

“The diversity in nature has long been and still is one of the biggest resources of pharmaceutical lead compounds.”
Oxytocin & vasopressin: an ancient signaling system
Oxytocin (OT), arginine-vasopressin (AVP) and their ancestral peptides in mammalian and non-mammalian vertebrates are multifunctional, closely related neurohypophyseal peptides that control important physiological and behavioral functions. Oxytocin (‘quick birth’ in Greek) was named after its uterine-contracting properties that were discovered by the British pharmacologist Sir Henry Hallett Dale at the beginning of the 20th century. In 1953 the amino acid sequence of OT was elucidated by Vincent du Vigneaud – an American chemist who later received the Nobel Prize in Chemistry for the first synthesis of a polypeptide hormone – and by the Austrian biochemist Hans Tuppy – the ‘father of peptide science’ in Austria. Although it was known by the late 18th and early 19th century that certain substances from the pituitary body have ‘pressor action’, it was Oliver Kamm in 1928 who started the isolation and purification of vasopressin. These scientists and their co-workers have established the basis for oxytocin and vasopressin research and several others have contributed over the years to advance the field. It is impossible to refer to all of them given the number of available publications (e.g., a PubMed search for vasopressin alone yields over 40,000 publications), nevertheless we attempt to provide a critical commentary on challenges in this research field with respect to therapeutic drug development and would like to introduce a new perspective to current peptide drug discovery efforts. We will give a brief overview on the evolution of OT and AVP, their diverse functions in humans, and the ‘selectivity dilemma’ that significantly hampers therapeutic development in this field. With natural product drug discovery in mind, we present our personal view as well as recent developments from our laboratories on how to utilize naturally occurring peptides as templates for ligand-based design. To conclude, we provide insights on traditional approaches to transform a natural ligand into a drug candidate and point out key factors that we believe are important for successful optimization and development of a preclinical peptide drug candidate.
The origin of the OT/AVP signaling system is thought to date back at least 600 million years [1,2]. All vertebrate OT/AVP-like peptides are considered to have evolved from the ancestral nonapeptide vasotocin (VT) [3] by gene duplication, and are today present in many different species, including non-mammalian vertebrates, fish, mammals and humans [1,4]. VT-like peptides have been identified in several invertebrate species, including molluscs, annelids, arthropods and several insects [2]. These nonapeptides all contain an N-terminal cyclic 6-residue ring structure, which is stabilized by an intramolecular disulfide bond, and a flexible C-terminal 3-residue tail.
In humans and other mammalian species these neuropeptides mediate a range of peripheral and central functions by signaling through four G-protein-coupled receptors (OTR, V1aR, V1bR and V2R) [5,6]. In the periphery, OT is involved in uterine smooth muscle contraction during parturition, ejaculation, and milk ejection during lactation. In the CNS, it functions as a neurotransmitter involved in complex social behaviors such as trust, maternal care and bonding, as well as stress and anxiety. AVP regulates peripheral fluid balance and blood pressure and is centrally implicated in memory and learning, stress-related disorders and aggressive behavior (summarized in [6]).
The structural similarity of OT and AVP, together with the high sequence homology of the extracellular binding domain of the OT-receptor and the AVP-receptors, results in significant cross talk, which becomes evident by the binding constants (Kd) and half maximal effective concentration (EC50) of OT and AVP to the four receptors (table 1) [7,8]. Specific receptor functionality is thus not controlled by ligand selectivity, but by a complex interplay comprising the enzymes oxytocinase and vasopressinase, controlled release, rapid clearance and by cell-specific up- and down-regulation of individual receptor expression (summarized in [6]). Additionally, there exist different signaling pathways upon activation of the GPCRs that can lead to a diverse range of functions depending on cell type and receptor interaction. Low receptor correlation between mammalian species complicates the situation further and several compounds selective in rat or mice were found to be unspecific for the human receptors, restricting translation into the clinic significantly. Novel drug-development programs are needed to discover selective agonists and antagonists for all four human receptors.
Table 1. Binding affinities and functional efficacies of oxytocin and arginine-vasopressin on the human receptor family.
The oxytocin and vasopressin space: a niche for peptide drugs?
