

Abstract
Introduction
Pregnancy is associated with decreased insulin sensitivity, which is usually overcome by a compensatory increase in insulin secretion. Some pregnant women are not able to increase their insulin secretion sufficiently, and consequently develop gestational diabetes mellitus (GDM). The disease normally disappears after delivery. Nevertheless, women with previous GDM have a high risk of developing type 2 diabetes (T2D) later in life. We aim to investigate the early development of T2D in women with previous GDM and to evaluate whether treatment with the glucagon-like peptide-1 receptor (GLP-1R) agonist, liraglutide, may modify their risk of developing T2D.
Methods and analyses
100 women with previous GDM will be randomised to either liraglutide or placebo treatment for 1 year (blinded) with an open-label extension for another 4 years. Additionally, 15 women without previous GDM will constitute a baseline control group. Women will be tested with an oral glucose tolerance test (primary endpoint: area under the curve for plasma glucose) and an isoglycaemic intravenous glucose infusion at baseline, after 1 year and after 5 years. Additional evaluations include a glucagon test, dual-energy X-ray absorptiometry, imaging of the liver (ultrasound elastography and fibroscanning), an ad libitum meal for food intake evaluation and questionnaires related to appetite, quality of life and alcohol consumption habits.
Ethics and dissemination
The protocol has been approved by the Danish Medicines Agency, the Scientific-Ethical Committee of the Capital Region of Denmark, and the Danish Data Protection Agency and will be carried out under the surveillance and guidance of the GCP unit at Copenhagen University Hospital Bispebjerg in compliance with the ICH-GCP guidelines and in accordance with the Helsinki Declaration. Positive, negative and inconclusive results will be published at scientific conferences and as one or more scientific manuscripts in peer-reviewed journals.
Registrations
The trial is registered at https://eudract.ema.europa.eu (2012-001371-37) and http://www.clinicaltrials.gov (NCT01795248).
Keywords: DIABETES & ENDOCRINOLOGY
Article summary.
Strengths and limitations of this study
Participants will be monitored extensively during a 1-year blinded phase and throughout a 4-year extension period.
Although all eligible women from a large part of Denmark have been invited to the study, the study population may not be fully representative.
The study may not be powered to conclude on the secondary endpoints.
Introduction
Women with previous gestational diabetes mellitus represent a high-risk group
Pregnancy is associated with insulin resistance.1 This is normally compensated for by increased pancreatic insulin secretion, resulting in retained normal glucose tolerance (NGT).2 Women with gestational diabetes mellitus (GDM) are not capable of increasing their insulin secretion enough to compensate for pregnancy-induced insulin resistance, resulting in abnormal glucose tolerance. In the majority of women, glucose tolerance is re-established after delivery. Nevertheless, most women with previous GDM are at high risk of developing type 2 diabetes (T2D) later in life.3 4 Bellamy et al5 state that up to 63% of women with GDM develop T2D within 16 years, Bian et al6 describe a risk of 33% for the development of T2D from 5 to 10 years after GDM and a Danish study reports a risk of 40% for the development of T2D 7 years after diet-treated GDM.3 Insulin dependency, high plasma glucose (PG) levels during an oral glucose tolerance test (OGTT), difficult controllable glycaemia (need of insulin treatment) and high body mass index during pregnancy are strong predictors of the risk of T2D after GDM.4
Early pathophysiology of T2D remains elusive
Studies show that, on average, patients with T2D are diagnosed when their relative β-cell function is approximately 50% of normal.7 The delayed onset of symptoms makes it difficult to characterise the pathogenesis of T2D. There is general agreement that insulin resistance and abnormal insulin secretion coexist in T2D. Nevertheless, the order in which the pathophysiological characteristics of T2D occur remains unclear.8 Studies of first-degree relatives of patients with T2D have yielded conflicting results regarding whether abnormal insulin secretion or insulin resistance is the primary defect in the disease.9–17 Other physiological defects have been suggested as primary pathogenic defects. These include decreased first-phase insulin response and impaired incretin effect (decreased β-cell response to the incretin hormones, glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide 1 (GLP-1)). In healthy participants, both incretin hormones possess strong glucose-dependent insulinotropic properties and enhance glucose-induced insulin secretion from the beginning of a meal.18–20 This potentiation of insulin secretion after oral glucose ingestion is called the incretin effect, and is illustrated when orally administered glucose elicits a greater insulin response than intravenous glucose at identical PG profiles. Patients with T2D exhibit an impaired incretin effect. The cause of this pathophysiological trait is unclear.21–23 Understanding the pathophysiology in the early stages of T2D may provide new and effective interventions aiming at preventing the development of the disease. One approach to achieve this is to prospectively follow a high-risk group.
