

Significance
Ten eleven translocation (TET) enzymes are recently identified proteins that mediate DNA demethylation, but their regulation is still unknown. This paper indicates that a microRNA, miR-26a, is able to directly target TETs and then modulate 5-hydroxymethylcytosine levels. Furthermore, regulation of TETs by miR-26a is shown to promote pancreatic cell differentiation both in vitro and in vivo. These results highlight a link between a miRNA, DNA demethylation, and stem/progenitor cell differentiation. It also suggests a potential approach to increase the efficiency of generating pancreatic endocrine cells in vitro through modulating miR-26a.
Keywords: epigenetics, DNA methyaltion, 5hmC, posttranscriptional regulation, stem cell
Abstract
Ten eleven translocation (TET) enzymes (TET1/TET2/TET3) and thymine DNA glycosylase (TDG) play crucial roles in early embryonic and germ cell development by mediating DNA demethylation. However, the molecular mechanisms that regulate TETs/TDG expression and their role in cellular differentiation, including that of the pancreas, are not known. Here, we report that (i) TET1/2/3 and TDG can be direct targets of the microRNA miR-26a, (ii) murine TETs, especially TET2 and TDG, are down-regulated in islets during postnatal differentiation, whereas miR-26a is up-regulated, (iii) changes in 5-hydroxymethylcytosine accompany changes in TET mRNA levels, (iv) these changes in mRNA and 5-hydroxymethylcytosine are also seen in an in vitro differentiation system initiated with FACS-sorted adult ductal progenitor-like cells, and (v) overexpression of miR-26a in mice increases postnatal islet cell number in vivo and endocrine/acinar colonies in vitro. These results establish a previously unknown link between miRNAs and TET expression levels, and suggest a potential role for miR-26a and TET family proteins in pancreatic cell differentiation.
Ten eleven translocation (TET) enzymes and thymine DNA glycosylase (TDG) are implicated in active DNA demethylation (1–3). The three TET family enzymes oxidize 5-methylcytosine (5mC) in DNA to 5-hydroxymethylcytosine (5hmC), and subsequently to 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) (1, 2, 4, 5). TDG, a base excision repair glycosylase, replaces 5fC and 5caC with an unmodified cytosine via DNA repair (5, 6). Despite these advances, the molecular mechanisms underlying TETs/TDG regulation are still not known. In addition, although recent data suggest a role of TET and 5hmC in embryonic stem cells and primordial germ cells (2, 7–12), evidence for enzymatic demethylation by TET enzymes during differentiation of cells of later stages, such as the postnatal and adult stem cells of various organs including pancreas, remains very limited (13–16).
MicroRNAs (miRNAs) are an abundant class of small, highly conserved noncoding RNAs that bind the 3′-untranslated regions (UTRs) of protein-coding genes to suppress gene expression. Accumulating data have demonstrated that miRNAs are critical for many developmental and cellular processes, including organogenesis and differentiation (17). However, the role of miRNAs in TET expression and active DNA demethylation remains unclear.
Three major cell lineages exist in the adult pancreas—duct, acinar, and endocrine cells. The endocrine pancreas is composed of several hormone-releasing cells, including the insulin-secreting beta cells and glucagon-secreting alpha cells. Many transcription factors are known to control pancreas development (18). For example, the expression of pancreatic and duodenal homeobox 1 (Pdx1) in embryonic foregut region induces pancreas commitment (19, 20), and those early progenitor cells have the potential to give rise to all three pancreatic lineages (21, 22). Subsequent activation of another transcription factor, neurogenin 3 (Ngn3), restricts the lineage potential to endocrine cells (21, 23).
Expression of TETs and the genomic content of 5hmC vary across tissues (13, 14). Interestingly, TET2 and TET3 are highly expressed in the murine adult pancreas (2), yet the pancreas has lower genomic 5hmC levels than other adult tissues derived from endoderm, including liver and lung (4), suggesting dynamic DNA demethylation during pancreas development. Additionally, the miR-26 family is unique to vertebrates (24) and correlates with the emergence of the pancreas during evolution (25). Therefore, we have begun to explore the potential involvement of miR-26a/TET in pancreas development.
