

Significance
Magnesium-bearing carbonate minerals play critical roles in the health and function of the Earth system because they constitute a significant fraction of lithosphere carbon reservoir and build skeletal structures for the majority of marine invertebrate organisms. Despite wide occurrence, high-Mg and sole-Mg phases such as dolomite ([Ca,Mg]CO3) and magnesite (MgCO3) prove virtually impossible to be crystallized under ambient conditions. It has long been believed that Mg2+ hydration is the cause for such a geological mystery. Here, we probe this hypothesis by investigating Ca–Mg–CO3 precipitation in the absence of water and find direct proof suggesting the existence of a more intrinsic crystallization barrier. These findings provide a perspective augmenting our understanding in carbonate mineralogy, biomineralization, and mineral-carbonation processes.
Keywords: sedimentary geology, carbon sequestration, nonaqueous solvent
Abstract
Dolomite and magnesite are simple anhydrous calcium and/or magnesium carbonate minerals occurring mostly at Earth surfaces. However, laboratory synthesis of neither species at ambient temperature and pressure conditions has been proven practically possible, and the lack of success was assumed to be related to the strong solvation shells of magnesium ions in aqueous media. Here, we report the synthesis of MgCO3 and MgxCa(1−x)CO3 (0 < x < 1) solid phases at ambient conditions in the absence of water. Experiments were carried out in dry organic solvent, and the results showed that, although anhydrous phases were readily precipitated in the water-free environment, the precipitates’ crystallinity was highly dependent on the Mg molar percentage content in the solution. In specific, magnesian calcite dominated in low [Mg2+]/[Ca2+] solutions but gave way to exclusive formation of amorphous MgxCa(1−x)CO3 and MgCO3 in high-[Mg2+]/[Ca2+] and pure-Mg solutions. At conditions of [Mg2+]/[Ca2+] = 1, both nanocrystals of Ca-rich protodolomite and amorphous phase of Mg-rich MgxCa(1−x)CO3 were formed. These findings exposed a previously unrecognized intrinsic barrier for Mg2+ and CO32− to develop long-range orders at ambient conditions and suggested that the long-held belief of cation-hydration inhibition on dolomite and magnesite mineralization needed to be reevaluated. Our study provides significant insight into the long-standing “dolomite problem” in geochemistry and mineralogy and may promote a better understanding of the fundamental chemistry in biomineralization and mineral-carbonation processes.
Rarely is there a geological challenge that has endured as long a search for answers as the “dolomite problem” has. Identified more than 220 y ago by the French mineralogist Déodat de Dolomieu, the calcium magnesium carbonate mineral known as dolomite [CaMg(CO3)2] has since been synthesized repeatedly but exclusively at high-temperature and, in some instances, high-pressure conditions (1–4). However, in many cases other than igneous-originated carbonatite and deep-burial dolomitization, the formation of dolomitic phases (e.g., microbial and cave deposits) are certainly not associated with such high-temperature and/or -pressure environments (5–8). This obvious discrepancy, compounded by the sharp contrast of massive ancient dolomite formations to the scarcity of modern observations, prompted an arduous search for answers for the past two centuries and, because of a lack of success, has been referred to as the dolomite problem (6, 9–16). Our inability to form dolomite at ambient conditions is hardly the only challenge in mineralogy; a similar situation exists in the case of pure magnesium carbonate [magnesite (MgCO3)], which has posed the same level of difficulty to nucleate and grow at room temperature and atmospheric pressure in laboratories but frequently occurs in natural sedimentary settings at earth (sub)surfaces. Interestingly, both dolomite and magnesite belong to the mineral class of anhydrous carbonates. Thus, it becomes logical to surmise that the dolomite problem and the magnesite problem probably share the same root, that is, the difficulty to incorporate unhydrated magnesium ions into carbonate from aqueous solutions without appealing to temperature and/or pressure changes. Given the 50% abundance of dolomites in the world’s carbonate reservoirs (6, 17) and the potential of magnesium carbonate for carbon sequestration (18, 19), as well as the critical role of Mg in carbonate biomineralization (20–22), it is safe to assume that scientific interests in the Ca–Mg–CO3 system will remain for years to come.
