

Abstract
It has recently been shown that expression of the rate-limiting GABA-synthesizing enzyme glutamic acid decarboxylase (GAD) is decreased in Brodmann area 9 (BA9) of the dorsolateral prefrontal cortex (DLPFC) in Parkinson’s disease (PD) compared to control brains (Lanoue, A.C., Dumitriu, A., Myers, R.H., Soghomonian, JJ., 2010. Exp Neurol. 206(1), 207–217). A subpopulation of cortical GABAergic interneurons expresses the calcium-binding protein parvalbumin and plays a critical role in the control of pyramidal neuron excitability and the generation of cortical gamma frequency oscillations. In view of its key role in the physiology of the cerebral cortex, we sought to determine whether the expression of parvalbumin and the number of parvalbumin-expressing neurons are altered in BA9 of PD brains. First, isotopic in situ hybridization histochemistry was used to examine mRNA expression of parvalbumin on post-mortem brain sections. Second, immunohistochemistry and design-based stereology were used to determine the density of parvalbumin-positive interneurons in BA9. Quantification of mRNA labeling at the single cell level showed a significant decrease in parvalbumin expression in PD cases. In contrast, neuronal density of parvalbumin-positive neurons was not significantly different between PD and controls. Results confirm that the GABAergic system is altered in the DLPFC in PD and identify the contribution of parvalbumin-expressing neurons in these alterations. We speculate that these effects could contribute to altered cortical excitability and oscillatory activity previously documented in PD.
Keywords: Parvalbumin, Parkinson’s disease, Prefrontal cortex, mRNA, GABA, Gene expression
1. Introduction
Parkinson’s disease (PD) is a neurodegenerative disease characterized by a substantial loss of dopaminergic neurons in the substantia nigra pars compacta (SNc). However, pathology is also observed in several other brain regions including the frontal and prefrontal cortex (for review see Braak et al., 2004), and several imaging studies have reported grey matter atrophy in the frontal lobe in PD patients (e.g. Ibarretxe-Bilbao et al., 2010; Lyoo et al., 2010; Melzer et al., 2012; Nishio et al., 2010; Weintraub et al., 2011). Decreases in regional cerebral blood flow in the dorsolateral prefrontal cortex (DLPFC) in PD patients were shown to correlate with disease severity (Kikuchi et al., 2001). On the other hand, gene-profiling studies have reported alterations in the expression of genes involved in mitochondrial pathways and the ubiquitin-proteosomal system in the prefrontal cortex of PD brains (Duke et al., 2006; Moran et al., 2006; Zhang et al., 2005). Furthermore, a meta-analysis of several gene-profiling studies of post-mortem PD brain tissue indicates that GABA receptor-signaling pathways are also overrepresented in Brodmann area 9 (BA9) of the DLPFC (Sutherland et al., 2009). This later finding suggests that altered GABAergic neurotransmission in the DLPFC is a pathophysiological feature of PD. This possibility is further supported by a recent report that mRNA expression levels of the major isoform of the GABA-synthesizing enzyme, glutamic acid decarboxylase (GAD67) are decreased in BA9 in PD (Lanoue et al., 2010).
Several subpopulations of GAD67-expressing GABAergic interneurons exist in the human DLPFC and can be distinguished by their specific expression of different calcium-binding proteins (for review see Benes and Berretta, 2001). One class of GABAergic interneurons, which includes small and large basket cells and chandelier cells, selectively expresses the calcium-binding protein parvalbumin, is fast-spiking, and modulates the activity of pyramidal neurons through axo-somatic, axo-axonic or axo-dendritic synapses (for review see Benes and Berretta, 2001). Fast-spiking parvalbumin-expressing interneurons also contribute to the generation and maintenance of gamma frequency oscillations in the cerebral cortex (e.g. for reviews see Bartos et al., 2007; Freund and Katona, 2007). In a mouse model of PD, gamma oscillations in the cortex were nearly absent in the dopamine-depleted state and replaced by beta and delta oscillations in the 10–30 Hz and 1.5–4 Hz range, respectively (Costa et al., 2006), a finding consistent with clinical studies showing that the predominance of cortical oscillations in the 10–35 Hz frequency range correlates with symptom severity of PD (Silberstein et al., 2005). In view of the key functions played by parvalbumin-expressing GABAergic neurons in cortical excitability and the generation of cortical oscillations, we investigated the possibility that parvalbumin expression is altered in BA9 of the DLPFC in PD. First, in situ hybridization histochemistry was used to measure parvalbumin mRNA levels in PD and control samples. Second, immunohistochemistry and design-based stereology were used to compare the density of parvalbumin-expressing neurons in PD and control samples from BA9.
2. Results
2.1. Parvalbumin immunohistochemistry
Light microscopic examination of sections immunolabeled with the parvalbumin antiserum revealed the presence of numerous positively-labeled soma as well as processes and puncta (example of labeled cells in layer III-IV are illustrated on Figures 1A and B). Some of the puncta were observed in the vicinity of unlabeled neuronal soma (Figures 1A and B), an observation consistent with earlier observations that parvalbumin-labeled axon terminals can establish synapses on the proximal portions of pyramidal neurons in the primate frontal cortex (for reviews see Lewis et al., 2005; Lewis et al., 2012). In order to determine the localization of parvalbumin-immunolabeled soma in different cortical layers and illustrate the cortical region were neurons were quantified (Figures 2A and B), their distribution was mapped and compared to the position of cortical layers identified on adjacent sections stained with thionin (Figure 2C). First, thionin-stained sections were viewed at low magnification and the layering pattern was compared to that reported in previous cytoarchitectural studies of human prefrontal cortex (Petrides and Pandya, 1999; Rajkowska and Goldman-Rakic, 1995). Our thionin-stained sections demonstrated the key features of BA9 reported in previous studies (Petrides and Pandya, 1999; Rajkowska and Goldman-Rakic, 1995), including a narrow and sparsely populated layer IV, a lower part of layer III characterized by the presence of large pyramidal neurons and large stained pyramidal neurons in layer V (Figure 2C). Based on comparisons with thionin-stained sections, we determined that parvalbumin-immunolabeled neurons were distributed throughout layers II to VI, were noticeably absent from layer I and were mostly concentrated in layer IV (Figure 2B). This distribution is consistent with previous studies (e.g. Beasley et al., 2002; Reynolds and Beasley, 2001) and did not appear to differ between control and PD cases. Design-based stereology indicated that the average density of parvalbumin-immunoreactive neurons in BA9 was not significantly different between control and PD brains (mean±SEM: control group=7841±388.5 neurons/mm3; PD group=8324±353.7 neurons/mm3; p=0.3791, two-tailed unpaired t-test). Further regression analyses showed no significant correlation between age at death or PMI and numbers of parvalbumin-labeled neurons (Pearson correlation coefficient: age at death: r=−0.130, p=0.6869; PMI: r=0.4172, p=0.1773).
Figure 1.
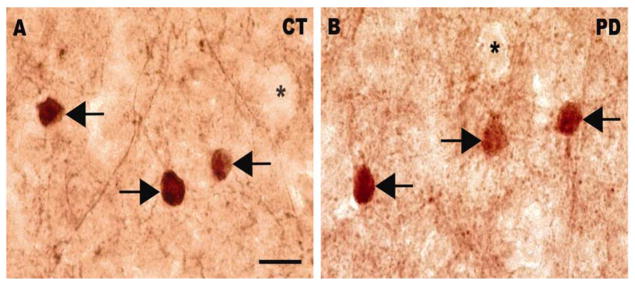
Bright-field photomicrographs illustrating immunoreactivity for parvalbumin in a normal control (A) and a PD case (B). Arrows indicate parvalbumin-positive neurons. Stars indicate unlabeled soma surrounded by immunolabeled puncta. Scale bar: 20 μm.