Liraglutide—a once-daily GLP-1 receptor agonist
GLP-1 receptor (GLP-1R) agonists are used in the treatment of overweight patients with T2D. Preclinical studies show that GLP-1R agonists may have β-cell protective properties (reduced β-cell apoptosis). Clinical studies have described sustained effects (2 years) on glucose tolerance, β cell function and body weight in obese patients without diabetes.24 25
Objective
Owing to their high risk of developing T2D, overweight and obese non-diabetic women with previous GDM provide an opportunity for investigating how the early stages of T2D develop and whether GLP-1R agonists may prevent or delay the onset of T2D. The primary objective of the present protocol was to assess whether treatment with the GLP-1R agonist liraglutide reduces the risk of T2D in women with previous GDM. Simultaneously, the developmental trajectories of pathophysiological defects known from patients with T2D will be evaluated. Such information will ultimately contribute to a clarification of the pathogenesis of T2D and provide a basis for preventive measures and interventions. We hypothesise that women with previous GDM receiving liraglutide treatment for a year will have a smaller area under the curve (AUC) for PG during an OGTT carried out after 1 week of washout (primary endpoint) than women with previous GDM treated with placebo. The primary endpoint will be re-evaluated after another 4 years of liraglutide treatment or no intervention. Secondary endpoints include metabolic measures, biomarkers and questionnaires as described below.
Methods and analyses
Study design, randomisation and blinding
This is an investigator-initiated, randomised, placebo-controlled, double-blinded, two-arm parallel group intervention trial carried out in non-diabetic women with previous GDM. Participants with previous GDM will be randomised to treatment with liraglutide or placebo after the baseline experiments. Adequate randomisation will be ensured by stratification based on a computer-generated random number sequence (1:1) with stratified block (permuted-block size) with participants stratified according to the baseline glucose tolerance: NGT versus non-NGT (impaired fasting glucose (IFG), impaired glucose tolerance (IGT) or combined IFG/IGT). Participants with T2D at baseline will not be included in the trial. The randomisation will be carried out through a central independent unit through an unblinded data manager at Public Health and Quality Improvement, Central Denmark Region, Denmark. The allocation sequence will be concealed from the investigators and healthcare staff enrolling and assessing participants. Trial medication will be allocated through the electronic case report form (eCRF). The unblinded person will be impartial and have no influence or knowledge of the treatment of the participants following randomisation. Participants, investigators and healthcare staff will remain blinded to the allocated treatment and kept masked until the afternoon of visit 10 (OGTT after 52 weeks). Data analysis will be carried out blinded.
Study population and study sites
Ninety-eight participants will participate in the randomised trial and 15 healthy women without previous GDM will make up a baseline control group. The trial will be conducted at the Diabetes Research Division, Copenhagen University Hospital Gentofte, Denmark. Recruitment will take place by an invitation letter sent to all women who have been diagnosed with GDM at either the Center for Pregnant Women with Diabetes, Rigshospitalet, Copenhagen, Denmark or the Department of Gynaecology-Obstetrics, Copenhagen University Hospital Herlev, Denmark, within the past 10 years. Moreover, participants will be recruited from advertisements in newspapers and on the internet. The total number of participants in the following categories will be recorded: invited, excluded before screening (including reasons for exclusion), declined to participate, not reached, received participants’ information and declined, screened, excluded at screening, randomised, allocated for intervention, received allocated intervention, completed intervention period, withdrew/dropped out during the study period (including reason for withdrawal/dropping out), lost to follow-up and analysed. Recruitment is planned to proceed until 49 participants in each group have been randomised. Reasons for withdrawal or exclusion will be reported in detail. Inclusion and exclusion criteria are presented in boxes 1 and 2.
Box 1. Inclusion criteria.
Women with previous gestational diabetes mellitus (GDM)
Informed oral and written consent
Age ≥18 years
Body mass index (BMI) between 25 and 45 kg/m2
Previous diagnosis of GDM according to current Danish guidelines (plasma glucose (PG) concentration at 120 min after 75 g oral glucose tolerance test (OGTT) ≥9 mmol/L during pregnancy) within the last 10 years
Normal glucose tolerance (NGT), impaired fasting glucose (IFG) and/or impaired glucose tolerance (IGT)
Use of safe contraception or sterilisation
Negative pregnancy test
Women without previous GDM
Informed oral and written consent
Age ≥18 years
BMI between 25 and 45 kg/m2
NGT (fasting PG (FPG) ≤6 mmol/L and PG concentration at 120 min after 75 g OGTT <7.8 mmol/L)
Pregnancy within the last 10 years without GDM
No family history of type 2 diabetes
Negative pregnancy test
Box 2. Exclusion criteria.