Aqueous-phase reactions between Mg2+ cations and CO32− anions at ambient temperature and pressure conditions yield exclusively hydrated [e.g., barringtonite MgCO3⋅2H2O; nesquehonite MgCO3⋅3H2O; and lansfordite, MgCO3⋅5H2O] or basic forms [e.g., hydromagnesite, Mg5(CO3)4(OH)2⋅4H2O; dypingite, Mg5(CO3)4(OH)2⋅5H2O; and artinite, Mg2(CO3)(OH)2⋅3H2O] of Mg-carbonate (23, 24). Nesquehonite is the most commonly formed phase under low temperature and pressure [e.g., 25 °C and a pCO2 of ∼10−2 atm (25–27)], whereas hydromagnesite and other basic forms become dominant in an intermediate temperature range [e.g., 40–60 °C (28–30)]. Despite being the most stable phase at all conditions, the anhydrous salt, magnesite, was reported to form only at a much elevated temperature and pressure [e.g., ∼90–180 °C and pCO2 > 50 atm (31–34)].
The difficulty in precipitating magnesite (and dolomite) from ambient aqueous solutions was first suggested to be related to the strong hydration status of Mg2+ ions in a hypothesis (35) that has since been corroborated by numerous pieces of circumstantial evidence (15, 22, 36). Indeed, both experimental and computational studies revealed the presence of a stable inner-sphere hydration shell around Mg2+ ions that contains six water molecules in an octahedral arrangement and has a water exchange rate five orders of magnitude slower than that for Ca2+ (37–39). An additional outer shell composed of up to 12 water molecules also exists through hydrogen bonding to the inner shell. The presence of tight hydration shells around Mg2+ is consistent with the results of molecular-orbital calculations that show the solvation free energy (per water molecule) for Mg2+ ions is as much as 60% higher than that of Ca2+ in both the inner and outer shells (40, 41). The strong Mg2+–water association is rationalized by the high charge density of Mg2+ ions resulting from their small ionic radius. In addition, the net charge on the central magnesium ion of a stabilized [Mg(H2O)6]2+ complex was found to be only ∼1.18 (42), indicating the occurrence of significant charge transfer between Mg2+ and the surrounding water molecules and a more covalent nature of the Mg–O bonding within the hydration shells. Such finding is further validated by recent experimental observations that collision between [Mg(H2O)n]2+ clusters can generate solvated magnesium hydroxide complexes MgOH+(H2O)n or monocations Mg+(H2O)n by inducing electron transfer from the associated H2O molecules to the Mg2+ cations (39, 43–45).
Despite the large body of literature documenting the assumed inhibitory role of cation hydration, little work has been found to directly test the solvation effect. Thus, the current understanding of the part that water plays in obstructing magnesite (and dolomite) formation leads to an intriguing and yet fundamental question: will anhydrous Mg carbonate salts precipitate readily if the cation solvation is nonaqueous and weaker than hydration shells? To the best of our knowledge, no data are available in the literature concerning the crystallization of magnesite (or dolomite) in nonaqueous solutions. In the present study, we explored the possibility of magnesite (MgCO3) and dolomite [CaMg(CO3)2] formation under ambient conditions in dry formamide [an organic solvent (O=CH–NH2)]. We chose formamide because it has a high dielectric constant (ε = 109.8) and resembles water in many physical properties, such as density, dipole moment, and surface tension. The strong polarity of formamide allows us to prepare high-concentration solutions containing Mg2+ (and Ca2+) cations or CO32− anions. In addition, the hydrogen bonding between formamide molecules is more poorly developed than that between water molecules because of the stronger steric effect (46), indicative of a much weaker solvation around the solute ions.