Peptides make great tools for basic science studies and many drug targets were identified using peptide ligands, yet when it comes to peptide drug development one has to be careful when selecting the right target. Not only have peptides a range of significant drug-development challenges to face, including efficient drug delivery, oral bioavailability and central penetration, but it also remains disputable whether a peptide drug is commercially viable if the same target can be modulated by small molecule drugs that usually have lower production costs. Hence, drug targets need to be selected on a basis that can leverage the advantages of peptides over small molecule drugs. These include the peptide’s ability to interact with proteins on a large surface area as well as the chiral and structural complexity of a peptide, a feature that directly defers to increased specificity and potency as well as to less side effects.
We strongly believe that the OT and AVP space is such a niche and that we are just at the beginning of exploring the pharmacological potential of receptor-subtype selective ligands for the treatment of a wide range of challenging, but yet untreated, diseases. OT and AVP peptides have already proven their efficacy and potential in the pharmaceutical market (table 2): unmodified OT was introduced in 1980 under the brand names Pitocin® and Syntocinon® and is still the drug-of-choice for labor induction as well as being used for the treatment of post-partum hemorrhage. AVP and lysopressin were initially used clinically to treat central diabetes insipidus until they were replaced by the more stable analogue desmopressin. Other available peptide drugs include terlipressin for the treatment of hypotension, carbetocin for the prevention of uterine atony and control of post-partum hemorrhage, and atosiban, approved in Europe to delay premature birth. On the other hand the great promise of nonpeptide OT and AVP drugs has not been realized; the only two exceptions are conivaptan and tolvaptan, which were approved by the US FDA, yet have not found widespread use in the clinic. Sanofi, Merck and Pfizer also abandoned their non-peptide OT/AVP drug development programs, leaving only GlaxoSmithKline with retosiban, which is currently undergoing Phase II clinical trials.
Table 2. Peptide drugs targeting oxytocin and vasopressin receptors with their chemical modification and therapeutic indication.
| Peptide | Modification | Therapeutic indication |
|---|---|---|
| Oxytocin | Induction of labor; treatment of post-partum hemorrhage | |
| Arginine–vasopressin | Central diabetes insipidus | |
| Lysopressin | [Lys8]-vasopressin | Central diabetes insipidus |
| Desmopressin | Desamino-[D-Arg8]-vasopressin | Central diabetes insipidus |
| Terlipressin | GGG-[Lys8]-vasopressin | Treatment of hypotension |
| Phenypressin | [Phe2]-arginine-vasopressin | Treatment of stomatitis and pharyngitis |
| Carbetocin | Desamino-thioether-[Tyr(Me)2]-oxytocin | Prevention of uterine atony and control of post-partum hemorrhage |
| Atosiban | Desamino-[D-Tyr(Et)2, Thr4, Orn8]-oxytocin | Delay of premature childbirth |
The plethora of potential targets and therapeutic indications makes this field particularly interesting for drug development. The receptors are found in the periphery as well as in the CNS; depending on receptor subtype, cell-type and type of receptor interaction (agonism, antagonism, inverseagonism), different functional responses can be elucidated. Here is just a short list of potential therapeutic applications, where agonists (+) or antagonists (−) with a high selectivity and bioavailability profile are needed: treatment of congestive heart failure (−), blood pressure (−), anxiety (−), stress (−), anger (−), depression (−), autism (−/+), hypotension (+), hypoosmolality (+), hyponatremia (−), diabetes insipidus (+) and bleeding disorders (+) [9,10]. The key to a successful drug development program will be high receptor specificity of the ligand paired with a valid drug target that has a well-understood mechanism and appropriate animal models. Ideally, the drug candidate should retain its selectivity profile over at least two mammalian species to enable translational studies. Hence, it is crucial to screen candidates against all four receptors early on – a process that only recently became high-throughput compatible due to the availability of fluorescent imaging plate readers and commercial kits that support 384-well-plate format that allow rapid screening of functional receptor responses, a feature that greatly facilitates early-stage drug discovery.
Last but not least, OT displays another intriguing property that makes it stand out over many other peptide drugs: a few high-impact studies demonstrated that intranasal delivered OT elicits functional effects in the CNS [5,11]; how much OT actually gets across the blood–brain barrier or the exact mechanism-of-action has yet to be elucidated. Nevertheless, many scientific studies in humans were conducted with intranasal OT, and a recent meta study evaluated intranasal OT administration in 38 randomized controlled trials to have no reliable side effects supporting a safe and long-term option for therapeutic treatment [12].