Although the existence of adult pancreatic progenitor cells in vivo remains debatable (26–29), we recently identified rare progenitor cells in the adult pancreas that possess extensive self-renewal and differentiation capacities in vitro (30). These adult progenitor cells, which are CD133+Sox9+, are enriched in the ductal cell fraction and can activate Pdx1 and Ngn3 expression before terminal differentiation into endocrine-like cells (30). Whether TETs, TDG, and miRNAs may affect differentiation of pancreatic cell lineages has not been tested. Here, we report that TETs and TDG are direct targets of miR-26a and the expression of TETs and miR-26a change in opposite directions during in vivo and in vitro pancreatic cell differentiation.
Results
TETs and TDG Are Direct Targets of miR-26a.
miRNA target genes are likely to have relatively long and conserved 3′-UTRs (31). We noticed that TETs, as well as TDG, have long, evolutionarily conserved 3′-UTRs (Figs. S1 and S2), so we used the TargetScan algorithm (32) to search for miRNAs that could potentially regulate TETs and TDG. Strikingly, the miR-26 family (miR-26a and miR-26b) has at least three putative target binding sites in TETs and one in TDG, in both the human and mouse genomes (Fig. 1A and Fig. S1). This putative regulation is also present in zebrafish, suggesting strong conservation (Fig. 1A and Fig. S1). Notably, miR-26 target sites are preferentially located near both ends of the 3′-UTR of TETs (Fig. S1), implying effective targeting (33). Moreover, some miR-26 target sites are close together (within 40 nt), which falls within the optimal distance for cooperative regulation (33, 34) (Fig. S1).
Fig. 1.

TET and TDG are direct targets of miR-26a. (A) Number of miR-26a target sites predicted by TargetScan for human, mouse, and zebrafish TET1/2/3 and TDG. (B) Relative luciferase activity in HEK293T cells transfected with reporter constructs containing the 3′-UTRs of TETs or TDG and cotransfected with either miR-26a precursor (Pre-26a), or negative control (Control). The 3′-UTR of TET2 and the position of the miR-26a binding sites in four TET2 reporter constructs (TET2-1, TET2-2, TET2-3+4, TET2-5+6) are indicated. The luciferase activity of reporter construct cotransfected with the negative control was set to 1. (C and D) Relative luciferase activity in HEK293T cells transfected with reporter constructs containing the wild-type or mutant 3′-UTRs of TET2 (C) or TDG (D) and cotransfected with either miR-26a precursor (Pre-26a), or negative control (Control). The Tet2 and TDG 3′-UTRs carry six and one putative miR-26a binding sites, respectively (putative pairing as shown in Fig. S2). Schematics of wild-type and mutant (M) constructs are shown along with the relative luciferase activities associated with each construct. Data are shown as mean ± SD; ***P < 0.001; *P < 0.05.
To directly test whether miR-26a targets TETs and TDG, we cloned the 3′-UTRs of TET1, TET2, TET3, and TDG downstream of a luciferase reporter, and cotransfected these reporter constructs along with miRNA precursors into the human cell line HEK293T. Coexpression of miR-26a was found to effectively down-regulate luciferase expression in constructs with these 3′-UTRs (Fig. 1B). Overexpression of miR-181a, an miRNA that does not have a putative target site in these 3′-UTRs, showed no repression on luciferase expression (Fig. S3). Mutations in the seed sequence of the predicted miR-26a binding sites within TET2 (Fig. 1C) or TDG (Fig. 1D) abolished the inhibitory effects of miR-26 on luciferase expression, indicating TET2 and TDG are directly regulated by miR-26a.
miR-26a Modulates Levels of TETs/TDG and 5hmC in HEK293T Cells.
By transfection of the miR-26 family inhibitor (designated 26 FI) and the miRNA precursor (pre-26a) into HEK293T cells (Fig. 2A), we found that expression of the endogenous TETs and TDG was inhibited by miR-26a overexpression (Fig. 2B) and enhanced by miR-26 knockdown (Fig. 2C). TET2 was the most responsive and TDG the least responsive to miR-26a regulation, which correlates with the number of miR-26a target sites within their 3′-UTRs (Fig. 1A). Consistently, levels of TET1, TET2, and TDG protein were decreased in miR-26a–overexpressing cells, but increased in miR-26a–depleted cells (Fig. 2 D and E).
Fig. 2.

miR-26a represses TETs and TDG in HEK293T cells. (A–C) qRT-PCR analysis of miR-26a (A) and TETs/TDG expression in HEK293T cells transfected with miR-26a precursor (Pre-26a) (B), miR-26a family inhibitor (26 FI) (C), or corresponding negative control (Control) for 24 h. (D and E) Western blot (D) and quantification (E) of TET1, TET2, and TDG in HEK293T cells transfected with Pre-26a, 26 FI, or Control for 48 h. (F) Quantification of 5hmC, as determined by dot blot analysis, in HEK293T cells transfected with Pre-26a, 26 FI, or Control for 48 h. Data are shown as mean ± SD; ***P < 0.001; *P < 0.05.