Women with previous gestational diabetes mellitus (GDM)
Pregnancy or breast feeding
Anaemia (haemoglobin <7 mmol/L)
Diabetes (fasting plasma glucose (FPG) >7 mmol/L or plasma glucose (PG) concentration at 120 min after 75 g oral glucose tolerance test ≥11.1 mmol/L or glycated haemoglobin (HbA1c) ≥48 mmol/mol (6.5%))
Previous pancreatitis
Cancer within the last 5 years
Desire to become pregnant within the next 5 years
Treatment with statins, corticosteroids or other hormone therapy (except oestrogens and gestagens)
Ongoing abuse of alcohol or narcotics
Impaired hepatic function (liver transaminases >3 times the upper normal limit)
Impaired renal function (se-creatinine >120 μM and/or albuminuria)
Uncontrolled hypertension (systolic blood pressure >180 mm Hg, diastolic blood pressure >100 mm Hg)
Receiving any investigational drug within the last 3 months
Any condition that the investigator feels would interfere with trial participation
Women without previous gestational diabetes mellitus
Pregnancy or breastfeeding
Anaemia (haemoglobin <7 mmol/L)
Experimental design
Eligible participants will receive detailed oral and written information about the study. Sufficient time for reflection will be allowed before written informed consent and authorisation are obtained. Both groups will attend the same visits unless stated otherwise. An initial screening visit will be followed by a baseline OGTT and an isoglycaemic intravenous glucose infusion (IIGI; figure 1).
Figure 1.
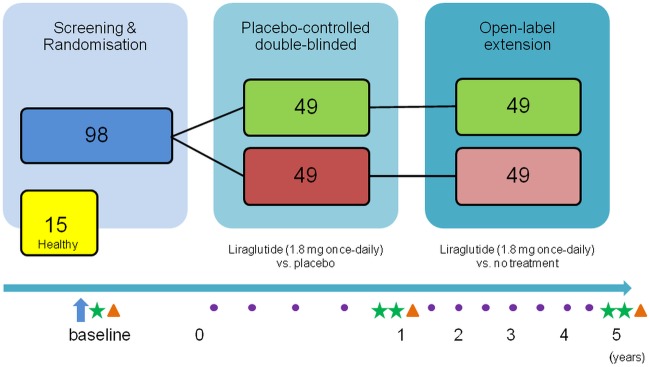
Schematic illustration of study design. The first year is blinded and the remaining 4 years are open-label. Arrow: screening visit; star: oral glucose tolerance tests (OGTT); triangle: isoglycaemic intravenous glucose infusion, full body dual energy X-ray absorptiometry scanning, ultrasound scanning of the liver, fibroscanning, glucagon test and ad libitum meal test; dot: clinical control visits.
Following a baseline OGTT, IIGI and other baseline procedures (including blood sampling, full body dual energy X-ray absorptiometry (DXA) scanning, glucagon test, imaging of the liver (ultrasound elastography and fibroscanning; figure 1)), the participants are randomised to once-daily injections of either liraglutide 1.8 mg or placebo. In the 1-year intervention period, the participants will be monitored at regular clinical control visits and phone calls. At the end of the intervention period, the OGTT will be repeated, as well as all other procedures made at baseline, with an additional OGTT after a 1-week washout in the liraglutide-treated group. Finally, an IIGI, matching the blood glucose profile of the latter OGTT, will be conducted in both groups. Both groups will attend biannual control visits in the 4-year follow-up period and terminate the study with the same experimental days as at the end of the intervention period. The women without previous GDM will only participate in the study for the baseline OGTT and IIGI experimental days and their related baseline procedures. An outline of the trial visits and experimental procedures is shown in table 1.
Table 1.
Trial visits and examinations
| Informed consent | Physical examination | Full body DXA scan | Ultrasound and fibroscan of liver | Blood samples | Glucagon test | Ad libitum meal and questionnaire | Appetite questionnaire | Quality of life questionnaire | Adverse event assessment | |
|---|---|---|---|---|---|---|---|---|---|---|
| Screening, week 1 | X | X | X | |||||||
| OGTT, week 0 | X | X | ||||||||
| IIGI, week 0 | X | X | X | X | X | X | X | |||
| Telephone, week 1 | X | |||||||||
| Control, week 4 | X | X | X | |||||||
| Telephone, week 8 | X | |||||||||
| Control, week 12 | X | X | X | |||||||
| Control, week 26 | X | X | X | |||||||
| Control, week 38 | X | X | X | |||||||
| OGTT, week 52 | X | X | X | X | X | |||||
| OGTT, week 53 (only liraglutide group) | X | X | X | |||||||
| IIGI, week 53 | X | X | X | X | X | X | X | X | ||
| Control, week 78 | X | X | X | |||||||
| Control, week 104 | X | X | X | |||||||
| Control, week 130 | X | X | X | |||||||
| Control, week 156 | X | X | X | |||||||
| Control, week 182 | X | X | X | |||||||
| Control, week 208 | X | X | X | |||||||
| Control, week 234 | X | X | X | |||||||
| OGTT, week 260 | X | X | X | X | X | |||||
| OGTT, week 261 (only liraglutide group) | X | X | ||||||||
| IIGI, week 261 | X | X | X | X | X | X | X |
DXA, dual energy X-ray absorptiometry; IIGI, isoglycaemic intravenous glucose infusion; OGTT, oral glucose tolerance test.