Figure 2.

Bright-field photomicrographs of adjacent sections of BA9 from a control case. Sections were labeled by immunohistochemistry with an antibody against parvalbumin (A, B) or Nissl-stained (C). In order to provide better visualization of the distribution of parvalbumin-labeled neurons in panel A, black dots were drawn over labeled neurons on the same photomicrograph in panel B. Cortical layers are demarcated in an adjacent Nissl-stained section (C) with black dotted lines. Note that parvalbumin-positive neurons appear in layers II-VI and their density is greatest in layer IV. Roman numerals indicate cortical layers and WM indicates white matter. Scale bar: 200 μm.
2.2. In situ hybridization analyses of parvalbumin gene expression
The observation of X-ray film radioautographs from sections processed for in situ hybridization with the parvalbumin antisense cRNA revealed labeled neurons throughout most of the cortical width. In contrast, sections hybridized with the sense probe showed no labeling. Parvalbumin mRNA-labeled neurons were concentrated in the middle layers of the cerebral cortex (Figure 3A). At the light microscope level, parvalbumin mRNA labeling in control and PD sections was detected as clusters of silver grains over hematoxylin-eosin-stained soma (Figures 3B and C). The average density of parvalbumin-mRNA labeled neurons was not different between control and PD cases (mean±SEM: control cases=19.80±2.331 neuronal profiles/mm2; PD cases=18.00±1.781 neuronal profiles/mm2; two-tailed unpaired t-test: p=0.5572). Quantitative analysis of silver grains on emulsion radioautographs at the single cell level indicated that the average level of parvalbumin mRNA labeling per neuron was significantly lower in PD compared to control cases (Figure 4) (26.0% less in PD cases; p=0.0102; two-tailed unpaired t-test). Because the mRNA values were more spread out in control than in PD cases, we determined if there were outliers in either group using the ROUT method (GraphPad™ software) with a Q value of 5%, which allows for a maximum desired false discovery rate. This analysis indicated that there were no outliers in either group. When the intensity of labeling in individual neurons (expressed as a number of pixels) was plotted against the relative proportion of labeled neurons, the distribution histogram from PD cases was uniformly shifted to the left compared to control cases (Figures 5A and B). A two-sample Kolmogorov-Smirnov test comparing the frequency distributions showed that this difference was significant (D=0.1739; p<0.0001). This latter observation suggests that the decrease in parvalbumin mRNA levels in PD cases was not restricted to a subset of parvalbumin-expressing neurons. Furthermore, we did not observe laminar differences in mRNA labeling between controls and PD cases, indicating that the decrease in parvalbumin mRNA levels in PD cases included different layers. We carried out an ANCOVA using the three covariates, age at death, PMI and tissue pH, to determine if these covariates could have impacted parvalbumin mRNA levels. The ANCOVA showed that neither pH (F(1,21)=1.317; p=0.264) nor age at death (F(1,21)=0.801; p=0.381) nor PMI (F(1,21)=1.947; p=0.177) had a significant effect on parvalbumin mRNA labeling and the main effect of group was significant (F(1,21)=5.519; p=0.034) after controlling for the effects of the three covariates. Linear regression analyses illustrate the lack of correlation between mRNA levels and age at death (Pearson correlation coefficient r=−0.1576, p=0.4420) (Figure 6A), pH (Pearson correlation coefficient r=0.2940, p=0.1449) (Figure 6B), PMI (Pearson correlation coefficient r=−0.1746, p=0.3937) (Figure 6C) and disease duration (Pearson correlation coefficient r=−0.1325, p=0.6516) (Figure 6D).
Figure 3.
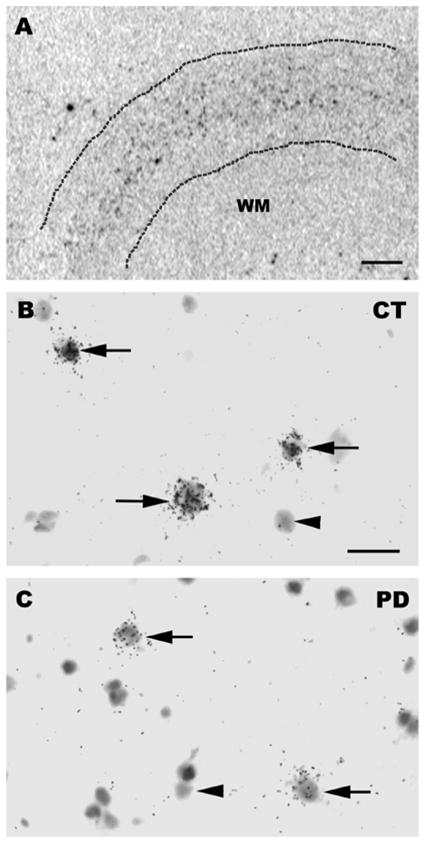
(A) Photograph from an X-ray film illustrating the distribution of parvalbumin mRNA positive neurons. The dotted lines delineate the pial and white matter boundaries and silver grain clusters representing parvalbumin mRNA positive neurons are represented by black dots between these dotted lines. Note that parvalbumin mRNA positive neurons are concentrated in the middle layers. (B, C) Bright-field photomicrographs illustrating parvalbumin mRNA labeling on emulsion radioautographs in BA9. Arrows indicate parvalbumin mRNA-labeled neurons. Arrowheads illustrate examples of unlabeled neuronal profiles. B: Control case. C: PD case. Scale bar: A=1 mm; B, C=20 μm. WM=white matter.
Figure 4.

Relative levels of parvalbumin mRNA labeling per single neuron in BA9. Labeling was measured by computerized image analysis of emulsion radioautographs. Values are the mean of the mean number of pixels per profile and are expressed as percent of controls. Data are from 11 control and 15 PD cases (*p<0.05 vs. controls; two-tailed unpaired t-test).
Figure 5.

Histograms of relative frequency distribution of parvalbumin mRNA labeling in BA9 of all control (A) and PD cases (B). Parvalbumin mRNA labeling was expressed as a number of pixels. In control cases (A), the median number of pixels per neuron is 40.5 and the mean is 59.7 pixels, while in PD cases (B), the median number of pixels per neuron is 32.0 and the mean is 45.5 pixels per neuron. This illustrates that the level of labeling per neuron is shifted to the left in the PD cases. The level of labeling was significantly decreased in the PD cases (data not shown; p<0.0001; two-tailed unpaired t-test). Med.: median.
Figure 6.

Linear regression analyses between (A) age at death, (B) pH, (C) PMI or (D) disease duration and mean parvalbumin mRNA labeling per neuron measured on emulsion radioautographs. Analyses include all 11 control and 15 PD samples from the in situ hybridization study for parvalbumin. There was no significant correlation between parvalbumin mRNA labeling and (A) age at death (Pearson correlation coefficient r=−0.1576, p=0.4420), (B) pH (Pearson correlation coefficient r=0.2940, p=0.1449), (C) PMI (Pearson correlation coefficient r=−0.1746, p=0.3937) or disease duration (Pearson correlation coefficient r=−0.1325, p=0.6516).