Because all three TET proteins can convert 5mC to 5hmC, we investigated whether miR-26a affects 5hmC levels. Dot blot analysis of global 5hmC levels in genomic DNA revealed that miR-26a overexpression reduced 5hmC levels, whereas miR-26 depletion significantly increased its levels (Fig. 2F). In contrast, global 5mC levels were not affected by miR-26a overexpression or depletion (Fig. S4), consistent with the previous report that small hairpin RNA (shRNA)-mediated knockdown of TET1 had no obvious effect on 5mC levels (2). The levels of 5hmC in HEK293 cells are less than 0.5% of total 5mC (4), so the changes in level of 5hmC that we see would not be expected to affect global 5mC levels. Collectively, these data demonstrate that miR-26a can directly regulate TETs/TDG.
miR-26a/TET Is Potentially Involved in Differentiation and Proliferation of the Developing Pancreas in Vivo.
The mouse and human genomes harbor three distinct miR-26 loci (miR-26a-1, miR-26a-2, and miR-26b), which renders genetic loss-of-function analysis difficult. We therefore turned to a gain-of-function approach and generated a miR-26a transgenic mouse line (Fig. S5). Expression of miR-26a in the adult pancreas of heterozygous miR-26a transgenic mice (TG) was about 10 times greater than that in the pancreas of wild-type (WT) littermates (Fig. 3A).
Fig. 3.

Overexpression of miR-26a promotes differentiation and proliferation of postnatal pancreas in vivo. (A) Expression of miR-26a in adult pancreas from miR-26a TG (TG) and WT mice. (B) Number of islets in P10 pancreas of WT and TG mice, as determined by counting the total number of islets from serial sections of whole pancreas. (C) Hematoxylin and eosin (H&E) images of P10 pancreas from miR-26a TG or WT mice. Islets are outlined in dots. (Scale bars, 100 µm.) (D) Expression of miR-26a in islet and nonislet fractions from P4 versus P7 WT mice. (E) Comparison of TETs and TDG gene expression in P4 and P7 islets from WT and TG mice. (F) Quantification of 5hmC, as determined by dot blot analysis, in P4 and P7 islets from WT and TG mice. (G–I) Expression of endocrine (G), progenitors or mature beta cells (H), and cell proliferation (I) markers in P7 islets of WT and TG mice. Data are shown as mean ± SD; *P < 0.05, **P < 0.01, and ***P < 0.001.
In mice, islet cells in the pancreas rapidly expand and differentiate between embryonic day 9.5 (E9.5) and birth, and postnatal day 2 (P2) and P9 (35), but not in adults (36). The pancreata of TG mice at P10 were not obviously abnormal, but did have a significantly greater number of islets (Fig. 3B). To confirm the involvement of miR-26a/TET in vivo, we determined the gene expression profiles of isolated primary islet and nonislet fractions from P4 and P7 murine pancreata. In islets, the expression of miR-26a was up-regulated at P7 compared with P4, whereas nonislets showed an opposite pattern (Fig. 3D). TETs and TDG were also preferentially expressed in islets (Fig. 3E) compared with nonislet cells (Fig. S6). In WT mice, TET2 transcripts were about 20-fold more abundant than TET1/3 transcripts in P4 islets, and maintained an approximately fourfold greater abundance in P7 islets (Fig. 3E), suggesting that TET2 may be the dominant TET during pancreas development. Remarkably, expression of TETs and TDG was greatly reduced in P7 compared with P4 islets, e.g., up to 15-fold decrease for TET2 transcripts (Fig. 3E). Even in miR-26a TG mice, down-regulation of TETs/TDG expression in older (P7) islets was seen (Fig. 3E). Importantly, expression of TETs and TDG was reduced, up to threefold for TET2 transcripts, in islets (both P7 and P4) from miR-26a TG mice compared with WT littermates (Fig. 3E), consistent with the results from studies of HEK293T cells (Fig. 2 B–E). Similar to the decreased TETs/TDG expression, global 5hmC levels were reduced in older islets in both WT controls and miR-26a TG mice (Fig. 3F). Both genotypes had similar low levels of 5hmC in P7 islets; however, P4 islets from miR-26a TG mice exhibited considerably less 5hmC than did WT controls (Fig. 3F), suggesting that miR-26a–mediated 5hmC loss correlates with endocrine cell differentiation in the developing pancreas. Gene expression and immunohistochemical analyses further confirmed an enhanced expression of endocrine genes in P7 islets (Fig. 3G) and E18.5 pancreata (Fig. S7A; insulin staining), respectively, in TG compared with WT mice. Cell proliferation, indicated by Ki67 expression, was also enhanced in P7 TG islets (Fig. 3I) and E18.5 beta cells (0.21 ± 0.03% TG vs. 0.09 ± 0.01% WT Ki67+ cells among insulin+ cells; P < 0.05) (Fig. S7A). Finally, transcription factors, indicative of progenitors or endocrine cells, were also enhanced in TG P7 islets (Fig. 3H) and E15.5 pancreas (Fig. S7B; 12 ± 2% TG vs. 5 ± 2% WT Ngn3+ pancreatic cells; P < 0.05). Collectively, these results suggest that miR-26a may affect both expansion and differentiation of the developing endocrine pancreas.