On the initial screening day, fasting blood samples (table 2) will be collected to verify that the participant fulfils the inclusion and exclusion criteria, medical history will be recorded and a full physical examination will be performed. All participants are required to use adequate contraceptive methods throughout the intervention and follow-up period. Intrauterine devices will be offered.
Table 2.
Blood samples
| Sampling period | Analysis |
|---|---|
| Screening and control visits | ▸ Albumin ▸ ALT, AST, amylase ▸ Cholesterol, triglycerides ▸ Creatinine, potassium, sodium ▸ Erythrocytes ▸ Glycated haemoglobin ▸ Haemoglobin ▸ Leucocytes ▸ TSH |
| Screening, control visits, OGTT, IIGI and glucagon test | ▸ Glucose ▸ Insulin, proinsulin, C-peptide |
| OGTT and IIGI | ▸ Glucose-dependent insulinotropic polypeptide (GIP) ▸ Glucagon ▸ Glucagon-like peptide 1 (GLP-1) ▸ Glucagon-like peptide 2 (GLP-2) |
| Screening | ▸ β-hCG ▸ Coagulation factors 2, 7 and 10 ▸ Urine albumin:creatinine ratio |
ALT, alanine aminotransferase; AST, aspartate aminotransferase; hCG, human chorionic gonadotrophin; IIGI, isoglycaemic intravenous glucose infusion; OGTT, oral glucose tolerance test; TSH, thyroid-stimulating hormone.
Within 10 days of the screening visit, a 4 h 75 g OGTT will be performed. The participant will ingest 75 g glucose dissolved in 300 mL water over 5 min and, subsequently, repeated blood samples for measurements of PG, insulin, C-peptide, glucagon, GLP-1 and GIP will be drawn throughout the test. At baseline and once an hour afterwards, the participants will answer a questionnaire on appetite using a visual analogue scale (VAS) on a tablet computer. If the 2 h PG value is below 11.1 mmol/L, the participant will be randomised. Within 10 days of the OGTT, the participants will attend an IIGI. In the morning of the IIGI, they are first DXA scanned, and their liver will be assessed by ultrasound scanning and fibroscanning.26 Over the next 4 h, the PG curve from the washed-out OGTT day will be mimicked using a 20% glucose intravenous infusion. Again, repeated blood samples will be drawn and questionnaires answered as on the OGTT day, although PG may be sampled more frequently to adjust the glucose infusion rate. Two additional questionnaires, one regarding the quality of life (QoL) and one regarding alcohol consumption habits, will be filled out once during the IIGI. At the end of the IIGI, a glucagon test will be performed; at time point 0 min, 1 mg glucagon will be injected and at time points 2, 6 and 10 min, blood will be sampled for measurements of PG, insulin and C-peptide. Next, the participants are offered an ad libitum meal consisting of pasta, minced meat, vegetables and cream, which they are instructed to eat as much of ‘until comfortably satisfied’ in addition to drinking 500 mL water. The distribution of energy in the ad libitum meal is 50 energy per cent (E%) carbohydrates, 37 E% fat and 13 E% protein. Additional palatability ratings (palatability, taste, aftertaste, smell and visual appeal) of the meal are filled in immediately after the first few bites and after consumption of the ad libitum meal using VAS scores on a tablet computer. The participants are then instructed in how to inject the trial medication, and how to measure blood glucose (Contour; Bayer HealthCare, Copenhagen, Denmark) twice a month and record it in a diary. At one and 8 weeks, respectively, after the IIGI day, the participants will be contacted by phone for clarification of any questions they may have and any adverse events (AEs) are recorded in the eCRF. Four, 12, 26 and 38 weeks after the IIGI, and biannually in the follow-up period, participants will meet in the fasting state for clinical control visits. Measurements of weight, waist–hip ratio, blood pressure and pulse, compliance and AEs will be recorded, and trial medication will be dispensed. At 52 weeks, the OGTT will be repeated for both groups and placebo will be initiated for 1 week in both groups. An additional OGTT day will be carried out in the liraglutide group to evaluate glucose tolerance off trial medication. Finally, an IIGI, as described above, will be conducted in both groups, matching the blood glucose profile of the latest OGTT. The exact same set-up will be repeated at the end of the 4-year follow-up period.