3. Discussion
3.1. Methodological considerations
The laminar distribution of parvalbumin-labeled neurons in our experiments is consistent with that of previous studies in human post-mortem prefrontal cortex, with parvalbumin-positive neurons reported in layers II-VI and a denser distribution in middle layers III and IV (e.g. Beasley et al., 2002; Reynolds and Beasley, 2001). The quantification of labeled neurons using design-based stereology did not reveal differences in the density of parvalbumin-positive neurons between PD and controls. This result was consistent with the quantification of mRNA-labeled neuronal profiles, which showed no differences in their density between control and PD using different cases. We were not able to determine the cortical volume in our experiments because we did not have access to the whole brains. Therefore, it cannot be ruled out that a parallel loss of neurons and brain volume occurred in PD relative to controls, which would have resulted in similar neuronal density. This possibility is unlikely, however, since earlier studies have shown that neocortical volume is not decreased in PD brains (Pedersen et al., 2005). The conclusion that there is no loss of parvalbumin-expressing neurons in BA9 in PD is consistent with earlier studies showing no global loss of neurons in the cerebral cortex (Pedersen et al., 2005) or in Brodmann area 6 (dorsolateral premotor region) in PD (MacDonald and Halliday, 2002). It should be mentioned, however, that imaging studies have reported some reduced grey matter volume or cerebral atrophy in certain cortical regions in PD (e.g. Ibarretxe-Bilbao et al., 2010; Lyoo et al., 2010; Nishio et al., 2010; Ramirez-Ruiz et al., 2005). It is also unlikely that the difference in age at death between our PD and control cases that were used in the quantification of parvalbumin immunostained neurons in fixed tissue was a confounding factor since we found no correlation between age at death and parvalbumin neuronal density in these samples.
3.2. Pathology of the dorsolateral prefrontal cortex in PD
Imaging studies have consistently found hypoactivity of the supplementary motor area and the DLPFC in PD (Buhmann et al., 2003; Haslinger et al., 2001; Jenkins et al., 1992; Playford et al., 1992; Rascol et al., 1994). The significance and mechanisms involved in these changes remain hypothetical. In particular, it is unclear if these changes are related to alterations in the balance between excitation and inhibition or to other cellular mechanisms. Several reports have documented mitochondrial dysfunction in the prefrontal cortex in PD. In particular, deficits in mitochondrial complex I (NADH-ubiquinone reductase) activity and protein levels have been reported (Arthur et al., 2009; Bueler, 2009; Navarro et al., 2009; Parker et al., 2008) while deficits in mRNA expression of multiple electron transport chain complexes are found in BA9 and other cortical regions in PD (Moran et al., 2006; Zhang et al., 2005). This suggests that depressed activation of the DLPFC in PD could reflect a general cellular metabolic deficit. In this regard, it is interesting that parvalbumin-expressing interneurons are fast-spiking and presumably have high metabolic demands. It is thus possible that deficient mitochondrial activity may especially affect these interneurons. Other pathological changes documented in the DLPFC are altered afferent inputs that could affect the overall activity of intrinsic cortical neuronal networks. For instance, loss of dopamine innervation has been documented and earlier studies found decreased density of fibers immunolabeled with tyrosine hydroxylase in the primary motor, premotor, and prefrontal cortical regions in PD (Fukuda et al., 1999; Gaspar et al., 1991). An alteration of the mesocortical dopaminergic system in the prefrontal cortex in PD is also supported by imaging studies showing a decrease in DOPA uptake and binding to dopamine D2 receptors (for review see Nikolaus et al., 2009). In rodents, loss of dopamine neurons has been shown to reduce GAD67 mRNA levels in the medial frontal cortex and fronto-parietal cortex (Lindefors et al., 1989; Reteaux et al., 1994). This suggests that the decreases in the expression of GAD67 and parvalbumin mRNA levels in PD patients may be triggered by a dopamine deficiency rather than dopamine-replacement therapy and medication history. On the other hand, decreased activity of thalamocortical excitatory projections has been documented in experimental models of PD (Galvan and Wichmann, 2008), which would also lead to depressed activity of cortical circuits in PD. In support of this possibility, levels of the vesicular glutamate transporter VGLUT1 were dramatically decreased in the prefrontal cortex of post-mortem brains from PD patients (Kashani et al., 2007), suggesting a depression of intrinsic prefrontal cortical excitatory circuits in PD. In the same study, levels of VGLUT1 and another vesicular glutamate transporter, VGLUT2, were increased in the putamen in PD. Because VGLUT1 is associated with corticostriatal projections, these studies of vesicular glutamate transporters suggest that corticostriatal glutamatergic projections and intrinsic glutamatergic cortico-cortical projections are differentially altered in the prefrontal cortex in PD.
Our findings suggest that inhibitory mechanisms are also depressed in the DLPFC in PD. The possibility of altered cortical inhibitory mechanisms in PD is otherwise supported by transcranial magnetic stimulation studies (TMS) in patients (Bares et al., 2003; Cantello et al., 2002; Hanajima and Ugawa, 2000; Lefaucheur, 2005). In particular, both the silent period and the short-stimulus interval cortical inhibition are reduced in PD and are restored by dopaminergic agents (Lefaucheur, 2005; Nakashima et al., 1995; Priori et al., 1994; Ridding et al., 1995). The silent period corresponds to the brief suppression of voluntary contraction following a single stimulus to the motor cortex while the short-stimulus interval intracortical inhibition (SICI) measures the decrease of motor evoked potentials elicited by a stimulation pulse that is preceded by a conditioning pulse. GABA has been implicated in the modulation of the cortical silent period and SICI (Lefaucheur, 2005; Ziemann et al., 1996). A contribution of the DLPFC to the silent period and SICI measured in TMS studies is not documented but our findings suggest that deficient inhibition may be widespread in the frontal lobe in PD and may occur in both primary motor and prefrontal cortices. This raises the intriguing possibility that abnormal activation of primary motor, premotor and dorsolateral prefrontal cortex seen in patients with PD (Buhmann et al., 2003; Haslinger et al., 2001; Jenkins et al., 1992; Rascol et al., 1994; Tessa et al., 2012) may be linked to deficient intracortical inhibition.
We previously reported decreased GAD67 mRNA expression in BA9 of PD brains (Lanoue et al., 2010). The finding that parvalbumin expression is also decreased suggests that the same population of GABAergic neurons may have decreased expression of the two markers. GAD67 is highly expressed in neurons with a high firing rate such as cortical parvalbumin-expressing neurons (Feldblum et al., 1993). In the prefrontal cortex, axons from chandelier cells co-express GAD67 and parvalbumin but not GAD65 while both GAD isoforms are co-expressed in axons of basket cells, which correspond to the other population of parvalbumin-expressing neurons (Fish et al., 2011). This raises the possibility that the decrease in GAD67 mRNA expression in BA9 in PD preferentially affects chandelier cells. Because chandelier cells exert a key control on pyramidal neuron excitability, depressed GABAergic activity in these neurons would have major consequences on corticofugal ouputs. There are several lines of evidence that suggest that parvalbumin may be affecting cortical inhibition through its relationship with GAD67. Parvalbumin is a calcium-binding protein that plays a key role in GABAergic neurotransmission (Collin et al., 2005; Muller et al., 2007). GABA release is increased in mice with reduced parvalbumin expression (Vreugdenhil et al., 2003; Collin et al., 2005) and in cultured parvalbumin interneurons, a decrease in GAD67 induced by the nonselective NMDA receptor antagonist, ketamine, was followed by a decrease in parvalbumin immunoreactivity (Kinney et al., 2006). Based on these studies, it has been proposed that reduced parvalbumin expression may represent a compensatory response to reduced GABA levels and enhance GABA release. This hypothesis is interesting and warrants more detailed physiological and anatomical studies in experimental models of PD.