Results and Discussion
We began our experiments by first making stock solutions of 0.1 M MgCl2 (stock 1), 0.1 M (Ca,Mg)Cl2 (stock 2), and 0.1 M Cs2CO3 (stock 3) in formamide. Stock 2 was made such that [Mg2+] + [Ca2+] = 0.1 M, whereas the Mg/Ca (molar) ratio varied from 1:5 to 5:1. We then carried out the synthesis by mixing or titrating stock 3 into stock 1 or stock 2 at room temperature in an open (to air) setting (Methods). Consistent with the prediction of cation-hydration hypothesis, precipitation occurred either immediately upon the mixing or gradually during the titration. X-ray diffraction (XRD) analyses of the isolated solid phases (Fig. 1), however, showed that the precipitates’ crystallinity was highly dependent of the Mg molar percentage content in the solution. In the pure-Mg system (combination of stocks 1 and 3) (Fig. 1A), exclusively amorphous material was formed. Infrared (IR) spectroscopy analyses of these amorphous phases (Fig. 2) confirmed that they were composed of disordered carbonate with characteristic peaks in the 870, 1,080, and 1,430 cm−1 regions and were anhydrous in nature (no water absorption near 3,400 cm−1). In the high-Mg, mixed-cation experiments (the combination of stocks 2 and 3), the lack of crystallinity remained as the precipitates from solutions with a Mg/Ca ratio of 5:1 and 2:1 showed no diffraction peaks in their XRD spectra (Fig. 1 B and C). We note that the amorphous nature of the precipitates from high-Mg systems was not altered by changing the mixing rate of the stock solutions (e.g., by titrating stock 3 into stock 1 or 2 over a period of ∼4 h or mixing the two within 1 min). Continuous decline in the solution Mg content finally led to the development of crystalline phases in the precipitates. For example, at a Mg/Ca ratio of 1:1, disordered protodolomite of moderate crystallinity was observed (Fig. 1D); at a Mg/Ca ratio of 1:2 and 1:5, magnesian calcite of good crystallinity was formed (Fig. 1 E and F). Using a previously established method (47), we determined the Mg contents in the crystalline phases to be ∼37 mol % (1:1), ∼23–25 mol % (1:2), and 9–10 mol % (1:5), respectively. Inductively coupled plasma–optical emission spectroscopy (ICP-OES) analyses of the dissolved precipitates in 1% HNO3 solution (Table 1) fully corroborated the XRD results, showing that Mg accounts for 71, 60, 40, 26, and ∼15 mol % of the total cations in the 5:1, 2:1, 1:1, 1:2, and 1:5 samples, respectively. It is noteworthy that the amorphous solids formed in the high-Mg systems (e.g., 2:1, 5:1, and pure-Mg) were stable as no discernible XRD patterns were observed in these samples ∼120 d after the synthesis.
Fig. 1.

XRD analyses of the precipitated solids in formamide solutions at 22 °C, atmospheric conditions. The corresponding peak positions for calcite (green) and dolomite (red) are indicated.
Fig. 2.

IR spectroscopy analysis of the amorphous solids formed in pure-magnesium and high-[Mg2+]/[Ca2+] solutions.
Table 1.