This leaves us with the burning question of where we can obtain selective ligands from. To answer this question we first have to take a closer look at the structure of OT and its receptor interaction. NMR spectroscopy and X-ray crystallography studies revealed that OT has two β-turns important for receptor recognition. It is known that both the N- and C-terminus of OT are involved in binding and receptor activation, and truncations or modifications of the termini are not well tolerated (with the exception of the deamination of the N-terminal cysteine residue of OT, which creates a more stable super-agonist) [9,13]. When comparing the sequences of OT and AVP peptides from different organisms it is obvious that certain positions are highly variable whereas others are highly conserved. For example, positions 2, 5, 7 and 9 are conserved, whereas position 4 and 8 are highly variable (tables 3 & 4). Changes in the disulfide-ring structure are also not well tolerated, suggesting that the peptide might undergo a conformational change upon receptor interaction [14]. Therefore we argue that combinatorial synthetic peptide libraries will not be a good approach towards the identification of novel drug leads, unless they contain a strategy that retains the β-turn structure (summarized in [6]), while exploring other positions for better selectivity. On the other hand, considering the ancient origin of OT/AVP peptides, one can assume other organisms exist that utilize a related signaling system with similar peptide ligands that have retained the conformational geometry and flexibility required for receptor recognition and activation, hence providing an evolutionary selection advantage over synthetic libraries. The post-genomic era greatly facilitates the search for such OT/AVP-like peptide sequences due to the steadily increasing number of completed genome sequencing projects as well as advanced bioinformatics tools. Our laboratories have started to process available genomic data and literature-known sequences to test this hypothesis with the anticipation to discover novel and interesting ligands for the human receptors.
Table 3. Sequence diversity and distribution of naturally occurring oxytocin-like nonapeptides.
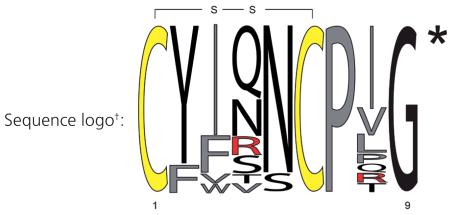
| Peptide | Sequence‡ | Organism(s) | Ref.§ |
|---|---|---|---|
| Oxytocin |
CYIQNCPLG* CYIQNCPPG* |
Eutherian mammals Monkeys |
P01178 G0ZNF6 |
| Valitocin | CYIQNCPVG* | Squalus acanthias | P43000 |
| Aspartocin | CYINNCPLG* | Squalus acanthias | P42999 |
| Asvatocin | CYINNCPVG* | Cartilaginous fish | P42996 |
| Phasitocin | CYFNNCPIG* | Triakis scyllium | Q6F4F6 |
| Phasvatocin | CYFNNCPVG* | Scyliorhinus caniculus | P42997 |
| Cephalotocin | CYFRNCPIG* | Octopus vulgaris | P80027 |
| Octopressin | CFWTSCPIG* | Octopus vulgaris | Q76DN1 |
| Seritocin | CYIQSCPIG* | Amietophrynus regularis | P42995 |
| Glumitocin | CYISNCPQG* | Cartilaginous fish | P42994 |
| Mesotocin |
CYIQNCPIG* CYFQNCPIG* |
Lungfishes and marsupials Setonix brachyurus |
P08162 0909223A¶ |
| Val4-mesotocin | CYIVNCPIG* | Plethodon shermani | A5A4K4 |
| Phe-mesotocin | CFIQNCPIG* | Neoceratodus forsteri | O57388 |
| Isotocin | CYISNCPIG* | Osteichthyes | P42993 |
| Annetocin# | CFVRNCPTG* | Eisenia foetida | P42998 |
| Vasotocin (ancestral)†† | CYIQNCPRG* | Nonmammalian vertebrates | P69129 |
Relative frequency sequence logo plot, conserved cysteine residues are highlighted in yellow, hydrophobic, aromatic and non-polar amino acids are labeled in gray and positively charged residues are labeled in red. All sequences are amidated at the C-terminus, as indicated by the asterisk.
Conserved cysteine residues forming a disulfide bond are highlighted in bold. All sequences are amidated at the C-terminus, as indicated by the asterisk.
UniProtKB entry, unless otherwise stated.
GenBank protein accession number.
Denoted also as Annepressin [1].
Presumed ancestral oxytocin/vasopressin nonapeptide [2].
Table 4. Sequence diversity and distribution of naturally occurring vasopressin-like nonapeptides.