4. Experimental Procedure
4.1. Human tissue and tissue sectioning
For the in situ hybridization study 11 control and 15 PD samples of BA9 from male subjects were obtained from the Harvard Brain Tissue Resource Center (HBTRC; McLean Hospital, Belmont, MA) and the Sun Health Research Institute (SHRI; Sun City, AZ) (Table 1). All subjects in the PD group had a clinical diagnostic of PD and pathological evidence of PD determined by the presence of Lewy bodies in the SNc, but cognitive measures were not available. The pH of all samples used in mRNA expression studies was measured as previously described (Harrison et al., 1995). For each sample, at least two pH readings were taken, and the mean of these values was used. Because lower tissue pH is associated with greater RNA degradation (Harrison et al., 1995; Kingsbury et al., 1995; Catts et al., 2005), we selected cases so that the average pH between groups was not significantly different (Table 1). Age at death and post-mortem interval (PMI) were also available for all samples (Table 1). Disease duration, defined as the time elapsed between the onset of motor symptoms and death, was available for all but four PD cases (Table 1). For the in situ hybridization studies, tissue blocks were stored at −80°C. From these blocks, 10 μm-thick sections were cut at −20°C using a cryostat. Each section was thaw-mounted onto a chromalum gelatin-coated glass slide and stored at −80°C until further processing.
Table 1.
Human subjects data for the in situ hybridization experiments.
| Age at Death (years) | PMI (hours) | pH | Disease Duration (years) | Cause of Death |
|---|---|---|---|---|
| Harvard Brain Tissue Resource Center (HBTRC) Control Cases | ||||
| 66 | 18.7 | 6.75 | N/A | Myocardial infarction |
| 69 | 15.3 | 7.323 | N/A | Respiratory Failure; Chronic obstructive pulmonary disease |
| 40 | 16.6 | 6.593 | N/A | Cardiac |
| 106 | 21 | 6.708 | N/A | Congestive heart failure; Acute renal failure; Myocardial infarction |
| 44 | 28.17 | 6.965 | N/A | Cardiac arrest |
| 43 | 14.68 | 6.985 | N/A | Myocardial infarction |
| 52 | 22.95 | 6.373 | N/A | Heart Attack |
| 60.00 | 19.63 | 6.814 | ||
| Harvard Brain Tissue Resource Center (HBTRC) PD Cases | ||||
| 68 | 22.87 | 6.848 | 5 | Cardiac Arrest |
| 79 | 11.77 | 6.415 | 13 | Aspiration Pneumonia |
| 74 | 15.15 | 6.67 | N/A | End Stage PD; Bladder Infection |
| 89 | 30.75 | 6.675 | 17 | End Stage PD |
| 66 | 11.21 | 6.735 | 11 | PD |
| 75.20* | 18.35* | 6.669* | 11.50 | |
| Sun Health Research Institute (SHRI) Control Cases | ||||
| 91 | 1.5 | 6.29 | N/A | Metastatic Bladder Cancer |
| 97 | 1.5 | 7.137 | N/A | Metastatic Colon Cancer |
| 79 | 2 | 6.915 | N/A | Cardiac and/or respiratory failure |
| 63 | 1.5 | 6.603 | N/A | Acute intracerebral hemorrhage |
| 82.50 | 1.625 | 6.736 | ||
| Sun Health Research Institute (SHRI) PD Cases | ||||
| 85 | 2.16 | 6.728 | 6 | Lung cancer |
| 77 | 1.66 | 6.438 | 13 | PD |
| 72 | 2 | 6.5 | 10 | Brain cancer - glioblastoma |
| 83 | 2 | 6.58 | 7 | Pneumonia; hypertension; vascular dementia |
| 72 | 3.5 | 6.72 | 17 | Possible Cerebrovascular accident; End stage PD |
| 77 | 1.16 | 6.593 | 22 | End stage PD; Complications from fall |
| 83 | 2.16 | 6.838 | 4 | End stage PD |
| 80 | 2.25 | 6.65 | 25 | End stage PD; Inanition |
| 84 | 2.5 | 6.47 | 4 | N/A |
| 88 | 2 | 6.71 | 3 | End stage chronic obstructive pulmonary disease |
| 80.10* | 2.139* | 6.623* | 11.10 | |
PMI: post-mortem interval
indicates no significant difference from controls, two-tailed unpaired t-test
Bold values indicate means for each group in each cohort
For the immunohistochemical and Nissl-staining studies, formalin-fixed tissue blocks of BA9 from 6 control and 6 PD cases were obtained from the SHRI. These cases were different from those used in the mRNA expression studies because fixed and unfixed matching samples from the same cases were not available. Tissue blocks were cryoprotected by immersing them sequentially in a solution of 2% dimethylsulfoxide (DMSO) and 10% glycerol at 4°C for three days and in a solution of 2% DMSO and 20% glycerol at 4°C for three days (Rosene et al., 1986). Blocks were rapidly frozen by immersion in 2-methyl-butane at −75°C for one hour, then stored in a freezer at −80°C for at least 2 days. Blocks were sectioned in series of 9 sections on a freezing microtome at a thickness of 30 μm or 60 μm and then placed in wells filled with a solution of 15% glycerol in 0.1 M phosphate buffer (PB) pH 7.4 and stored at −80°C. Subjects were matched for PMI (control: 3.250±0.258 hours; PD: 3.080±0.337 hours) but the mean age at death of control cases (87.17±2.65 years) was significantly higher than the mean age at death of PD cases (77.33±2.69 years) (two-tailed unpaired t-test between controls and PD: p=0.0264).
4.2. Immunohistochemistry
Thirty μm-thick sections were removed from the −80°C freezer, thawed and mounted onto chromalum gelatin-coated glass slides and air-dried for 3 days at room temperature. For antigen retrieval, slides were loaded into stainless steel racks and lowered into a pressure cooker filled with a boiling diluted citrate-based antigen unmasking solution (Vector Laboratories, Burlingame, CA). The pressure cooker was locked. One minute after reaching high pressure, the cooker was quickly depressurized and the racks were placed in cold water for five minutes. For all subsequent steps, the slides were processed in vertical staining wells under gentle agitation on an orbital shaker. The staining wells were filled with 0.1 M Tris-buffered saline (TBS) pH 7.4. After two ten-minute washes in TBS, endogenous peroxidase activity was quenched by treatment with a solution of 3% hydrogen peroxide in 50% ethanol for 45 minutes. To block non-specific binding of the secondary antibody, sections were then placed in a blocking solution of 10% horse serum in 0.1 M TBS plus 0.4% Triton-X for 2 hours. Sections were then incubated for 48 hours at 4°C with a primary monoclonal antibody for parvalbumin (Swant, Switzerland). Sections were washed three times in a solution of 1% horse serum and 0.2% Triton-X in 0.1 M TBS pH 7.4 for 10 minutes, followed by incubation with a biotinylated anti-mouse secondary antibody raised in horse (Vector Laboratories, Burlingame, CA) for 2 hours at room temperature. Sections were then incubated at room temperature for 1 hour and 45 minutes in an avidin–biotinylated peroxidase complex solution. The antigenic sites were visualized by incubation of the sections in a solution containing 3,3′-Diaminobenzidine (DAB) (ImmPact DAB Peroxidase Substrate kit, Vector Laboratories, Burlingame, CA). The reaction was stopped by rinsing in distilled water. Sections were air-dried overnight at room temperature, dehydrated in graded alcohols and coverslipped with Permount (Fisher Scientific, Pittsburgh, PA).