Intervention
Trial medication will be initiated at the end of the IIGI day in a dosage of 0.6 mg subcutaneously. All participants will be instructed to inject the medicine in the abdomen once daily. The trial medication dose will be escalated over 3 weeks with once weekly dose adjustments in increments of 0.6 mg to a maximal dose of 1.8 mg. If intolerable AEs occur, the dose may be reduced to 1.2 mg, or the participant may remain on 1.2 mg for more than a week before increasing the dose. Trial medication will be delivered in boxes of five prefilled, disposable pen injectors, each containing 3 mL of a premixed colourless solution. The active pens will contain liraglutide (6 mg/mL) mixed with sterile water, disodium phosphate dihydrate, propylene glycol and phenol. Pens containing placebo will be visually identical to those with the active component and be composed of sterile water, disodium phosphate dihydrate, propylene glycol and phenol.
Outcome measures and analysis methods
Primary endpoint
The primary endpoint is the change in glucose tolerance from baseline to week 52 as measured by the AUC for the PG excursion following a 4 h 75 g OGTT. Additional endpoints regarding glucose tolerance include changes from baseline to week 53 (after trial medication washout), to week 260 (end of extension period) to week 261 (after trial medication washout). The primary endpoint will be analysed by the intention-to-treat approach. In case of missing data points, values will be imputed. Safety measures will be analysed in the per protocol population. In case the full intervention period has not been completed, we will use the ‘last observation carried forward’ method.
Secondary endpoints
All secondary endpoints will be assessed as changes from baseline until 52 weeks, and until the end of the extension period (260 weeks). In addition, most endpoints will also be assessed as changes from baseline until weeks 53 and 261 (after trial medication washout). The secondary endpoints are listed in boxes 3 and 4.
Box 3. Secondary endpoints related to glycaemic status.
Worsening of glycaemia assessed by plasma glucose (PG), expressed as a percentage of participants in each treatment arm with normal glucose tolerance (NGT) at inclusion who develop
– Prediabetes
– Type 2 diabetes (T2D)
Progression, assessed by PG, expressed as a percentage of participants in each treatment arm with isolated impaired fasting glucose (IFG) or isolated impaired glucose tolerance (IGT) who progress to
– Combined IFG/IGT
– T2D
Progression, assessed by PG, expressed as a percentage of participants with combined IFG/IGT who progress to
– T2D
Improvement in glycaemic status, assessed by PG, expressed as a percentage of participants in each treatment arm going from
– IFG to NGT
– IGT to NGT
– Combined IFG/IGT to NGT or isolated IFG or IGT
Worsening of glycaemia, assessed by changes in HbA1c, expressed as a percentage of participants who progress from
– Normoglycaemia to prediabetes
– Normoglycaemia to T2D
– Prediabetes to T2D
Improvement of glycaemia, assessed by changes in HbA1c, expressed as a percentage of participants who progress from prediabetes to normoglycaemia
NGT: normal glucose tolerance: FPG ≤6 mmol/L and 2 h PG <7.8 mmol/L; normoglycaemia: HbA1c ≤42 mmol/mol (6%); prediabetes: IFG and/or IGT or 43≤ HbA1c ≤47 mmol/mol (6.1–6.4%); IFG: impaired fasting glucose: 6.1 mmol/L ≤FPG <7 mmol/L and 2 h PG after 75 g OGTT <7.8 mmol/L; IGT: impaired glucose tolerance: FPG ≤6 mmol/L and 7.8 mmol/L ≤2 h PG after 75 g OGTT ≤11 mmol/L; T2D: FPG ≥7 mM or 2 h PG ≥11.1 mmol/L; HbA1c ≥48 mmol/mol; OGTT: oral glucose tolerance test; IIGI: isoglycaemic intravenous glucose infusion; PG: plasma glucose; FPG: fasting plasma glucose.
Box 4. Additional secondary endpoints.
Changes in
Anthropometric measurements (BMI, absolute body weight and waist–hip ratio)
β-cell secretory responses during OGTT, IIGI and glucagon test (AUC for plasma insulin, C-peptide and pro-insulin)
Insulin sensitivity (assessment of insulin resistance (HOMAIR) and Matsuda insulin sensitivity index)
Hormone secretion (fasting plasma concentrations and AUC for plasma GLP-1, GLP-2, GIP and glucagon) during OGTT
Incretin effect and insulin secretory rate (AUC for plasma insulin and C-peptide responses after OGTT and IIGI)
Cardiometabolic risk measures (changes in ELF test, intrahepatic fat, whole body and visceral fat mass/fat-free mass, circulating lipids and cardiovascular biomarkers (hs-CRP, NT-proBNP, TNF-α, adiponectin and PAI-1)
Subjective appetite (measured by VAS)
QoL (measured by a validated questionnaire)
Alcohol consumption (measured by a validated questionnaire)
AUC, area under the curve; BMI, body mass index; ELF, enhanced liver fibrosis; GIP, glucose-dependent insulinotropic peptide; GLP-1, glucagon-like peptide-1; GLP-2, glucagon-like peptide-2; HOMA, homeostasis model assessment; hs-CRP, highly sensitive C reactive protein; IIGI, isoglycaemic intravenous glucose infusion; NT-proBNP, N-terminal prohormone of brain natriuretic peptide; OGTT, oral glucose tolerance test; PAI-1, plasminogen activator inhibitor-1; QoL, quality of life; TNF-α, tumour necrosis factor-α; VAS, visual analogue scale.