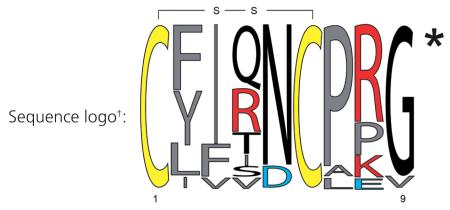
| Peptide | Sequence‡ | Organism(s) | Ref.§ |
|---|---|---|---|
| Vasopressin | CYFQNCPRG* | Eutherian mammals | P01185 |
| Lysopressin | CYFQNCPKG* | Marsupials and pigs | P01183 |
| Phenypressin | CFFQNCPRG* | Marsupials | 0909223B¶ |
| Vasotocin |
CFITNCPPG* CFVRNCPPG* CFISDCARG* |
Daphnia pulex
Platynereis dumerilii Saccoglossus kowalevskii |
E9H8T6 A6YIC5 D1LXH6 |
| Ile4-vasotocin | CYIINCPRG* |
Branchiostoma
floridae |
[23] |
| Conopressin# | CFIRNCPP* | Conus textile | P86255 |
| γ-conopressin vil# | CLIQDCPEG* | Conus villepinii | P85141 |
| [Arg]-conopressin S# | CIIRNCPRG* | Conus striatus | P05487 |
| [Lys]-conopressin G | CFIRNCPKG* | Leeches and molluscs | P05486 |
| Conopressin T# | CYIQNCLRV* | Conus tulipa | [15] |
| Inotocin |
CLITNCPRG* CLIVNCPRG* |
Arthropods, ants Camponotus floridanus |
A3RE83 [2] |
| Vasotocin (ancestral)†† | CYIQNCPRG* | Nonmammalian vertebrates |
P69129 |
Relative frequency sequence logo plot, conserved cysteine residues are highlighted in yellow, hydrophobic, aromatic and non-polar amino acids are labeled in gray, negatively charged residues are labeled in blue and positively charged residues are labeled in red. All sequences are amidated at the C-terminus, as indicated by the asterisk.
Conserved cysteine residues forming a disulfide bond are highlighted in bold. All sequences are amidated at the C-terminus, as indicated by the asterisk.
UniProtKB entry, unless otherwise stated.
GenBank protein accession number.
Peptides from Conus spp. have been isolated from venom ducts, glutamic acid represents γ-carboxyglutamate.
Presumed ancestral oxytocin/vasopressin nonapeptide [2].
Discovery & pharmacology of natural oxytocin & vasopressin peptides
The diversity in nature has long been and still is one of the biggest resources of pharmaceutical lead compounds. Many natural products often exhibit biological activity against unrelated biological targets, thus providing us with starting points for drug development [6].
Endogenous analogues of OT and AVP have been reported in several groups and lineages of the animal kingdom (tables 3 & 4)[1,2]. Surprisingly, several variants were also found in the venom of predatory cone snails. The original discovery of two of these AVP analogues was based on the characteristic grooming and scratching behavior upon intracerebral injection into mice. Although the sequences of conopressins are similar to human AVP, they have an additional positive charge in position 4, which is only found in two other endogenous vasopressin analogues, Cephalotocin (Octopus vulgaris) and Annetocin (Eisenia foetida). Conopressin-S was isolated from Conus striatus, whereas Conopressin-G was first isolated from Conus geographus venom but later also found to be present in the venom of Conus imperialis, as well as in tissue extracts of the nonvenomous snails Lymnea stagnalis and Aplysia californica and the leech Erpobdella octoculata ([15] and references cited therein). It is not clear yet what evolutionary advantage is conferred by the presence of these peptides in the venom of the cone snail. Nevertheless, the discovery and characterization of Conopressin-T in comparison with the human neuropeptides AVP and OT led to the identification of an interesting agonist/antagonist switch, which is currently being investigated towards novel antagonist design for the human receptors [15].
Another source of endogenous OT/AVP-like peptides are insects, in particular social ants. We have recently discovered inotocin sequences and their putative receptors in the genomes of a South American leaf-cutter ant (Atta cephalotes), the Florida carpenter ant (Camponotus floridanus) and the Jerdon’s jumping ant (Harpegnathos saltator) [2]. Two newly identified ant inotocin sequences display high similarity to VT. These novel sequences show amino acid variations in position 2 and position 4 (table 4), and structure-activity studies are underway to reveal if these modifications provide any novel selectivity leads for the human receptors.