4.3. Design-based stereology
The density of parvalbumin-immunoreactive neurons in BA9 was estimated using a design-based stereological approach. Nine serial sections per case were used for this analysis. The number of sections and counting parameters were determined using data from a previous pilot study to obtain a coefficient of error (CE) (Gundersen et al., 1999) less than 0.1 for each case. Because the tissue blocks were not uniform in size, the number of series sectioned varied between cases. The interval between sections varied from 180 μm to 390 μm. Nevertheless, all cases included nine sections and a minimum of 300 neurons per case was counted. A Nikon Eclipse E600 microscope (Nikon Instruments Inc., Melville, NY) equipped with a motorized staged and joystick and StereoInvestigator software (version 8.21.7, MBF Bioscience, Williston, VT) were used. The StereoInvestigator software system is based on the optical disector method, which allows the estimation of a number of objects in a known volume without introducing biases due to size, shape, or section thickness (Gundersen et al., 1988; Mouton, 2002). In order to avoid edge and lost cap effects, a 1 μm guard volume was implemented above the disector probe and a floating guard volume (range 1–5 μm) was left at the bottom of the disector. The thickness of sections used for the analysis was not significantly different between groups (control group: 13.25±0.1057 μm, PD group: 13.55±0.1544; p=0.1399). The region of interest was the portion of the section comprised of the largest available continuous area of grey matter. The region of interest was viewed and outlined with the contour function of Stereoinvestigator using the 4x objective. The 40x objective was used for cell counts. The grid size for the study was 1142.3 μm × 1003.1 μm. A three-dimensional counting box was used with the following dimensions: 100 μm × 100 μm × 9 μm. The counting objects for this study were stained parvalbumin-immunoreactive cell bodies. Any soma meeting criteria within the counting frame or in contact with the inclusionary line was counted; any soma touching an exclusionary line was ignored. These rules ensure that all objects regardless of size, shape and/or orientation have an equal chance of being counted once (Gundersen et al., 1988; Mouton, 2002). The number of parvalbumin-immunoreactive cells counted was divided by the total volume of the disector probes in each case. This provided a sample estimate of the total number of objects counted in the volume of reference space, also known as the numerical density (Gundersen et al., 1988; Mouton, 2002). The density was expressed in numbers of neurons per cubic millimeter. The mean coefficient of error (Gundersen et al., 1999) for each case in every study was 0.051. The investigator who performed this quantification was blind to the experimental groups.
4.4. Thionin staining
Sixty μm-thick sections were mounted onto chromalum gelatin-coated glass slides and air-dried for 3 days at room temperature. Slides were then loaded into stainless steel racks. The tissue was defatted in a solution of 50% ethanol and 50% chloroform for 5 hours. Tissue was then hydrated in a series of graded alcohols followed by deionized water. Sections were then placed in a 0.5% thionin solution (pH 3.6) for 90 seconds. Sections were dehydrated in deionized water followed by a series of graded ethanol washes then cleared in xylene and coverslipped with Permount (Fisher Scientific, Pittsburgh, PA).
4.5. In situ hybridization histochemistry
A 35S radiolabeled complementary RNA (cRNA) probe was transcribed in vitro from human parvalbumin (PVALB gene) complementary DNA (cDNA) (GenBank: BC069300). The 418 base pair parvalbumin cDNA was inserted into the transcription vector pPCR-Script Amp SK(+). The vector was linearized with the restriction enzyme, SacI. In vitro transcription of the radioactive cRNA sense and antisense probes was performed for 2 hours at 37°C in the presence of 2.5 μM 35S-uracil triphosphate (UTP; specific activity 1250 Ci/mmol; PerkinElmer Life Sciences, Boston, MA) and 10 μM unlabeled UTP with adenosine triphosphate (ATP), cytosine triphosphate (CTP), and guanine triphosphate (GTP) in excess. The cDNA template was then digested with DNAse I. The labeled cRNAs were purified by phenol/chloroform extraction and ethanol precipitation. The length of the cRNA probes was reduced by partial alkaline hydrolysis to increase the sensitivity of the hybridization reaction (Cox et al., 1984).
Two adjacent sections per subject were used and all sections used for the quantitative analyses were strictly processed in parallel under identical conditions. The hybridization protocol has been previously published (Soghomonian et al., 1994). Briefly, sections were fixed in 3% paraformaldehyde, washed in 2×SSC, phosphate buffer saline, 0.25% acetic anhydride and triethanolamine, Tris-glycine, and dehydrated in ethanol. Sections were hybridized for 4 hours at 52°C with 8 ng of radiolabeled cRNA probe in hybridization solution (40% formamide, 10% dextran sulfate, 4×SSC, 10 mM dithiothreitol, 1.0% sheared salmon sperm DNA, 1.0% yeast tRNA, 1×Denhardt’s solution). Post-hybridization washes were in 50% formamide and RNAseA. After dehydration and defatting, sections were apposed to Kodak BioMax MR films in light-tight cassettes for 11 days. After the films were developed, slides were processed for emulsion radioautography by dipping in Kodak NTB3 nuclear emulsion, air-dried for 3 hours, and stored in light-tight boxes for 19 days. Emulsion was then developed in Kodak D-19, the sections were lightly counterstained with eosin and hematoxylin, and mounted with Eukitt (Sigma-Aldrich Chemicals, St. Louis, MO)
4.6. Quantification of parvalbumin mRNA labeling on emulsion radioautographs
Individual labeled neurons were visualized under bright-field illumination with a 60x objective on a Nikon E600 microscope connected to a Sony CCD camera, which was connected to a Macintosh computer. Microscope images were observed live with NIH Image 1.61. The area covered by silver grains over each neuron was measured by computerized image analysis using the density slice function of NIH image. The labeled cellular profiles were selected with the drawing tool of NIH image and the area covered by silver grains in the selection was calculated and expressed as a number of pixels per cellular profile. Before this analysis was carried out, we determined that there was a linear relationship between numbers of silver grains and the area expressed in pixels on a sample of neurons in which the numbers of grains were counted manually and compared to the area covered by grains as measured with NIH image. For each section, 50 neuronal profiles were sampled and analyzed and the labeling was reported as a number of pixels per profile. Sampling of neurons was carried out throughout the cortical width by systematically scanning the tissue from layer VI to the pial surface and back from the pial surface to layer VI until a minimum of 50 neurons per section was analyzed. Every labeled neuron encountered while scanning the tissue was analyzed. Because artifactual scattered silver grains were occasionally present in the neuropil, the level of background was determined by measuring the average density of silver grains in the neuropil. Based on this quantification and in order to ensure that only labeled neurons were included in the quantification, neurons were considered parvalbumin mRNA positive if their soma was covered by 5 or more silver grains. For each subject, two adjacent sections were analyzed so that the mean number of pixels per neuronal profile per subject was the mean of 100 profiles. In the control group, labeling was measured in 11 cases for a total of 1100 neurons and in the PD group labeling was measured in 15 cases for a total of 1500 neurons. The investigator who performed this quantification was blind to the experimental groups in order to prevent unintentional bias in the selection of silver grains. Finally, the density of parvalbumin mRNA-labeled neurons per mm2 of tissue was determined on emulsion radioautographs by systematically scanning sections on the Nikon Eclipse E600 microscope at 60x throughout the cortical thickness and counting all labeled profiles. A grid size of 0.02779 mm2 was chosen and an average of 190 grids per section were counted.