Levels of TETs/TDG and 5hmC Are Reduced During Pancreatic Cell Differentiation in Vitro.
To confirm and further extend our observation that miR-26a/TET may be involved in endocrine cell differentiation, we used a recently reported in vitro differentiation assay (30) and asked whether the miR-26/TET circuit participates in differentiation toward endocrine cells. This recently published colony-forming assay depends on the growth of progenitor cells (enriched in the CD133+Sox9/EGFP+ sorted ductal cells) in semisolid media containing Matrigel into multicellular colonies, which contain mostly ductal-like cells and are organized as a hollow sphere. Based on the morphology under phase-contrast illumination, we refer to these ductal-like colonies as “ring” colonies. When replated in a laminin hydrogel medium (Fig. 4A), these ductal-like ring colonies differentiate to colonies containing mostly endocrine and acinar-like cells (Fig. 4A). These colonies are termed “endocrine/acinar” colonies.
Fig. 4.

Reduced levels of TETs/TDG and 5hmC during pancreatic cell differentiation in vitro. (A) Schematic for generation of ductal-like ring colonies and the more differentiated endocrine/acinar colonies. CD133+Sox9/EGFP+ cells were sorted from pancreata of adult Sox9/EGFP transgenic mice and plated sequentially in two 3D pancreatic colony assays, one containing Matrigel and the other laminin hydrogel. In both of these gels, the cells do not migrate, but grow into colonies containing up to 3,000 cells. (B and C) Expression of miR-26a (B), TETs and TDG (C), determined by qRT-PCR. Designated colonies were handpicked and pooled for analysis. (D) Quantification of 5hmC, as determined by dot blot analysis. (E) Confocal images of immunostaining for 5hmC in whole-mounted individual colonies. (Scale bars, 100 µm.) (F) Confocal images of double immunostaining for insulin and 5hmC in whole-mounted individual endocrine/acinar colonies. (Scale bars, 50 µm.) Data are shown as mean ± SD; *P < 0.05, **P < 0.01, and ***P < 0.001.
We found that endocrine/acinar colonies express 10-fold more miR-26a than ring colonies (Fig. 4B), in agreement with our finding for postnatal islets. However, miR-26b, the other miR-26 family member, was barely detectable in either type of colony, suggesting that miR-26a primarily contributes to this differentiation process (Fig. S8). Consistent with being direct targets of miR-26a, and with our analysis of developing islets, TETs and TDG were significantly down-regulated in endocrine/acinar colonies (Fig. 4C). In particular, expression of endogenous TET2, which was much higher than expression of TET1 and TET3, was reduced by as much as 75% in endocrine/acinar colonies. In line with reduced TETs/TDG expression, dot blot analysis revealed significantly lower 5hmC levels in endocrine/acinar colonies (Fig. 4D). Whole-mount immunostaining analysis also confirmed less 5hmC staining in endocrine/acinar colonies compared with ring colonies (Fig. 4E). As in postnatal islets, there was no apparent difference in 5mC levels between the two colony types, as determined by dot blot analysis (Fig. S9). Furthermore, we found that most insulin-expressing cells within endocrine/acinar colonies exhibited low staining for 5hmC (Fig. 4F), supporting the idea that the decrease in 5hmC accompanies endocrine differentiation from pancreatic progenitor-like cells.
Overexpression of miR-26a Enhances Endocrine Cell Differentiation in Vitro.