From natural ligand to peptide drug candidate
The traditional approach to transform an identified drug lead into a pharmaceutical drug candidate requires, ideally, a highthroughput bioassay of a validated drug target that allows for a fast turnaround of structure–activity relationship (SAR) studies. Functional bioassays monitoring the intracellular secondary messengers (accumulation of IP-one, PIP2 or cAMP; Ca2+-ion transients) are preferred over radioligand binding/displacement assays, not only due to their high-throughput compatibility, but also due to a more comprehensive readout of mechanism- of-action (agonism vs antagonism), including allosteric modulators and maximal functional response (Emax). Additionally, a robust stability assay should be in place to monitor chemical modifications towards proteolytic stability. Typically, peptides are incubated in different tissue homogenates and membrane preparations, as well as in plasma and serum at physiological pH and temperature. The kinetics of peptide degradation and the appearance of new metabolites as a function of time can easily be monitored by HPLC and LC–MS, feeding back important information for the design of further analogues. The beauty of peptide synthesis in contrast to the synthesis of complex small molecules is that it is a highly automated process facilitating SAR studies. Hence, a typical starting point is N- and C-terminal truncation to identify the pharmacophore and the minimal sequence that still displays activity and selectivity. Alanine and d-amino acid scans are used to identify residues that can either be modified to improve proteolytic stability or can be hijacked for conjugation chemistry to improve circulation time. We believe it is important to establish early if the N- and C-termini can be altered to prevent degradation by exopeptidases. Medicinal chemists often modify or expand the termini of the shortest active sequence in order to maintain or stabilize secondary structural features important for activity. If the termini are located in close spatial proximity to each other, a cyclization strategy should be pursued [16]. NMR structure determination can help to select appropriate linker lengths for such a cyclization approach. Many peptides contain disulfide bonds, which are easily reduced in vivo by oxidoreductases, serum albumin or glutathione, resulting in the inactivation of the peptide. A lot of work has been carried out on disulfide bond mimetics to prevent such inactivation by reduction, with thioether, selenoether and diselenide bonds being the most isosteric strategies to date [14]. At this stage, synthetically more challenging modifications can be introduced to transform the peptide into a more drug-like peptidomimetic. Such approaches include N-methylation, pseudopeptides, azapeptides, peptoids, β-amino acids and turn-mimetics, to name a few that have been described in more detail in the literature [17,18].
With a more drug-like ligand in hand, it is time to address renal clearance and circulation time. This is often done by introducing functional groups to enable conjugation. PEGylation is a validated approach that can increase a peptide’s size to avoid renal clearance and keep it in circulation. Other approaches include the introduction of fatty acids (C14, C16) that facilitates binding to serum albumin in the blood, as well as formulation strategies such as hydrophobic depoting, which leads to a slow release of the drug from the injection site providing increased time-of-action.
Since OT and AVP were frontrunners in the peptide drug-development process, many of these strategies have already been implemented successfully: desmopressin (1-desamino-8-D-Arg-vasopressin), differs from AVP by its N-terminal deamination and the replacement of l-Arg by D-Arg at position 8 (table 2), which protects the peptide against N-terminal deactivation by exopeptidases and against the trypsin-like cleavage of the proline–arginine bond. These modifications improved the proteolytic stability so remarkably that desmopressin was one of the first peptides to be administered orally as well as intranasally with a half-life of 1–2 h. Terlipressin has a triple-glycine acting as a prodrug strategy to increase its half-life and carbetocin is a desamino OT analogue with a methylated tyrosine on position 2 and a thioether instead of disulfide bond to improve proteolytic stability (table 2).
We believe we are just beginning to tap the potential of peptides as drug sources. The ease of synthesis, the vast natural abundance of bioactive peptides and their immense potential as therapeutics are so convincing that it is just a question of time until an efficient delivery platform is developed that will make the above-described tedious ligand to drug candidate process redundant. We envision orally bioavailable or even CNS-penetrating scaffolds [19] where bioactive peptide sequences or their functional geometry can be grafted onto, preserving their selectivity and interaction with extracellular targets, yet simultaneously retain stability to enzymatic degradation.
Acknowledgments
Work on bioactive peptides was financially supported by the Austrian Science Fund (FWF): P22889-B11 and the European Union Seventh Framework Program (FP7/2007–2013) under grant agreement no. [254897].