Analysis of chemical compositions for the precipitated solids using ICP-OES
| Nominal Ca/Mg ratios | [Ca2+] molar percentage in the precipitates | [Mg2+] molar percentage in the precipitates | [Cs+] molar percentage in the precipitates |
| 5:1 | 81.0 | 15.4 | 3.6 |
| 2:1 | 69.5 | 25.7 | 4.8 |
| 1:1 | 56.0 | 39.7 | 4.3 |
| 1:2 | 34.6 | 60.0 | 5.4 |
| 1:5 | 23.9 | 71.3 | 4.8 |
The evolving crystallinity and composition in the precipitates from different solutions were further verified by imaging and selected-area electron diffraction (SAED) analyses using scanning-electron microscopy (SEM) and high-resolution transmission electron microscopy (HR-TEM) coupled with energy-dispersive spectroscopy (EDS). The SEM and TEM micrographs indicated that the solids formed in pure-Mg solutions (Figs. 3A and 4A1) and high Mg/Ca (5:1, 2:1; Figs. 3B and 4B1) were composed of nano-sized aggregates with no specific crystal habit, whereas those in low Mg/Ca conditions (1:5, 1:2; Fig. 3D and 4D1) appeared as (sub)micron-sized crystals, in agreement with the XRD analyses. Imaging analyses also identified a morphological change in the crystal shape with increasing [Mg2+] in the solution [e.g., from well-developed euhedral in the 1:5 system (Fig. 3D) to that elongated in the c-direction in the 1:2 system (Fig. 4D1, arrow)], consistent with a previous atomic force microscopy observation (48) of corner rounding at the +/− intersection sites on calcite rhombohedra attributable to the Mg2+ effect. The SAED and EDS data collected at various locations within each sample provided additional information concerning the compositional homogeneity and Mg/Ca ordering of the obtained solids. Specifically, the precipitates in high-Mg/Ca and pure-Mg solutions exhibited highly homogeneous amorphicity with predominant Mg contents (95 mol % for pure Mg and 73 mol % for 5:1 Mg/Ca) (Fig. 4 A1 and B1, and insets). In contrast, the 1:1 samples displayed inhomogeneity by comprising both amorphous and crystalline phases (Fig. 4C). It was confirmed that the crystalline phase in the 1:1 samples consist of nanoscale protodolomite crystals (Fig. 4C2) with randomly distributed Mg2+ and Ca2+ in the lattice. Intriguingly, there seemed to be a threshold level of Mg (in molar percentage) that determines the development of crystallinity in these solids (Fig. 4C1 and insets) because the polycrystalline phase contained consistently lower molar percentages (∼38 mol %) of MgCO3 than their amorphous counterparts (∼47 mol %) formed in the same 1:1 solution. The MgCO3 content in the rhombohedral to oval-shaped crystals (Fig. 4D1) formed in the low-Mg systems (i.e., 1:2) was estimated to be ∼30 mol % by EDS (Fig. 4D2 and inset), in line with the XRD (23–25%) and ICP-OES (26%) results.
Fig. 3.

SEM images of the precipitated solids in formamide solutions. (A) Pure magnesium. (B) [Mg2+]/[Ca2+] = 5:1. (C) [Mg2+]/[Ca2+] = 1:1. (D) [Mg2+]/[Ca2+] = 1:5. (Scale bars: 200 nm.) Note the euhedral crystal shape in the 1:5 samples and the lack of morphological patterns in the 5:1 and pure-Mg precipitates.
Fig. 4.

TEM and EDS analyses of the precipitated solids. (A, 1) Micrograph of the precipitates in the pure-Mg system. The SAED (Lower Left) and EDS (Upper Right) analyses indicated that these solids were amorphous and composed of ∼94 mol % MgCO3. (B, 1 and B, 2) Micrograph displaying fine-grained (B, 1) and amorphous (B, 2) structure of the precipitates in the 5:1 system. EDS spectrum (B, 1, Inset) confirmed the predominant occurrence of MgCO3 (73 mol %) with minor components in Ca, Na, Cl, and Cs. SAED pattern (B, 2, Inset) revealed the homogeneity of the amorphous nature. (C, 1) Amorphous and oval-shaped-crystalline phases formed in the 1:1 solution. EDS analyses identified 47 mol % of MgCO3 in the amorphous phase (upper spectrum for the dashed circle) and 37 mol % in the crystals (lower spectrum for the solid circle). Trace amounts of Cs, Na, and Cl were also detected. (C, 2) Close examination and SAED analyses of an oval-shaped unit revealing its polycrystalline nature. Note the characteristic diffraction arcs for Ca-rich protodolomite and the lack of (003) superlattice reflections, suggesting the random distribution of Ca2+ and Mg2+ in these crystals. (C, 3) High-resolution images of the oval-shaped crystals with a fast Fourier transform pattern for the [010] zone axis showing the lattice fringe of Ca-rich protodolomite. (D, 1) Round-cornered rhombohedral crystals containing ∼32 mol % of MgCO3 (estimated from EDS spectrum Inset) collected in the 1:2 solution. (D, 2) EDS spectrum for a selected area of the 1:2 sample.