4.7. Statistical analyses
Differences in mRNA labeling values, densities of parvalbumin-immunoreactive neurons, age at death, PMI, and pH between control and PD groups were analyzed with two-tailed unpaired t-tests. Correlations between parvalbumin mRNA labeling or density of parvalbumin-positive neurons and age at death, PMI, pH or disease duration were analyzed by linear regression. An analysis of covariance (ANCOVA) was used to determine and control for the possible effects of the covariates pH, age at death and PMI on mRNA labeling. In all cases, a value of p<0.05 was considered significant.
Highlights.
Parvalbumin mRNA expression is decreased in Brodmann area 9 in Parkinson’s disease
Parvalbumin neurons are not lost in Brodmann area 9 in Parkinson’s disease
Parvalbumin-positive neurons are concentrated in middle cortical layers
Acknowledgments
The authors would like to thank the following institutions for providing us with brain tissue used in this study: the Sun Health Research Institute in Sun City, AZ, which is supported by the National Institute on Aging (P30 AG19610 Arizona Alzheimer’s Disease Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer’s Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011 and 05-901 to the Arizona Parkinson’s Disease Consortium) and the Prescott Family Initiative of the Michael J. Fox Foundation for Parkinson’s Research, and the Harvard Brain Tissue Resource Center, which is supported in part by PHS grant number R24 MH 068855. We also acknowledge the support of the American Parkinson’s Disease Association (APDA).
Footnotes
Conflict of Interest
The authors have no conflict of interest to report.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arthur CR, Morton SL, Dunham LD, Keeney PM, Bennett JP., Jr Parkinson’s disease brain mitochondria have impaired respirasome assembly, age-related increases in distribution of oxidative damage to mtDNA and no differences in heteroplasmic mtDNA mutation abundance. Mol Neurodegener. 2009;4:37. doi: 10.1186/1750-1326-4-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bares M, Kanovsky P, Klajblova H, Rektor I. Intracortical inhibition and facilitation are impaired in patients with early Parkinson’s disease: a paired TMS study. Eur J Neurol. 2003;10:385–9. doi: 10.1046/j.1468-1331.2003.00610.x. [DOI] [PubMed] [Google Scholar]
- Bartos M, Vida I, Jonas P. Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat Rev Neurosci. 2007;8:45–56. doi: 10.1038/nrn2044. [DOI] [PubMed] [Google Scholar]
- Beasley CL, Zhang ZJ, Patten I, Reynolds GP. Selective deficits in prefrontal cortical GABAergic neurons in schizophrenia defined by the presence of calcium-binding proteins. Biol Psychiatry. 2002;52:708–15. doi: 10.1016/s0006-3223(02)01360-4. [DOI] [PubMed] [Google Scholar]
- Benes FM, Berretta S. GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology. 2001;25:1–27. doi: 10.1016/S0893-133X(01)00225-1. [DOI] [PubMed] [Google Scholar]
- Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004;318:121–34. doi: 10.1007/s00441-004-0956-9. [DOI] [PubMed] [Google Scholar]
- Bueler H. Impaired mitochondrial dynamics and function in the pathogenesis of Parkinson’s disease. Exp Neurol. 2009;218:235–46. doi: 10.1016/j.expneurol.2009.03.006. [DOI] [PubMed] [Google Scholar]
- Buhmann C, Glauche V, Sturenburg HJ, Oechsner M, Weiller C, Buchel C. Pharmacologically modulated fMRI--cortical responsiveness to levodopa in drug-naive hemiparkinsonian patients. Brain. 2003;126:451–61. doi: 10.1093/brain/awg033. [DOI] [PubMed] [Google Scholar]
- Cantello R, Tarletti R, Civardi C. Transcranial magnetic stimulation and Parkinson’s disease. Brain Res Brain Res Rev. 2002;38:309–27. doi: 10.1016/s0165-0173(01)00158-8. [DOI] [PubMed] [Google Scholar]
- Catts VS, Catts SV, Fernandez HR, Taylor JM, Coulson EJ, Lutze-Mann LH. A microarray study of post-mortem mRNA degradation in mouse brain tissue. Brain Research: Molecular Brain Research. 2005;138:164–177. doi: 10.1016/j.molbrainres.2005.04.017. [DOI] [PubMed] [Google Scholar]
- Collin T, Chat M, Lucas MG, Moreno H, Racay P, Schwaller B, et al. Developmental changes in parvalbumin regulate presynaptic Ca2+ signaling. Journal of Neuroscience. 2005;25:96–107. doi: 10.1523/JNEUROSCI.3748-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa RM, Lin SC, Sotnikova TD, Cyr M, Gainetdinov RR, Caron MG, Nicolelis MA. Rapid alterations in corticostriatal ensemble coordination during acute dopamine-dependent motor dysfunction. Neuron. 2006;52:359–69. doi: 10.1016/j.neuron.2006.07.030. [DOI] [PubMed] [Google Scholar]
- Cox KH, DeLeon DV, Angerer LM, Angerer RC. Detection of mrnas in sea urchin embryos by in situ hybridization using asymmetric RNA probes. Dev Biol. 1984;101:485–502. doi: 10.1016/0012-1606(84)90162-3. [DOI] [PubMed] [Google Scholar]
- Duke DC, Moran LB, Kalaitzakis ME, Deprez M, Dexter DT, Pearce RK, Graeber MB. Transcriptome analysis reveals link between proteasomal and mitochondrial pathways in Parkinson’s disease. Neurogenetics. 2006;7:139–48. doi: 10.1007/s10048-006-0033-5. [DOI] [PubMed] [Google Scholar]
- Feldblum S, Erlander MG, Tobin AJ. Different distributions of GAD65 and GAD67 mRNAs suggest that the two glutamate decarboxylases play distinctive functional roles. J Neurosci Res. 1993;34:689–706. doi: 10.1002/jnr.490340612. [DOI] [PubMed] [Google Scholar]
- Fish KN, Sweet RA, Lewis DA. Differential distribution of proteins regulating GABA synthesis and reuptake in axon boutons of subpopulations of cortical interneurons. Cereb Cortex. 2011;21:2450–60. doi: 10.1093/cercor/bhr007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Katona I. Perisomatic inhibition. Neuron. 2007;56:33–42. doi: 10.1016/j.neuron.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Fukuda T, Takahashi J, Tanaka J. Tyrosine hydroxylase-immunoreactive neurons are decreased in number in the cerebral cortex of Parkinson’s disease. Neuropathology. 1999;19:10–3. doi: 10.1046/j.1440-1789.1999.00196.x. [DOI] [PubMed] [Google Scholar]
- Galvan A, Wichmann T. Pathophysiology of parkinsonism. Clin Neurophysiol. 2008;119:1459–74. doi: 10.1016/j.clinph.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar P, Duyckaerts C, Alvarez C, Javoy-Agid F, Berger B. Alterations of dopaminergic and noradrenergic innervations in motor cortex in Parkinson’s disease. Ann Neurol. 1991;30:365–74. doi: 10.1002/ana.410300308. [DOI] [PubMed] [Google Scholar]