Biographies
Author photographs
Christian W. Gruber 
Johannes Koehbach 
Markus Muttenthaler 
Footnotes
The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Contributor Information
Christian W Gruber, Medical University of Vienna, Center for Physiology & Pharmacology, Schwarzspanierstr. 17, A-1090 Vienna, Austria. Tel.: +43 1 4277 62511. Fax: +43 1 4277 9623. christian.w.gruber@meduniwien.ac.at.
Johannes Koehbach, Medical University of Vienna, Center for Physiology & Pharmacology, Schwarzspanierstr. 17, A-1090 Vienna, Austria.
Markus Muttenthaler, Departments of Chemistry & Cell Biology, The Scripps Research Institute, La Jolla, CA, USA.
References
Papers of special note have been highlighted as:
■ of interest
■■ of considerable interest
- 1.Donaldson ZR, Young LJ. Oxytocin, vasopressin, and the neurogenetics of sociality. Science. 2008;322:900–904. doi: 10.1126/science.1158668. [DOI] [PubMed] [Google Scholar]; ■ Provides a detailed overview of the neurobiology of oxytocin and vasopressin and their role in social behavior and cognition. Also contains a comprehensive illustration and description of the evolution of the oxytocin/vasopressin nonapeptide family.
- 2.Gruber CW, Muttenthaler M. Discovery of defense- and neuropeptides in social ants by genome-mining. PLoS ONE. 2012;7:e32559. doi: 10.1371/journal.pone.0032559. [DOI] [PMC free article] [PubMed] [Google Scholar]; ■ Provides a methodological basis for the discovery of neuropeptides and in particular oxytocin/vasopressin-related peptides from available (insect) genomic data and describes ideas on how to utilize these peptides for drug design.
- 3.Goodson JL. Nonapeptides and the evolutionary patterning of sociality. Prog. Brain Res. 2008;170:3–15. doi: 10.1016/S0079-6123(08)00401-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoyle CH. Neuropeptide families and their receptors: evolutionary perspectives. Brain Res. 1999;848:1–25. doi: 10.1016/s0006-8993(99)01975-7. [DOI] [PubMed] [Google Scholar]
- 5.Gimpl G, Fahrenholz F. The oxytocin receptor system: structure, function, and regulation. Physiol. Rev. 2001;81:629–683. doi: 10.1152/physrev.2001.81.2.629. [DOI] [PubMed] [Google Scholar]; ■■ A highly cited and comprehensive article that describes the structure and physiology of the oxytocin receptor system.
- 6.Gruber CW, Muttenthaler M, Freissmuth M. Ligand-based peptide design and combinatorial peptide libraries to target G protein-coupled receptors. Curr. Pharm. Des. 2010;16:3071–3088. doi: 10.2174/138161210793292474. [DOI] [PMC free article] [PubMed] [Google Scholar]; ■■ Describes the use of combinatorial and natural peptide libraries as a rich source of drug leads for G-protein-coupled receptors and illustrates examples for ligand-based drug design of oxytocin and vasopressin receptor ligands.
- 7.Chini B, Mouillac B, Balestre MN, et al. Two aromatic residues regulate the response of the human oxytocin receptor to the partial agonist arginine vasopressin. FEBS Lett. 1996;397:201–206. doi: 10.1016/s0014-5793(96)01135-0. [DOI] [PubMed] [Google Scholar]
- 8.Kimura T, Makino Y, Saji F, et al. Molecular characterization of a cloned human oxytocin receptor. Eur. J. Endocrinol. 1994;131:385–390. doi: 10.1530/eje.0.1310385. [DOI] [PubMed] [Google Scholar]
- 9.Manning M, Stoev S, Chini B, Durroux T, Mouillac B, Guillon G. Peptide and non-peptide agonists and antagonists for the vasopressin and oxytocin V1a, V1b, V2 and OT receptors: research tools and potential therapeutic agents. Prog. Brain Res. 2008;170:473–512. doi: 10.1016/S0079-6123(08)00437-8. [DOI] [PubMed] [Google Scholar]; ■ Describes in great detail the progress until 2008 on the design and synthesis of peptide and non-peptide selective ligands and research tools for the oxytocin and vasopressin receptor family.