Both EDS (Fig. 4) and ICP-OES (Table 1) detected the presence of small quantities (3–5 mol %) of Cs in the precipitates, more in amorphous phases than in crystals. The higher level of Cs associated with lower crystallinity, along with the coupled appearance of Cs and Cl in the EDS spectra (Fig. 4, Inset), indicated that a significant portion of the Cs was surface-bound such as trapped residual solute or sorbed species.
These findings are of significance for a number of reasons. Firstly, the lack of crystallinity in the precipitates harvested in magnesium-only and high-Mg/Ca solutions suggests that long-range orders in the MgxCa(1−x)CO3 solid cannot be readily achieved at high magnesium percentage content (x > 0.38 in our experiments), exposing a previously unrecognized intrinsic difficulty for Mg2+ and CO32− to attain the R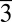 2/c (in magnesite) or R
2/c (in magnesite) or R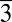 (in dolomite) arrangements at ambient conditions. It is noteworthy that such difficulty is unlikely caused by the reaction kinetics of Mg2+ and CO32− because varied mixing rates (i.e., slow titration versus quick mixing) showed little effect on the precipitate’s morphology and crystallinity. Secondly, in light of the widely reported crystallization of magnesium carbonate hydrates such as nesquehonite (MgCO3⋅3H2O) (monoclinic, P21/n) from aqueous environments at ambient conditions, the observed formation of amorphous MgCO3 anhydrates in the present study implies that water facilitates the nucleation process by direct incorporation into the lattice. Thirdly, comparing the partitioning behavior of Mg in this study (in formamide) with those in previous work (in water) (Fig. 5), we found that far higher molar percentage of Mg was incorporated into the crystalline solids (calcite) formed in nonaqueous environments. For example, the distribution coefficient for Mg, DMg = (Mg/Ca)solid/(Mg/Ca)solution, was estimated to be ∼0.6–0.8 in this study, about one order of magnitude higher than that in water at similar conditions (0.02–0.09, depending on the specific experiments) (49–52). This observation is consistent with the hypothesized role of water in limiting Mg-rich carbonate formation and likely constitutes direct evidence that quantifies the hindrance of cation hydration on Mg2+ incorporation into calcite lattice. Lastly, our experimental approach provided a synthetic route to produce anhydrous amorphous magnesium carbonate at room temperature and atmospheric pCO2, which may help to extend the current effort in understanding amorphous calcium carbonate (20, 53–55) to amorphous magnesium carbonate (AMC), both of which provide a low-energy pathway for carbonate mineralization in biological (organic-rich) environments. To the best of our knowledge, no literature report is available to date documenting the formation of anhydrous AMC, although the synthesis of hydrous forms, Ca(1−x)MgxCO3⋅nH2O (0 ≤ x ≤ 1), was achieved at elevated temperature (19).
(in dolomite) arrangements at ambient conditions. It is noteworthy that such difficulty is unlikely caused by the reaction kinetics of Mg2+ and CO32− because varied mixing rates (i.e., slow titration versus quick mixing) showed little effect on the precipitate’s morphology and crystallinity. Secondly, in light of the widely reported crystallization of magnesium carbonate hydrates such as nesquehonite (MgCO3⋅3H2O) (monoclinic, P21/n) from aqueous environments at ambient conditions, the observed formation of amorphous MgCO3 anhydrates in the present study implies that water facilitates the nucleation process by direct incorporation into the lattice. Thirdly, comparing the partitioning behavior of Mg in this study (in formamide) with those in previous work (in water) (Fig. 5), we found that far higher molar percentage of Mg was incorporated into the crystalline solids (calcite) formed in nonaqueous environments. For example, the distribution coefficient for Mg, DMg = (Mg/Ca)solid/(Mg/Ca)solution, was estimated to be ∼0.6–0.8 in this study, about one order of magnitude higher than that in water at similar conditions (0.02–0.09, depending on the specific experiments) (49–52). This observation is consistent with the hypothesized role of water in limiting Mg-rich carbonate formation and likely constitutes direct evidence that quantifies the hindrance of cation hydration on Mg2+ incorporation into calcite lattice. Lastly, our experimental approach provided a synthetic route to produce anhydrous amorphous magnesium carbonate at room temperature and atmospheric pCO2, which may help to extend the current effort in understanding amorphous calcium carbonate (20, 53–55) to amorphous magnesium carbonate (AMC), both of which provide a low-energy pathway for carbonate mineralization in biological (organic-rich) environments. To the best of our knowledge, no literature report is available to date documenting the formation of anhydrous AMC, although the synthesis of hydrous forms, Ca(1−x)MgxCO3⋅nH2O (0 ≤ x ≤ 1), was achieved at elevated temperature (19).