- Gundersen HJ, Bagger P, Bendtsen TF, Evans SM, Korbo L, Marcussen N, Moller A, Nielsen K, Nyengaard JR, Pakkenberg B, et al. The new stereological tools: disector, fractionator, nucleator and point sampled intercepts and their use in pathological research and diagnosis. Apmis. 1988;96:857–81. doi: 10.1111/j.1699-0463.1988.tb00954.x. [DOI] [PubMed] [Google Scholar]
- Gundersen HJ, Jensen EB, Kieu K, Nielsen J. The efficiency of systematic sampling in stereology--reconsidered. J Microsc. 1999;193:199–211. doi: 10.1046/j.1365-2818.1999.00457.x. [DOI] [PubMed] [Google Scholar]
- Hanajima R, Ugawa Y. Intracortical inhibition of the motor cortex in movement disorders. Brain Dev. 2000;22(Suppl 1):S132–5. doi: 10.1016/s0387-7604(00)00140-6. [DOI] [PubMed] [Google Scholar]
- Harrison PJ, Heath PR, Eastwood SL, Burnet PW, McDonald B, Pearson RC. The relative importance of premortem acidosis and postmortem interval for human brain gene expression studies: selective mRNA vulnerability and comparison with their encoded proteins. Neurosci Lett. 1995;200:151–4. doi: 10.1016/0304-3940(95)12102-a. [DOI] [PubMed] [Google Scholar]
- Haslinger B, Erhard P, Kampfe N, Boecker H, Rummeny E, Schwaiger M, Conrad B, Ceballos-Baumann AO. Event-related functional magnetic resonance imaging in Parkinson’s disease before and after levodopa. Brain. 2001;124:558–70. doi: 10.1093/brain/124.3.558. [DOI] [PubMed] [Google Scholar]
- Ibarretxe-Bilbao N, Ramirez-Ruiz B, Junque C, Marti MJ, Valldeoriola F, Bargallo N, Juanes S, Tolosa E. Differential progression of brain atrophy in Parkinson’s disease with and without visual hallucinations. J Neurol Neurosurg Psychiatry. 2010;81:650–7. doi: 10.1136/jnnp.2009.179655. [DOI] [PubMed] [Google Scholar]
- Jenkins IH, Fernandez W, Playford ED, Lees AJ, Frackowiak RS, Passingham RE, Brooks DJ. Impaired activation of the supplementary motor area in Parkinson’s disease is reversed when akinesia is treated with apomorphine. Ann Neurol. 1992;32:749–57. doi: 10.1002/ana.410320608. [DOI] [PubMed] [Google Scholar]
- Kashani A, Betancur C, Giros B, Hirsch E, El Mestikawy S. Altered expression of vesicular glutamate transporters VGLUT1 and VGLUT2 in Parkinson disease. Neurobiol Aging. 2007;28:568–78. doi: 10.1016/j.neurobiolaging.2006.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi A, Takeda A, Kimpara T, Nakagawa M, Kawashima R, Sugiura M, Kinomura S, Fukuda H, Chida K, Okita N, Takase S, Itoyama Y. Hypoperfusion in the supplementary motor area, dorsolateral prefrontal cortex and insular cortex in Parkinson’s disease. J Neurol Sci. 2001;193:29–36. doi: 10.1016/s0022-510x(01)00641-4. [DOI] [PubMed] [Google Scholar]
- Kingsbury AE, Foster OJ, Nisbet AP, Cairns N, Bray L, Eve DJ, et al. Tissue pH as an indicator of mRNA preservation in human post-mortem brain. Brain Research: Molecular Brain Research. 1995;28:311–318. doi: 10.1016/0169-328x(94)00219-5. [DOI] [PubMed] [Google Scholar]
- Kinney JW, Davis CN, Tabarean I, Conti B, Bartfai T, Behrens MM. A specific role for NR2A-containing NMDA receptors in the maintenance of parvalbumin and GAD67 immunoreactivity in cultured interneurons. Journal of Neuroscience. 2006;26:1604–1615. doi: 10.1523/JNEUROSCI.4722-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanoue AC, Dumitriu A, Myers RH, Soghomonian JJ. Decreased glutamic acid decarboxylase mRNA expression in prefrontal cortex in Parkinson’s disease. Exp Neurol. 2010;226:207–17. doi: 10.1016/j.expneurol.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefaucheur JP. Motor cortex dysfunction revealed by cortical excitability studies in Parkinson’s disease: influence of antiparkinsonian treatment and cortical stimulation. Clin Neurophysiol. 2005;116:244–53. doi: 10.1016/j.clinph.2004.11.017. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6:312–24. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Curley AA, Glausier JR, Volk DW. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 2012;35:57–67. doi: 10.1016/j.tins.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindefors N, Brene S, Herrera-Marschitz M, Persson H. Region specific regulation of glutamic acid decarboxylase mRNA expression by dopamine neurons in rat brain. Exp Brain Res. 1989;77:611–620. doi: 10.1007/BF00249614. [DOI] [PubMed] [Google Scholar]
- Lyoo CH, Ryu YH, Lee MS. Topographical distribution of cerebral cortical thinning in patients with mild Parkinson’s disease without dementia. Mov Disord. 2010;25:496–9. doi: 10.1002/mds.22975. [DOI] [PubMed] [Google Scholar]
- MacDonald V, Halliday GM. Selective loss of pyramidal neurons in the pre-supplementary motor cortex in Parkinson’s disease. Mov Disord. 2002;17:1166–73. doi: 10.1002/mds.10258. [DOI] [PubMed] [Google Scholar]
- Melzer TR, Watts R, MacAskill MR, Pitcher TL, Livingston L, Keenan RJ, Dalrymple-Alford JC, Anderson TJ. Grey matter atrophy in cognitively impaired Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2012;83:188–94. doi: 10.1136/jnnp-2011-300828. [DOI] [PubMed] [Google Scholar]
- Moran LB, Duke DC, Deprez M, Dexter DT, Pearce RK, Graeber MB. Whole genome expression profiling of the medial and lateral substantia nigra in Parkinson’s disease. Neurogenetics. 2006;7:1–11. doi: 10.1007/s10048-005-0020-2. [DOI] [PubMed] [Google Scholar]
- Mouton P. Principles And Practices of Unbiased Stereology: An Introduction For Bioscientists. The Johns Hopkins University Press; Baltimore and London: 2002. [Google Scholar]
- Muller M, Felmy F, Schwaller B, Schneggenburger R. Parvalbumin is a mobile presynaptic Ca2+ buffer in the calyx of held that accelerates the decay of Ca2+ and short-term facilitation. Journal of Neuroscience. 2007;27:2261–2271. doi: 10.1523/JNEUROSCI.5582-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima K, Wang Y, Shimoda M, Sakuma K, Takahashi K. Shortened silent period produced by magnetic cortical stimulation in patients with Parkinson’s disease. J Neurol Sci. 1995;130:209–14. doi: 10.1016/0022-510x(95)00029-2. [DOI] [PubMed] [Google Scholar]