To test whether ring colony-forming progenitor cells were affected by a change in miR-26a, individual 3-wk-old ring colonies derived from miR-26a TG and WT littermate mice were handpicked and examined for expression of a panel of lineage markers by microfluidic qRT-PCR analysis (Fig. 5A). Microfluidic quantitative RT-PCR (qRT-PCR) is a relatively new technology that can be used to determine gene expression in as little as one colony in a reaction volume in the nanoliter range (37). We found that the frequency of ring colonies derived from miR-26a TG mice that expressed endocrine markers (insulin 1, insulin 2, glucagon, somatostatin) was about three times higher compared with WT mice (Fig. 5B, arrows). The enhanced endocrine gene expression is accompanied by lowered expression of Ngn3 in TG compared with WT colonies (Fig. 5C), again implicating miR-26a in increasing endocrine cell differentiation.
Fig. 5.
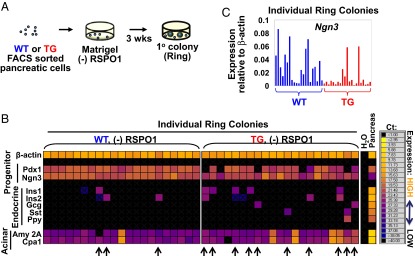
Overexpression of miR-26a enhances differentiation of pancreatic progenitor-like cells in vitro. (A) Schematic of ring colony formation. CD133+ cells were sorted from dissociated pancreata of miR-26a TG or WT mice, and plated in Matrigel-containing colony assay without exogenous R-Spondin1 (RSPO1). Ring colonies formed, consisting of mostly duct-like cells. (B) Heat map of microfluidic qRT-PCR analysis of gene expression in individual ring colonies that were handpicked 3 wk postplating. Each column indicates a signal colony. Amy 2A, amylase 2A; Cpa1, carboxypeptidase A1; Gcg, glucagon; Ins1, insulin 1; Ins2, insulin 2; Ngn3, neurogenin 3; Pdx1, pancreas and duodenal homeobox 1; Ppy, pancreatic polypeptide; Sst, somatostatin. (C) Relative gene expression (ΔCt) compared with β-actin as shown in Fig. 5B. Each column indicates a single colony.
Knockdown of TETs and TDG in WT adult pancreatic CD133+Sox9/EGFP+ cells resulted in a dramatic reduction of ring colony numbers, precluding further analysis (Fig. S10). Overexpression of TET2 in adult pancreatic CD133+Sox9/EGFP+ cells isolated from TG mice reduced the number of colonies expressing endocrine genes (Fig. S11). These results again suggest a role for miR-26a in endocrine cell differentiation.
Discussion
It is now known that genome-wide and locus-specific active DNA demethylation occurs extensively in mammals (38, 39). DNA demethylation is dynamically regulated during development, especially in certain rapid developmental stages including embryonic cell differentiation, germ cell, and zygote development. These observations imply that an accurate and coordinated regulation of TETs and TDG is required to ensure precise DNA demethylation. Here, we report that miR-26a can target and decrease mRNA levels of all members of the TET family, as well as TDG. Importantly, miR-26a–mediated targets in the UTRs of TET/TDG is highly conserved among vertebrates. miR-26a and TETs/TDG are expressed in a wide variety of tissues, so we speculate that the miR-26/TET circuit has a broad, evolutionarily conserved role in development and disease. Indeed, miR-26a has been shown to promote myogenic (40, 41) and neuronal (42) cell differentiation, although the involvement of TET and TDG in these processes awaits further investigation.
Down-regulation of TET1 is known to be required for embryonic stem cell differentiation (2), whereas overexpression of TET1 can mimic Oct4 and help dedifferentiate fibroblasts into pluripotent stem cells (43). These results suggest an important role of TET/TDG-dependent DNA demethylation in maintenance of cell identity. Our current study expands these previous findings and shows that, in addition to embryonic stem cell differentiation, TETs, together with miR-26a, may also play a role in later-stage cell differentiation, such as the endocrine pancreas. Beside TETs and TDG, other known miR-26a targets, including EZH2, PTEN, and pRB (40, 44), may also contribute to the function of miR-26a in cell differentiation.