Fig. 5.

Comparison of the Mg-distribution coefficients in the crystalline/amorphous Mg–Ca–CO3 phases formed in formamide (this study) and in water (previous studies). The results for this study are based on ICP-OES (solid triangle) and XRD (void triangle) analyses, respectively. The results for previous studies are average values (with error bars) of those from Mucci and Morse (49), Mucci (50), Hartley and Mucci, (51), and Huang and Fairchild (52).
We ascribe the major difficulty in anhydrous MgCO3 mineralization at ambient conditions to the reduced freedom of the CO3 groups in magnesite that, consequently, incurs a high-energy (chiefly from entropy decrease) penalty for the crystallization process. Previous studies on the crystal structure of carbonate minerals revealed that Mg and O form much more compact octahedra in magnesite than Ca and O do in calcite. As a result, the CO3 groups in magnesite have far less freedom of motion than those in calcite, although the C–O interatomic distance is nearly uniform in the two minerals because of the stronger interactions between the Mg–O octahedra (56–61). For example, the CO3 group within the calcite structure can undergo translational, librational (i.e., rotational), or screw motions depending on the energy level of the environment (59), whereas only translational motion is allowed for the CO3 group in magnesite (57). The enhanced lattice restriction in magnesite is also confirmed by the outcome of structural refinement for calcite and magnesite formed under different temperature/pressure conditions (57, 62–67). Specifically, the C–O and O–O interatomic distances in the magnesite-CO3 groups maintain a much higher stability when external conditions change, resulting in a lower overall distortion in the groups at higher temperature/pressure, presumably due to the stronger confinement by the highly compact and rigid Mg–O octahedra. Some researchers (67, 68) even suggested that the CO3-structural change is mainly responsive to the expansion/compaction of the Mg–O octahedra at the elevated temperature/pressure.
Building on these lines of previous understanding, we deduce that the specific configuration of Mg and CO3 in magnesite, where the CO3 groups are precisely and securely interlocked between the Mg–O octahedra, may be rather difficult to reach at ambient conditions. Unless the Mg–O octahedra are efficiently compressed so that the CO3 group can have more freedom in motion (Fig. 6), the energy barrier for forming an ordered Mg–CO3 arrangement may be too high to be overcome at low-temperature/pressure conditions because of the entropic loss associated with the atoms’ strict spatial distribution. Therefore, we attribute the main force inhibiting Mg–CO3 crystallization in water-free environments to the thermodynamic hindrance resulting from the entropy decrease associated with intensified restrictions on the CO3 groups in magnesite.
Fig. 6.

(Left) Ambient-pressure structure of magnesite. The arrows indicate the direction in which the MgO6 octahedra may rotate upon compression. (Right) High-pressure structure of magnesite. The MgO6 octahedra have rotated about c axis and are approaching alignment with one another, allowing for a lengthening of the C–O bond. This results in a lengthening of the C–O bond and a noticeable enlargement of the carbonate group relative to the MgO6 octahedra. [Modified from Santillan et al. (67).]