Sample size
The primary outcome measure was used in the sample size calculation. With expected end-of-treatment values of 1713 mmol/L min (SD 212) and 1853 mmol/L min (SD 212) in the intervention and placebo groups,27 respectively, and with α set to 5% and power to 90%, the estimated sample size was 98 participants (49 participants in each arm).
Data analyses
Continuous data will be presented by descriptive statistics with the number of observations (n), mean, SD, SE of the mean, minimum, median and/or maximum. Categorical data will be summarised in frequency tables using count and percentages. All participants will be presented in separate data listings. Data from participants screened but not randomised will not be presented in any tables or listings. Comparisons of data from the two treatment groups will be performed using the two-tailed t test (paired within groups, unpaired between groups) if the data are normally distributed. For data which are not normally distributed, the significance of differences between the groups will be tested using the Mann-Whitney U test. For within-group comparisons, the Wilcoxon test for paired differences will be used. One-way analysis of variance will be used to compare the means of several groups. Categorical data will be analysed by χ2 test or Fisher’s exact test. The relationship between an effect of liraglutide and hip, waist and weight measures, visceral fat mass, FPG, glycated haemoglobin (HbA1c), degree of steatosis, QoL, alcohol consumption, and appetite, respectively, will be examined by correlation analysis. Multivariable linear regression analysis will be performed to evaluate the potential influence of predictors of primary outcomes. All tests will be carried out at a significance level of 5%. AEs will be summarised qualitatively and quantitatively.
Ethics and dissemination
Ethics
The treatment is associated with minimal discomfort for the participants comprising a blood sample collection and a daily injection of liraglutide or placebo in the subcutis of the abdomen, in the thigh or the upper arm. Common AEs associated with the active comparator (liraglutide) are mild-to-moderate transient gastrointestinal symptoms (nausea, vomiting and diarrhoea), affecting around 10–15% of treated patients, and headache. The injection is practically pain-free but may leave a small haemorrhage which will resolve spontaneously. Less commonly, participants may experience stomach pain, constipation, fever, reflux, gastritis, dizziness, tiredness and upper airway infection. When collecting blood, some participants may experience minor discomfort when the needle penetrates the skin and rarely a small bleeding occurs. The volume of blood collected during the entire study period will amount to a maximum of 1800 mL (over 5 years) and only participants with a normal haemoglobin level will be included. Severe systemic AEs are not expected.
A DXA scan will be performed three times during the study with the objective of determining the distribution of bone and adipose tissue. DXA scanning takes 15 min and is a painless procedure with no expected side effects; it results in a modest radiation dose (approximately equivalent to 2–3 times the dose received from a dental X-ray). Scanning of the liver will be carried out using ultrasound. Ultrasound creates images using sound waves and no side effects are expected from this procedure.
The participants will receive thorough verbal and written information about the risk of developing the aforementioned AEs. Verbal and written informed consent will be obtained from participants prior to participation in accordance with the ICH-GCP guidelines. The declaration of consent will emphasise that participation in the project is voluntary and that participants may withdraw their consent to participate at any time without providing a reason and without any consequences for the patient's current or future treatment by the health service. The participants will receive a randomisation number after the baseline OGTT. All data forms and blood samples will only be labelled with the patient's initials and study number. The sponsor-investigator is responsible for keeping a list separately for all randomised participants containing patient numbers, full names and date of birth. Extra plasma and serum, white blood cells and urine will be stored for up to 15 years after the end of the study for repeated measurements in case of error analysis or the need for more analyses. The protocol is registered at the Danish Data Protection Agency (01714 GEH-2012-024).
The advantage for the participant, of participating in this trial, is the initial screening for diabetes and the subsequent monitoring of her blood glucose, which also allows for detection of diabetes and hence early treatment. Serious AEs are rare. Research will gain more understanding of the pathophysiology in the early stages of T2D. This may provide new and effective interventions aiming at preventing the development of the disease.
Dissemination
At the end of the trial, one or more manuscripts will be prepared for publication in scientific journals in accordance with the CONSORT 2010 Statement.28 Positive, negative, as well as inconclusive results will be published. Novo Nordisk will be given 4 weeks to review and comment on any manuscript/abstract or other means intended for publication or presentation of the data. The investigators will have the right to decide to publish the trial and the final responsibility for the result. All authors should qualify for authorship according to the International Committee of Medical Journal Editors, 1997. Each author should have participated sufficiently in the work to take public responsibility for the content. The final decision on the order of authorship will be decided when the study has been finalised. The results from the study may, moreover, be presented as posters or oral presentations at national and/or international conferences.