Mice overexpressing miR-26a are viable and overtly normal, as are TET2 homozygous knockout mice (45). This may suggest that miR-26a and TET2 are only involved in the fine-tuning of embryogenesis and development. Alternatively, it is more likely that the circuits controlling development are complex and compensation takes place during development. This is likely the case for TET knockout mice. The knockout of both TET1 and TET2 causes much perinatal lethality but some overtly normal mice are obtained (46, 47), suggesting compensation by TET3. Therefore, the function of miR-26a/TET in embryogenesis and development, especially for pancreatic cell differentiation in vivo, requires further investigation. For example, it will be important to determine whether miR-26a overexpression affects methylation status of specific promoters of key pancreas transcription factors, such as that of Pax4 and Arx, which are known to play essential roles in specifying endocrine cell types (48). In addition, the results from in vitro differentiation system presented here will need to be verified in vivo in future studies.
In summary, we firmly demonstrate that TETs and TDG can be directly down-regulated by miR-26a in a reporter assay system, and miR-26a expression varies inversely with TETs/TDG expression during in vivo and in vitro differentiation of pancreatic progenitor cells to endocrine cells. Given the broad expression patterns of miR-26a and TETs/TDG, we predict that the miR-26a/TET circuit could be involved in cellular differentiation of many organ systems in addition to pancreas.
Materials and Methods
Mice.
To generate miR-26a transgenic mice, a genomic DNA fragment encoding the miR-26a-1 locus, preceded by the synthetic CAG promoter and a loxP-flanked Neo-STOP cassette, was inserted into the Rosa26 locus (49). Mice were generated by injecting targeted ES cells into blastocysts and maintained in a mixed C57BL/6 and 129 background. Mice carrying the targeted allele were bred with hypoxanthine-guanine phosphoribosyltransferase-Cre mice (50), which caused deletion of the Neo-STOP cassette early during embryogenesis, including the germ line (50). Heterozygous miR-26a transgenic mice and littermate wild-type mice were used for experiments. Sox9/EGFP transgenic reporter mice (CD1 background) (51) were obtained from Mutant Mouse Regional Resource Centers and maintained at City of Hope. All mouse experiments were approved by the City of Hope Institutional Animal Care and Use Committee and followed National Institutes of Health and City of Hope guidelines for the care and use of laboratory animals.
Luciferase Reporter Assays.
The 3′-UTR fragments for TET1, TET2, TET3, and TDG were generated by PCR and cloned into the psiCHECK-2 vector (Promega) downstream from the Renilla luciferase cassette. The predicted miR-26a binding site was mutated using the QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene). HEK293T cells were grown in a 96-well plate and cotransfected with the luciferase reporter vector together with a miRNA precursor or a negative control (20 nM; Ambion) using Attractene (Qiagen) according to the manufacturer’s instructions. Activities of firefly and Renilla luciferase were analyzed using the Dual-Luciferase Reporter Assay System (Promega) 24 h after transfection.
Microfluidic qRT-PCR.
Microfluidic RT-PCR was performed using a BioMark 48.48 Dynamic Array system (Fluidigm). Individual ring colonies were lifted one by one from the methylcellulose medium by using a 10-µL Eppendorf pipette under direct microscopic visualization. Each colony was collected in 10 μL of reaction buffer, and preamplified (14 cycles) according to the manufacturer’s instructions (Fluidigm). Amplified cDNA was loaded onto a 48.48 Dynamic Array using the NanoFlex IFC controller (Fluidigm). Threshold cycle (Ct), as a measurement of fluorescence intensity, was determined with BioMark PCR analysis software (Fluidigm) and expressed as a heat map or relative expression (ΔCt) of the gene of interest to the internal control, β-actin. All reactions were performed along with negative (water) and positive (adult pancreatic cells) controls.
Statistical Analysis.
Data are displayed as mean ± SD. Two-tailed Student t test was used to determine differences. Unless otherwise indicated, the single asterisk (*) denotes P < 0.05, the double asterisk (**) denotes P < 0.01, and the triple asterisk (***) denotes P < 0.001.
Other Methods.
Please see SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank members of the W.H. and H.T.K. Laboratories for helpful discussions, Dr. David Tirrell (California Institute of Technology) for providing laminin hydrogel, Lucy Brown and Alexander Spalla from the Analytical Cytometry Core (City of Hope) for assistance in cell sorting, Dr. Maike Sander (University of California, San Diego) for providing anti-Ngn3 antibody, Dr. Qiang Lu (City of Hope) for providing TET2-overexpressing plasmid, and Dr. Keely Walker (City of Hope) for editing the manuscript. This work is supported in part by a pilot grant (to W.H.) from National Institutes of Health Grant P30CA033572, as well as by National Institute of Diabetes and Digestive and Kidney Diseases Grants R01DK081587 (to H.T.K.) and U01DK098533 (to A.D.R.), and American Cancer Society Grant RSG-11-132-01-CCE (to W.H.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1317397110/-/DCSupplemental.