- 10.Treschan TA, Peters J. The Vasopressin system: physiology and clinical strategies. Anesthesiology. 2006;105:599–612. doi: 10.1097/00000542-200609000-00026. [DOI] [PubMed] [Google Scholar]
- 11.Kosfeld M, Heinrichs M, Zak PJ, Fischbacher U, Fehr E. Oxytocin increases trust in humans. Nature. 2005;435:673–676. doi: 10.1038/nature03701. [DOI] [PubMed] [Google Scholar]
- 12.Macdonald E, Dadds MR, Brennan JL, Williams K, Levy F, Cauchi AJ. A review of safety, side-effects and subjective reactions to intranasal oxytocin in human research. Psychoneuroendocrinology. 2011;36:1114–1126. doi: 10.1016/j.psyneuen.2011.02.015. [DOI] [PubMed] [Google Scholar]
- 13.Manning M, Misicka A, Olma A, et al. Oxytocin and vasopressin agonists and antagonists as research tools and potential therapeutics. J. Neuroendocrinol. 2012;24:609–628. doi: 10.1111/j.1365-2826.2012.02303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; ■■ Provides an in-depth review of available data on the design and synthesis of selective agonists and antagonists and research tools for the oxytocin and vasopressin receptor family.
- 14.Muttenthaler M, Andersson A, De Araujo AD, Dekan Z, Lewis RJ, Alewood PF. Modulating oxytocin activity and plasma stability by disulfide bond engineering. J. Med. Chem. 2010;53:8585–8596. doi: 10.1021/jm100989w. [DOI] [PubMed] [Google Scholar]
- 15.Dutertre S, Croker D, Daly NL, et al. Conopressin-T from Conus tulipa reveals an antagonist switch in vasopressin-like peptides. J. Biol. Chem. 2008;283:7100–7108. doi: 10.1074/jbc.M706477200. [DOI] [PubMed] [Google Scholar]; ■ This article describes the discovery of a vasopressin analogue from cone snail venom and its potential for rational drug design for the oxytocin and vasopressin receptor family.
- 16.Clark RJ, Craik DJ. Native chemical ligation applied to the synthesis and bioengineering of circular peptides and proteins. Biopolymers. 2010;94:414–422. doi: 10.1002/bip.21372. [DOI] [PubMed] [Google Scholar]
- 17.Gentilucci L, De Marco R, Cerisoli L. Chemical modifications designed to improve peptide stability: incorporation of non-natural amino acids, pseudo-peptide bonds, and cyclization. Curr. Pharm. Des. 2010;16:3185–3203. doi: 10.2174/138161210793292555. [DOI] [PubMed] [Google Scholar]
- 18.Hruby VJ. Designing peptide receptor agonists and antagonists. Nat. Rev. Drug Discovery. 2002;1:847–858. doi: 10.1038/nrd939. [DOI] [PubMed] [Google Scholar]
- 19.Brasnjevic I, Steinbusch HW, Schmitz C, Martinez-Martinez P. Delivery of peptide and protein drugs over the blood–brain barrier. Prog. Neurobiol. 2009;87:212–251. doi: 10.1016/j.pneurobio.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 20.Chini B, Manning M. Agonist selectivity in the oxytocin/vasopressin receptor family: new insights and challenges. Biochem. Soc. Trans. 2007;35:737–741. doi: 10.1042/BST0350737. [DOI] [PubMed] [Google Scholar]; ■ This review article provides a critical insight into the ‘selectivity dilemma’ of oxytocin and vasopressin drug design.
- 21.Hicks C, Jorgensen W, Brown C, et al. The non-peptide oxytocin receptor agonist WAY 267,464: receptor binding profile, prosocial effects and distribution of c-Fos expression in adolescent rats. J. Neuroendocrinol. 2012;24:1012–1029. doi: 10.1111/j.1365-2826.2012.02311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wisniewski K, Galyean R, Tariga H, et al. New, potent, selective, and short-acting peptidic V1a receptor agonists. J. Med. Chem. 2011;54:4388–4398. doi: 10.1021/jm200278m. [DOI] [PubMed] [Google Scholar]
- 23.Gwee PC, Tay BH, Brenner S, Venkatesh B. Characterization of the neurohypophysial hormone gene loci in elephant shark and the Japanese lamprey: origin of the vertebrate neurohypophysial hormone genes. BMC Evol. Biol. 2009;9:47. doi: 10.1186/1471-2148-9-47. [DOI] [PMC free article] [PubMed] [Google Scholar]