Other than the high-energy barrier for establishing long-range order in anhydrous MgCO3 salt, the amorphism of the MgxCa(1−x)CO3 phases (x > 0.37) in our high-Mg systems may also result from the internal lattice strains and stresses induced by the significant mismatches of size and charge density between Mg2+ and Ca2+. Both experimental and computational investigations in the literature provided evidence indicating that calcite crystals with low concentrations of Mg are stable in structure (19, 69–71); these studies, however, predicted that high concentrations of Mg (≥ ∼50 atom %) may prevent crystal formation because of the increasing structural stiffness and distortion resulting from the random Mg substitution of Ca (72, 73). Recent studies (73) using computational methods reported that substituting Ca by Mg in calcite crystals can alter the cation–C and the cation–cation interatomic distances significantly and cause local tilt of the CO32− groups while maintaining the C–O (within the CO32−) and the cation–O bond lengths constant. Moreover, literature data indicated that pyramidal distortion of the CO3 group needs to occur in magnesite to satisfy the R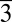 c space-group symmetry (74, 75). Altogether, these structural distortions at high lattice-Mg content will stiffen the crystal structure by reducing the equilibrium volume and raising the bulk modulus. Our experimental observations that no crystalline phase with a Mg molar percentage greater than ∼37% can precipitate directly from high-Mg solutions is largely in line with these previous lines of understanding.
c space-group symmetry (74, 75). Altogether, these structural distortions at high lattice-Mg content will stiffen the crystal structure by reducing the equilibrium volume and raising the bulk modulus. Our experimental observations that no crystalline phase with a Mg molar percentage greater than ∼37% can precipitate directly from high-Mg solutions is largely in line with these previous lines of understanding.
Conclusion
In summary, we have approached the long-standing geochemical question of why anhydrous high-Mg carbonate minerals (i.e., magnesite and dolomite) cannot crystallize at ambient conditions by exploring the formation of MgCO3 and MgxCa(1−x)CO3 in nonaqueous solutions. Data collected from our experiments suggest that a fundamental barrier, other than cation hydration, exists that prevents Mg2+ and CO32− ions from forming long-range ordered structures. We propose that this barrier mainly stems from the lattice limitation on the spatial configuration of CO3 groups in magnesite crystals. On the other hand, the measured higher distribution coefficients of Mg between magnesian calcites formed in the absence and presence of water give us direct proof to support and quantify the cation-hydration effect. These findings may expand our current understanding of the Mg(–Ca)–CO3 system and provide important insight into dolomite/magnesite formation, as well as the processes involved in biomineralization and mineral carbonation.
Methods
All chemicals and solvents used in the synthesis experiments were of American Chemical Society or higher grade and purchased from Sigma-Aldrich. Before use, the glassware was thoroughly cleaned and dried, and both the inorganic chemicals (including MgCl2, CaCl2, and CsCO3) and the glassware were maintained at 60 °C in an oven. Stock solutions containing 100 mM Mg2+ (stock 1), Mg2+/Ca2+ mixture (stock 2), or CO32− (stock 3) were prepared, respectively, by dissolving certain amounts of MgCl2, MgCl2, and CaCl2, or Cs2CO3 in 50 mL of formamide (99.5% purity) with the assistance of stirring bars. Cs2CO3 was chosen for the experiment mainly because of its higher solubility in formamide (than Na2CO3 or K2CO3), which allowed us to prepare uniform and clear solutions. For stock 2, the total concentration of MgCl2 and CaCl2 was fixed, whereas varied Mg-to-Ca concentration ratios ([Mg]/[Ca]) were created (5:1, 2:1, 1:1, 1:2, and 1:5). In the synthesis experiments, stock 3 was either slowly titrated or fast-poured into stock 1/2 to produce precipitates. Once the precipitates settled (which was rather slow and took ∼5–6 d), the supernatant was removed using glass pipettes. The remaining solids were then washed in fresh formamide three times to remove the residual Cs+ and Cl− ions in the solution, following which the washed samples were dried in a desiccator placed in the fume hood. All of the experiments were conducted in triplicates at room temperature (22 ± 1 °C) and atmospheric conditions.