References
- 1.Tahiliani M, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324(5929):930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ito S, et al. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466(7310):1129–1133. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cortellino S, et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell. 2011;146(1):67–79. doi: 10.1016/j.cell.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ito S, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333(6047):1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.He YF, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333(6047):1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maiti A, Drohat AC. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: Potential implications for active demethylation of CpG sites. J Biol Chem. 2011;286(41):35334–35338. doi: 10.1074/jbc.C111.284620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koh KP, et al. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell. 2011;8(2):200–213. doi: 10.1016/j.stem.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hackett JA, et al. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science. 2013;339(6118):448–452. doi: 10.1126/science.1229277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vincent JJ, et al. Stage-specific roles for tet1 and tet2 in DNA demethylation in primordial germ cells. Cell Stem Cell. 2013;12(4):470–478. doi: 10.1016/j.stem.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen ZX, Riggs AD. DNA methylation and demethylation in mammals. J Biol Chem. 2011;286(21):18347–18353. doi: 10.1074/jbc.R110.205286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Branco MR, Ficz G, Reik W. Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat Rev Genet. 2012;13(1):7–13. doi: 10.1038/nrg3080. [DOI] [PubMed] [Google Scholar]
- 12.Gu TP, et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature. 2011;477(7366):606–610. doi: 10.1038/nature10443. [DOI] [PubMed] [Google Scholar]
- 13.Cimmino L, Abdel-Wahab O, Levine RL, Aifantis I. TET family proteins and their role in stem cell differentiation and transformation. Cell Stem Cell. 2011;9(3):193–204. doi: 10.1016/j.stem.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tan L, Shi YG. Tet family proteins and 5-hydroxymethylcytosine in development and disease. Development. 2012;139(11):1895–1902. doi: 10.1242/dev.070771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu H, Zhang Y. Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes Dev. 2011;25(23):2436–2452. doi: 10.1101/gad.179184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang RR, et al. Tet1 regulates adult hippocampal neurogenesis and cognition. Cell Stem Cell. 2013;13(2):237–245. doi: 10.1016/j.stem.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ivey KN, Srivastava D. MicroRNAs as regulators of differentiation and cell fate decisions. Cell Stem Cell. 2010;7(1):36–41. doi: 10.1016/j.stem.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 18.Pan FC, Wright C. Pancreas organogenesis: From bud to plexus to gland. Dev Dyn. 2011;240(3):530–565. doi: 10.1002/dvdy.22584. [DOI] [PubMed] [Google Scholar]
- 19.Jonsson J, Carlsson L, Edlund T, Edlund H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature. 1994;371(6498):606–609. doi: 10.1038/371606a0. [DOI] [PubMed] [Google Scholar]
- 20.Offield MF, et al. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development. 1996;122(3):983–995. doi: 10.1242/dev.122.3.983. [DOI] [PubMed] [Google Scholar]
- 21.Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129(10):2447–2457. doi: 10.1242/dev.129.10.2447. [DOI] [PubMed] [Google Scholar]
- 22.Zhou Q, et al. A multipotent progenitor domain guides pancreatic organogenesis. Dev Cell. 2007;13(1):103–114. doi: 10.1016/j.devcel.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 23.Gradwohl G, Dierich A, LeMeur M, Guillemot F. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci USA. 2000;97(4):1607–1611. doi: 10.1073/pnas.97.4.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fu X, Adamski M, Thompson EM. Altered miRNA repertoire in the simplified chordate, Oikopleura dioica. Mol Biol Evol. 2008;25(6):1067–1080. doi: 10.1093/molbev/msn060. [DOI] [PubMed] [Google Scholar]
- 25.Slack JM. Developmental biology of the pancreas. Development. 1995;121(6):1569–1580. doi: 10.1242/dev.121.6.1569. [DOI] [PubMed] [Google Scholar]
