug Design & Discovery. Vol. 3. Bentham Science Publishers; Oak Park, IL, USA: 2007. Structures of HIV protease guide inhibitor design to overcome drug resistance; pp. 45–62. [Google Scholar]
- (11).Weber IT, Agniswamy J. HIV-1 protease: structural perspectives on drug resistance. Viruses. 2009;1:1110–1136. doi: 10.3390/v1031110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Velazquez-Campoy A, Kiso Y, Freire E. The binding energetics of first- and second-generation HIV-1 protease inhibitors: implications for drug design. Arch. Biochem. Biophys. 2001;390:169–175. doi: 10.1006/abbi.2001.2333. [DOI] [PubMed] [Google Scholar]
- (13).Tisdale M, Myers R, Randall S, Maguire M, Ait-Khaled M, Elston R, Snowden W. Resistance to the HIV protease inhibitor amprenavir in vitro and in clinical studies. Clin. Drug Invest. 2000;20:267–285. [Google Scholar]
- (14).Kim EE, Baker CT, Dwyer MD, Murcko MA, Rao BG, Tung RD, Navia MA. Crystal structure of HIV-1 protease in complex with VX-478, a potent and orally bioavailable inhibitor of the enzyme. J. Am. Chem. Soc. 1995;117:1181–1182. [Google Scholar]
- (15).Surleraux DL, Tahri A, Verschueren WG, Pille GME, de Kock HA, Jonckers THM, Peeters A, De Meyer S, Azijn H, Pauwels R, de Bethune M-P, King NM, Prabu-Jeyabalan M, Schiffer CA, Wigerinck PBTP. Discovery and selection of TMC114, a next generation HIV-1 protease inhibitor. J. Med. Chem. 2005;48:1813–1822. doi: 10.1021/jm049560p. [DOI] [PubMed] [Google Scholar]
- (16).Shen C-H, Wang Y-F, Kovalevsky AY, Harrison RW, Weber IT. Amprenavir complexes with HIV-1 protease and its drug-resistant mutants alerting hydrophobic clusters. FEBS J. 2010;277:3699–3714. doi: 10.1111/j.1742-4658.2010.07771.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kovalevsky AY, Liu F, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT. Ultra-high resolution crystal structure of HIV-1 protease mutant reveals two binding sites for clinical inhibitor TMC114. J. Mol. Biol. 2006;363:161–173. doi: 10.1016/j.jmb.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Niimura N, Arai S, Kurihara K, Chatake T, Tanaka I, Bau R. Recent results on hydrogen and hydration in biology studies by neutron macromolecular crystallography. Cell. Mol. Life Sci. 2006;63:285–300. doi: 10.1007/s00018-005-5418-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Blakeley MP. Neutron macromolecular crystallography. Crystal-lography Reviews. 2009;15:157–218. [Google Scholar]
- (20).Chen JC-H, Hanson BL, Fisher SZ, Langan P, Kovalevsky AY. Direct observation of hydrogen atom dynamics and interactions by ultrahigh resolution neutron protein crystallography. Proc. Natl. Acad. Sci. U.S.A. 2012;109:15301–15305. doi: 10.1073/pnas.1208341109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Shu F, Ramakrishnan V, Schoenborn BP. Enhanced visibility of hydrogen atoms by neutron crystallography on fully deuterated myoglobin. Proc. Natl. Acad. Sci. U.S.A. 2000;97:3872–3877. doi: 10.1073/pnas.060024697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Hazemann I, Dauvergne MT, Blakeley MP, Meilleur F, Haertlein M, van Dorsselaer A, Mitschler A, Myles DAA, Podjarny A. High-resolution neutron protein crystallography with radically small crystal volumes: application of perdeuteration to human aldose reductase. Acta Cryst. 2005;D61:1413–1417. doi: 10.1107/S0907444905024285. [DOI] [PubMed] [Google Scholar]
- (23).Blakeley MP, Ruiz F, Cachau R, Hazemann I, Meilleur F, Mitschler A, Ginell S, Afonine P, Ventura ON, Cousido-Siah A, Haertlein M, Joachimiak A, Myles D, Podjarny A. Quantum model of catalysis based on a mobile proton revealed by subatomic X-ray and neutron diffraction studies of h-aldose reductase. Proc. Natl. Acad. Sci. U.S.A. 2008;105:1844–1848. doi: 10.1073/pnas.0711659105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Howard EI, Blakeley MP, Haertlein M, Petit-Haertlein I, Mitschler A, Fisher SJ, Cousido-Siah A, Salvay AG, Popov A, Muller-Dieckmann C, Petrova T, Podjarny A. Neutron structure of type-III antifreeze protein allows the reconstruction of AFP-ice interface. J. Mol. Recognit. 2011;24:724–732. doi: 10.1002/jmr.1130. [DOI] [PubMed] [Google Scholar]