- Navarro A, Boveris A, Bandez MJ, Sanchez-Pino MJ, Gomez C, Muntane G, Ferrer I. Human brain cortex: mitochondrial oxidative damage and adaptive response in Parkinson disease and in dementia with Lewy bodies. Free Radic Biol Med. 2009;46:1574–80. doi: 10.1016/j.freeradbiomed.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Nikolaus S, Antke C, Muller HW. In vivo imaging of synaptic function in the central nervous system: I. Movement disorders and dementia. Behav Brain Res. 2009;204:1–31. doi: 10.1016/j.bbr.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Nishio Y, Hirayama K, Takeda A, Hosokai Y, Ishioka T, Suzuki K, Itoyama Y, Takahashi S, Mori E. Corticolimbic gray matter loss in Parkinson’s disease without dementia. Eur J Neurol. 2010;17:1090–7. doi: 10.1111/j.1468-1331.2010.02980.x. [DOI] [PubMed] [Google Scholar]
- Parker WD, Jr, Parks JK, Swerdlow RH. Complex I deficiency in Parkinson’s disease frontal cortex. Brain Res. 2008;1189:215–8. doi: 10.1016/j.brainres.2007.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen KM, Marner L, Pakkenberg H, Pakkenberg B. No global loss of neocortical neurons in Parkinson’s disease: a quantitative stereological study. Mov Disord. 2005;20:164–71. doi: 10.1002/mds.20289. [DOI] [PubMed] [Google Scholar]
- Petrides M, Pandya DN. Dorsolateral prefrontal cortex: comparative cytoarchitectonic analysis in the human and the macaque brain and corticocortical connection patterns. Eur J Neurosci. 1999;11:1011–36. doi: 10.1046/j.1460-9568.1999.00518.x. [DOI] [PubMed] [Google Scholar]
- Playford ED, Jenkins IH, Passingham RE, Nutt J, Frackowiak RS, Brooks DJ. Impaired mesial frontal and putamen activation in Parkinson’s disease: a positron emission tomography study. Ann Neurol. 1992;32:151–61. doi: 10.1002/ana.410320206. [DOI] [PubMed] [Google Scholar]
- Priori A, Berardelli A, Inghilleri M, Accornero N, Manfredi M. Motor cortical inhibition and the dopaminergic system. Pharmacological changes in the silent period after transcranial brain stimulation in normal subjects, patients with Parkinson’s disease and drug-induced parkinsonism. Brain. 1994;117(Pt 2):317–23. doi: 10.1093/brain/117.2.317. [DOI] [PubMed] [Google Scholar]
- Rajkowska G, Goldman-Rakic PS. Cytoarchitectonic definition of prefrontal areas in the normal human cortex: I. Remapping of areas 9 and 46 using quantitative criteria. Cereb Cortex. 1995;5:307–22. doi: 10.1093/cercor/5.4.307. [DOI] [PubMed] [Google Scholar]
- Ramirez-Ruiz B, Marti MJ, Tolosa E, Bartres-Faz D, Summerfield C, Salgado-Pineda P, Gomez-Anson B, Junque C. Longitudinal evaluation of cerebral morphological changes in Parkinson’s disease with and without dementia. J Neurol. 2005;252:1345–52. doi: 10.1007/s00415-005-0864-2. [DOI] [PubMed] [Google Scholar]
- Rascol O, Sabatini U, Chollet F, Fabre N, Senard JM, Montastruc JL, Celsis P, Marc-Vergnes JP, Rascol A. Normal activation of the supplementary motor area in patients with Parkinson’s disease undergoing long-term treatment with levodopa. J Neurol Neurosurg Psychiatry. 1994;57:567–71. doi: 10.1136/jnnp.57.5.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retaux S, Trovero F, Besson MJ. Role of dopamine in the plasticity of glutamic acid decarboxylase messenger RNA in the rat frontal cortex and the nucleus accumbens. European Journal of Neuroscience. 1994;6:1782–1791. doi: 10.1111/j.1460-9568.1994.tb00571.x. [DOI] [PubMed] [Google Scholar]
- Reynolds GP, Beasley CL. GABAergic neuronal subtypes in the human frontal cortex--development and deficits in schizophrenia. J Chem Neuroanat. 2001;22:95–100. doi: 10.1016/s0891-0618(01)00113-2. [DOI] [PubMed] [Google Scholar]
- Ridding MC, Inzelberg R, Rothwell JC. Changes in excitability of motor cortical circuitry in patients with Parkinson’s disease. Ann Neurol. 1995;37:181–8. doi: 10.1002/ana.410370208. [DOI] [PubMed] [Google Scholar]
- Rosene DL, Roy NJ, Davis BJ. A cryoprotection method that facilitates cutting frozen sections of whole monkey brains for histological and histochemical processing without freezing artifact. J Histochem Cytochem. 1986;34:1301–15. doi: 10.1177/34.10.3745909. [DOI] [PubMed] [Google Scholar]
- Silberstein P, Pogosyan A, Kuhn AA, Hotton G, Tisch S, Kupsch A, Dowsey-Limousin P, Hariz MI, Brown P. Cortico-cortical coupling in Parkinson’s disease and its modulation by therapy. Brain. 2005;128:1277–91. doi: 10.1093/brain/awh480. [DOI] [PubMed] [Google Scholar]
- Soghomonian JJ, Pedneault S, Audet G, Parent A. Increased glutamate decarboxylase mRNA levels in the striatum and pallidum of MPTP-treated primates. J Neurosci. 1994;14:6256–65. doi: 10.1523/JNEUROSCI.14-10-06256.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland GT, Matigian NA, Chalk AM, Anderson MJ, Silburn PA, Mackay-Sim A, Wells CA, Mellick GD. A cross-study transcriptional analysis of Parkinson’s disease. PLoS One. 2009;4:e4955. doi: 10.1371/journal.pone.0004955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessa C, Lucetti C, Diciotti S, Paoli L, Cecchi P, Giannelli M, Baldacci F, Ginestroni A, Vignali C, Mascalchi M, Bonuccelli U. Hypoactivation of the primary sensorimotor cortex in de novo Parkinson’s disease: a motor fMRI study under controlled conditions. Neuroradiology. 2012;54:261–8. doi: 10.1007/s00234-011-0955-y. [DOI] [PubMed] [Google Scholar]
- Vreugdenhil M, Jefferys JG, Celio MR, Schwaller B. Parvalbumin-deficiency facilitates repetitive IPSCs and gamma oscillations in the hippocampus. Journal of Neurophysiology. 2003;89:1414–1422. doi: 10.1152/jn.00576.2002. [DOI] [PubMed] [Google Scholar]
- Weintraub D, Doshi J, Koka D, Davatzikos C, Siderowf AD, Duda JE, Wolk DA, Moberg PJ, Xie SX, Clark CM. Neurodegeneration across stages of cognitive decline in Parkinson disease. Arch Neurol. 2011;68:1562–8. doi: 10.1001/archneurol.2011.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, James M, Middleton FA, Davis RL. Transcriptional analysis of multiple brain regions in Parkinson’s disease supports the involvement of specific protein processing, energy metabolism, and signaling pathways, and suggests novel disease mechanisms. Am J Med Genet B Neuropsychiatr Genet. 2005;137B:5–16. doi: 10.1002/ajmg.b.30195. [DOI] [PubMed] [Google Scholar]
- Ziemann U, Lonnecker S, Steinhoff BJ, Paulus W. The effect of lorazepam on the motor cortical excitability in man. Exp Brain Res. 1996;109:127–35. doi: 10.1007/BF00228633. [DOI] [PubMed] [Google Scholar]