Supplementary Material
Footnotes
Contributors: SF and LV contributed to the design of the study and wrote the manuscript. ERM, JAS, LLG, JJH and PD contributed to the design of the study and reviewed the manuscript. FKK and TV conceived and designed the study and reviewed the manuscript. TV is the sponsor of the trial. SF, LV and FKK are coinvestigators at the Diabetes Research Division, Copenhagen University Hospital Gentofte; ERM and PD are coinvestigators at the Center for Pregnant Women with Diabetes, Copenhagen University Hospital Rigshospitalet; JAS is coinvestigator at the Department of Obstetrics and Gynaecology, Copenhagen University Hospital Herlev; and JJH is coinvestigator at the Department of Biomedical Sciences, Faculty of Health Sciences, The NNF Center for Basic Metabolic Research, University of Copenhagen, Denmark. All authors have read and approved the final version of the manuscript.
Funding: This study was initiated by the authors and is supported as an investigator-initiated study (IIS) by an unrestricted grant from Novo Nordisk.
Competing interests: SF has received research support from Novo Nordisk. LLG has participated in a trial sponsored by Merck. FKK has received lecture fees from AstraZeneca, Boehringer Ingelheim Pharmaceuticals, Bristol-Myers Squibb, Eli Lilly and Company, Gilead Sciences, Merck Sharp & Dohme, Novo Nordisk, Ono Pharmaceuticals, Sanofi, and Zealand Pharma, is a member of the Advisory Boards of Eli Lilly, Bristol-Myers Squibb/AstraZeneca and Zealand Pharma, and has consulted for AstraZeneca, Gilead Sciences, Ono Pharmaceuticals and Zealand Pharma. ERM and PD have received lecture fees from Novo Nordisk and have been members of an Advisory Board of Novo Nordisk; JJH has consulted for Merck Sharp & Dohme, Novo Nordisk and Roche; Tina Vilsbøll has received lecture fees from AstraZeneca, Boehringer Ingelheim Pharmaceuticals, Bristol-Myers Squibb, Eli Lilly and Company, Merck Sharp & Dohme, Novo Nordisk, Novartis, Sanofi, and Zealand Pharma, and is a member of the Advisory Boards of Novo Nordisk, Merck Sharp & Dohme and Bristol-Myers Squibb/AstraZeneca.
Ethics approval: The study has been approved by the Danish Medicines Agency (EudraCT number: 2012-001371-37), the Scientific-Ethical Committee of the Capital Region of Denmark (H-2-2012-073) and the Danish Data Protection Agency (01714 GEH-2012-024). The study is registered at ClinicalTrials.gov (NCT01795248) and will be carried out under the surveillance and guidance of the GCP unit at the Copenhagen University Hospital in compliance with the ICH-GCP guidelines conducted in accordance with the Helsinki Declaration.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Catalano PM, Tyzbir ED, Wolfe RR, et al. Carbohydrate metabolism during pregnancy in control subjects and women with gestational diabetes. Am J Physiol 1993;264(1 Pt 1):E60–7 [DOI] [PubMed] [Google Scholar]
- 2.Damm P, Vestergaard H, Kühl C, et al. Impaired insulin-stimulated nonoxidative glucose metabolism in glucose-tolerant women with previous gestational diabetes. Am J Obstet Gynecol 1996;174:722–9 [DOI] [PubMed] [Google Scholar]
- 3.Lauenborg J, Hansen T, Jensen DM, et al. Increasing incidence of diabetes after gestational diabetes: a long-term follow-up in a Danish population. Diabetes Care 2004;27:1194–9 [DOI] [PubMed] [Google Scholar]
- 4.Xiang AH, Kjos SL, Takayanagi M, et al. Detailed physiological characterization of the development of type 2 diabetes in Hispanic women with prior gestational diabetes mellitus. Diabetes 2010;59:2625–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bellamy L, Casas J-P, Hingorani AD, et al. Type 2 diabetes mellitus after gestational diabetes: a systematic review and meta-analysis. Lancet 2009;373:1773–9 [DOI] [PubMed] [Google Scholar]
- 6.Bian X, Gao P, Xiong X, et al. Risk factors for development of diabetes mellitus in women with a history of gestational diabetes mellitus. Chin Med J (Engl) 2000;113:759–62 [PubMed] [Google Scholar]
- 7.UK Prospective Diabetes Study (UKPDS) Group Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998;352:837–53 [PubMed] [Google Scholar]
- 8.Porte D., Jr Mechanisms for hyperglycemia in the metabolic syndrome. The key role of beta-cell dysfunction. Ann N Y Acad Sci 1999;892:73–83 [DOI] [PubMed] [Google Scholar]