- (25).Adams PD, Mustyalimov M, Afonine PV, Langan P. Generalized X-ray and neutron crystallographic analysis: more accurate and complete structures for biological macromolecules. Acta Cryst. 2009;D65:567–573. doi: 10.1107/S0907444909011548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Hyland LJ, Tomaszek TA, Meek TD. Human immunodeficiency virus-1 protease. 2. Use of pH rate studies and solvent kinetic isotope effects to elucidate details of chemical mechanism. Biochemistry. 1991;30:8454–8463. doi: 10.1021/bi00098a024. [DOI] [PubMed] [Google Scholar]
- (27).Ido E, Han H, Kezdy FJ, Tang J. Kinetic studies of human immunodeficiency virus type 1 protease and its active-site hydrogen bond mutant A28S. J. Biol. Chem. 1991;266:24349–24366. [PubMed] [Google Scholar]
- (28).Smith R, Brereton IM, Chai RY, Kent SB. Ionization states of the catalytic residues in HIV-1 protease. Nat. Struct. Biol. 1996;3:946–950. doi: 10.1038/nsb1196-946. [DOI] [PubMed] [Google Scholar]
- (29).Wang YX, Freedberg DI, Yamazaki T, Wingfield PT, Stahl SJ, Kaufman JD, Kiso Y, Torchia DA. Solution NMR evidence that the HIV-1 protease catalytic aspartyl groups have different ionization states in the complex formed with the asymmetric drug KNI-272. Biochemistry. 1996;35:9945–9950. doi: 10.1021/bi961268z. [DOI] [PubMed] [Google Scholar]
- (30).Grabowski SJ. What is the covalency of hydrogen bonding? Chem. Rev. 2011;111:2597–2625. doi: 10.1021/cr800346f. [DOI] [PubMed] [Google Scholar]
- (31).Perrin CL, Nielson JB. “Strong” hydrogen bonds in chemistry and biology. Annu. Rev. Phys. Chem. 1997;48:511–544. doi: 10.1146/annurev.physchem.48.1.511. [DOI] [PubMed] [Google Scholar]
- (32).Adachi M, Ohhara T, Kurihara K, Tamada T, Honjo E, Okazaki N, Arai S, Shoyama Y, Kimura K, Matsumura H, Sugiyama S, Adachi H, Takano K, Mori Y, Hidaka K, Kimura T, Hayashi Y, Kiso Y, Kuroki R. Structure of HIV-1 protease in complex with potent inhibitor KNI-272 determined by high-resolution X-ray and neutron crystallography. Proc. Natl. Acad. Sci. U.S.A. 2009;106:4641–4646. doi: 10.1073/pnas.0809400106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Das A, Mahale S, Prashar V, Bihani S, Ferrer J-L, Hosur MV. X-ray snapshot of HIV-1 protease in action: observation of tetrahedral intermediate and short ionic hydrogen bond SIHB with catalytic aspartate. J. Am. Chem. Soc. 2010;132:6366–6373. doi: 10.1021/ja100002b. [DOI] [PubMed] [Google Scholar]
- (34).Shen C-H, Tie Y, Yu X, Wang Y-F, Kovalevsky AY, Harrison RW, Weber IT. Capturing the reaction pathway in near-atomic-resolution crystal structures of HIV-1 protease. Biochemistry. 2012;51:7726–7732. doi: 10.1021/bi3008092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Kovalevsky AY, Chumanevich AA, Liu F, Louis JM, Weber IT. Caught in the act: the 1.5 Å resolution crystal structures of the HIV-1 protease and the I54V mutant reveal a tetrahedral reaction intermediate. Biochemistry. 2007;46:14854–14864. doi: 10.1021/bi700822g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Ghosh AK, Gemma S, Baldridge A, Wang Y-F, Kovalevsky AY, Koh Y, Weber IT, Mitsuya H. Flexible cyclic ethers/polyethers as novel P2-ligands for HIV-1 protease inhibitors: design, synthesis, biological evaluation, and protein-ligand X-ray studies. J. Med. Chem. 2008;51:6021–6033. doi: 10.1021/jm8004543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Wang YF, Tie Y, Boross PI, Tozser J, Ghosh AK, Harrison RW, Weber IT. Potent new antiviral compound shows similar inhibition and structural interactions with drug resistant mutants and wild type HIV-1 protease. J. Med. Chem. 2007;50:4509–4515. doi: 10.1021/jm070482q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Zhang H, Wang YF, Shen C-H, Agniswamy J, Rao KV, Xu C-X, Ghosh AK, Harrison RW, Weber IT. Novel P2 Tristetrahydrofuran group in antiviral compound 1 (GRL-0519) fills the S2 binding pocket of selected mutants of HIV-1 protease. J. Med. Chem. 2013;56:1074–1083. doi: 10.1021/jm301519z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Ghosh AK, Sridhar PR, Leshchenko S, Hussain AK, Li J, Kovalevsky AY, Walters DE, Wedekind JE, Grum-Tokars V, Das D, Koh Y, Maeda K, Gatanaga H, Weber IT, Mitsuya H. Structure-based design of novel HIV-1 protease inhibitors to combat drug resistance. J. Med. Chem. 2006;49:5252–5261. doi: 10.1021/jm060561m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.