- 26.Inada A, et al. Carbonic anhydrase II-positive pancreatic cells are progenitors for both endocrine and exocrine pancreas after birth. Proc Natl Acad Sci USA. 2008;105(50):19915–19919. doi: 10.1073/pnas.0805803105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu X, et al. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell. 2008;132(2):197–207. doi: 10.1016/j.cell.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 28.Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429(6987):41–46. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- 29.Solar M, et al. Pancreatic exocrine duct cells give rise to insulin-producing beta cells during embryogenesis but not after birth. Dev Cell. 2009;17(6):849–860. doi: 10.1016/j.devcel.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 30.Jin L, et al. Colony-forming cells in the adult mouse pancreas are expandable in Matrigel and form endocrine/acinar colonies in laminin hydrogel. Proc Natl Acad Sci USA. 2013;110(10):3907–3912. doi: 10.1073/pnas.1301889110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bartel DP. MicroRNAs: Target recognition and regulatory functions. Cell. 2009;136(2):215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 33.Grimson A, et al. MicroRNA targeting specificity in mammals: Determinants beyond seed pairing. Mol Cell. 2007;27(1):91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saetrom P, et al. Distance constraints between microRNA target sites dictate efficacy and cooperativity. Nucleic Acids Res. 2007;35(7):2333–2342. doi: 10.1093/nar/gkm133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blum B, et al. Functional beta-cell maturation is marked by an increased glucose threshold and by expression of urocortin 3. Nat Biotechnol. 2012;30(3):261–264. doi: 10.1038/nbt.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Teta M, Rankin MM, Long SY, Stein GM, Kushner JA. Growth and regeneration of adult beta cells does not involve specialized progenitors. Dev Cell. 2007;12(5):817–826. doi: 10.1016/j.devcel.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 37.Sanchez-Freire V, Ebert AD, Kalisky T, Quake SR, Wu JC. Microfluidic single-cell real-time PCR for comparative analysis of gene expression patterns. Nat Protoc. 2012;7(5):829–838. doi: 10.1038/nprot.2012.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu SC, Zhang Y. Active DNA demethylation: Many roads lead to Rome. Nat Rev Mol Cell Biol. 2010;11(9):607–620. doi: 10.1038/nrm2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shen L, et al. Genome-wide analysis reveals TET- and TDG-dependent 5-methylcytosine oxidation dynamics. Cell. 2013;153(3):692–706. doi: 10.1016/j.cell.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wong CF, Tellam RL. MicroRNA-26a targets the histone methyltransferase Enhancer of Zeste homolog 2 during myogenesis. J Biol Chem. 2008;283(15):9836–9843. doi: 10.1074/jbc.M709614200. [DOI] [PubMed] [Google Scholar]
- 41.Dey BK, Gagan J, Yan Z, Dutta A. miR-26a is required for skeletal muscle differentiation and regeneration in mice. Genes Dev. 2012;26(19):2180–2191. doi: 10.1101/gad.198085.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dill H, Linder B, Fehr A, Fischer U. Intronic miR-26b controls neuronal differentiation by repressing its host transcript, ctdsp2. Genes Dev. 2012;26(1):25–30. doi: 10.1101/gad.177774.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao Y, et al. Replacement of Oct4 by Tet1 during iPSC induction reveals an important role of DNA methylation and hydroxymethylation in reprogramming. Cell Stem Cell. 2013;12(4):453–469. doi: 10.1016/j.stem.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 44.Kim H, et al. Integrative genome analysis reveals an oncomir/oncogene cluster regulating glioblastoma survivorship. Proc Natl Acad Sci USA. 2010;107(5):2183–2188. doi: 10.1073/pnas.0909896107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Z, et al. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood. 2011;118(17):4509–4518. doi: 10.1182/blood-2010-12-325241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dawlaty MM, et al. Tet1 is dispensable for maintaining pluripotency and its loss is compatible with embryonic and postnatal development. Cell Stem Cell. 2011;9(2):166–175. doi: 10.1016/j.stem.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dawlaty MM, et al. Combined deficiency of Tet1 and Tet2 causes epigenetic abnormalities but is compatible with postnatal development. Dev Cell. 2013;24(3):310–323. doi: 10.1016/j.devcel.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dhawan S, Georgia S, Tschen SI, Fan G, Bhushan A. Pancreatic β cell identity is maintained by DNA methylation-mediated repression of Arx. Dev Cell. 2011;20(4):419–429. doi: 10.1016/j.devcel.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thai TH, et al. Regulation of the germinal center response by microRNA-155. Science. 2007;316(5824):604–608. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- 50.Tang SH, Silva FJ, Tsark WM, Mann JR. A Cre/loxP-deleter transgenic line in mouse strain 129S1/SvImJ. Genesis. 2002;32(3):199–202. doi: 10.1002/gene.10030. [DOI] [PubMed] [Google Scholar]
- 51.Gong S, et al. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425(6961):917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.