- 9.Warram JH, Martin BC, Krolewski AS, et al. Slow glucose removal rate and hyperinsulinemia precede the development of type II diabetes in the offspring of diabetic parents. Ann Intern Med 1990;113:909–15 [DOI] [PubMed] [Google Scholar]
- 10.Leslie RD, Volkmann HP, Poncher M, et al. Metabolic abnormalities in children of non-insulin dependent diabetics. Br Med J Clin Res Ed 1986;293:840–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ishikawa M, Pruneda ML, Adams-Huet B, et al. Obesity-independent hyperinsulinemia in nondiabetic first-degree relatives of individuals with type 2 diabetes. Diabetes 1998;47:788–92 [DOI] [PubMed] [Google Scholar]
- 12.Gulli G, Ferrannini E, Stern M, et al. The metabolic profile of NIDDM is fully established in glucose-tolerant offspring of two Mexican-American NIDDM parents. Diabetes 1992;41:1575–86 [DOI] [PubMed] [Google Scholar]
- 13.O'Rahilly SP, Nugent Z, Rudenski AS, et al. Beta-cell dysfunction, rather than insulin insensitivity, is the primary defect in familial type 2 diabetes. Lancet 1986;2:360–4 [DOI] [PubMed] [Google Scholar]
- 14.Van Haeften TW, Dubbeldam S, Zonderland ML, et al. Insulin secretion in normal glucose-tolerant relatives of type 2 diabetic subjects. Assessments using hyperglycemic glucose clamps and oral glucose tolerance tests. Diabetes Care 1998;21:278–82 [DOI] [PubMed] [Google Scholar]
- 15.Alford FP, Henriksen JE, Rantzau C, et al. Impact of family history of diabetes on the assessment of beta-cell function. Metabolism 1998;47:522–8 [DOI] [PubMed] [Google Scholar]
- 16.Fernández-Castañer M, Biarnés J, Camps I, et al. Beta-cell dysfunction in first-degree relatives of patients with non-insulin-dependent diabetes mellitus. Diabet Med J Br Diabet Assoc 1996;13:953–9 [DOI] [PubMed] [Google Scholar]
- 17.Pimenta W, Korytkowski M, Mitrakou A, et al. Pancreatic beta-cell dysfunction as the primary genetic lesion in NIDDM. Evidence from studies in normal glucose-tolerant individuals with a first-degree NIDDM relative. JAMA 1995;273:1855–61 [PubMed] [Google Scholar]
- 18.Vilsbøll T, Krarup T, Madsbad S, et al. Both GLP-1 and GIP are insulinotropic at basal and postprandial glucose levels and contribute nearly equally to the incretin effect of a meal in healthy subjects. Regul Pept 2003;114:115–21 [DOI] [PubMed] [Google Scholar]
- 19.Deane AM, Nguyen NQ, Stevens JE, et al. Endogenous glucagon-like peptide-1 slows gastric emptying in healthy subjects, attenuating postprandial glycemia. J Clin Endocrinol Metab 2010;95:215–21 [DOI] [PubMed] [Google Scholar]
- 20.Rijkelijkhuizen JM, McQuarrie K, Girman CJ, et al. Effects of meal size and composition on incretin, α-cell, and β-cell responses. Metabolism 2010;59:502–11 [DOI] [PubMed] [Google Scholar]
- 21.Vilsbøll T, Holst JJ. Incretins, insulin secretion and Type 2 diabetes mellitus. Diabetologia 2004;47:357–66 [DOI] [PubMed] [Google Scholar]
- 22.Vilsbøll T. On the role of the incretin hormones GIP and GLP-1 in the pathogenesis of type 2 diabetes mellitus. Dan Med Bull 2004;51:364–70 [PubMed] [Google Scholar]
- 23.Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev 2007;87:1409–39 [DOI] [PubMed] [Google Scholar]
- 24.Astrup A, Rössner S, Van Gaal L, et al. Effects of liraglutide in the treatment of obesity: a randomised, double-blind, placebo-controlled study. Lancet 2009;374:1606–16 [DOI] [PubMed] [Google Scholar]
- 25.Astrup A, Carraro R, Finer N, et al. Safety, tolerability and sustained weight loss over 2 years with the once-daily human GLP-1 analog, liraglutide. Int J Obes 2005 2012;36:843–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Myers RP, Pollett A, Kirsch R, et al. Controlled Attenuation Parameter (CAP): a noninvasive method for the detection of hepatic steatosis based on transient elastography. Liver Int Off J Int Assoc Study Liver 2012;32:902–10 [DOI] [PubMed] [Google Scholar]
- 27.Bagger JI, Knop FK, Lund A, et al. Impaired regulation of the incretin effect in patients with type 2 diabetes. J Clin Endocrinol Metab 2011;96:737–45 [DOI] [PubMed] [Google Scholar]
- 28.Schulz KF, Altman DG, Moher D. CONSORT 2010 statement: updated guidelines for reporting parallel group randomised trials. BMJ 2010;340:c332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.