The structure and elemental composition of the precipitates were characterized by XRD, IR spectroscopy, SEM analyses, and high-resolution transmission electron microscopy (HR-TEM) coupled with an EDS, respectively. XRD analyses were carried out using a Rigaku Rapid II XRD system (Mo Kα radiation). The powder samples were contained in thin-wall glass capillaries, which do not introduce background on XRD patterns. Diffraction data were collected on a 2D image-plate detector. The 2D images were then integrated to produce conventional 2θ versus intensity patterns using Rigaku 2DP software. We note that most of the characteristic peaks for calcite and dolomite can be covered within a 2θ of 35° (∼1.18 Å) using Mo Kα radiation, which corresponds to a 2θ of ∼80° (∼1.2 Å) if the system used Cu Kα as radiation source (76). SEM samples were prepared by dispersing powders on carbon tapes and then lightly carbon-coated (50- to 100-Å coating). SEM observations were performed using a high-resolution field emission LEO 1530 SEM. TEM and SAED analyses were carried out using a Philips CM 200UT microscope (with a spherical aberration coefficient of 0.5 mm and a point-to-point resolution of 0.19 nm). TEM-based EDS analyzer (NORAN Voyager) was also used to measure the content ratio of MgCO3 to CaCO3 in the thin specimen of each sample, and the method has been described in detail elsewhere (47). Cliff–Lorimer k factors for Mg and Ca were obtained using a well-characterized dolomite standard from Delight (49.43 mol % CaCO3, 50.48 mol % MgCO3, and 0.09 mol % FeCO3 by electron microprobe analyses). The analysis errors for the chemical compositions quantified with the EDS method were estimated to be within ∼2 mol % for all of the samples. To accurately determine the molar percentage contents of Mg, Ca, and potential Cs in the precipitates, we dissolved each solid sample fully in 1% HNO3 solution and diluted certain times before measuring the concentrations of available ions (i.e., Mg2+, Ca2+, and Cs+) using ICP-OES (Perkin-Elmer; Elan 9000).
Acknowledgments
We thank the anonymous reviewers for thoughtful and thorough reviews and constructive comments. This research was financially supported by the Department of Energy Grant DE-FG02-02ER15366 (to H.T.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
References
- 1.Rosenberg PE, Holland HD. Calcite-dolomite-magnesite stability relations in solutions at elevated temperatures. Science. 1964;145(3633):700–701. doi: 10.1126/science.145.3633.700. [DOI] [PubMed] [Google Scholar]
- 2.Liebermann O. Synthesis of dolomite. Nature. 1967;213:241–245. [Google Scholar]
- 3.Gaines AM. Protodolomite synthesis at 100 °C and atmospheric pressure. Science. 1974;183(4124):518–520. doi: 10.1126/science.183.4124.518. [DOI] [PubMed] [Google Scholar]
- 4.Katz A, Matthews A. The dolomitization of CaCO3: An experimental study at 252–292°C. Geochim Cosmochim Acta. 1973;41:297–308. [Google Scholar]
- 5.Burn SJ, McKenzie JA, Vasconcelos C. Dolomite formation and biogeochemical cycles in the Phanerozoic. Sedimentology. 2000;47:49–61. [Google Scholar]
- 6.Warren J. Dolomite: Occurrence, evolution and economically important associations. Earth Sci Rev. 2000;52:1–81. [Google Scholar]
- 7.Vasconcelos C, McKenzie JA. Microbial mediation of modern dolomite precipitation and diagenesis under anoxic conditions (Lagoa Vermelha, Rio de Janeiro, Brazil) J Sediment Res. 1997;67:378–390. [Google Scholar]
- 8.Alonso-Zarza AM, Martín-Pérez A. Dolomite in caves: Recent dolomite formation in oxic, non-sulfate environments. Castañar Cave, Spain. Sediment Geol. 2008;205:160–164. [Google Scholar]
- 9.Weber JN. Trace element composition of dolostones and dolomites and its bearing on the dolomite problem. Geochim Cosmochim Acta. 1964;29:1817–1868. [Google Scholar]
- 10.Gaines AM. Protodolomite redefined. J Sediment Petrol. 1977;47:543–546. [Google Scholar]
- 11.Baker PA, Kastner M. Constraints on the formation of sedimentary dolomite. Science. 1981;213(4504):214–216. doi: 10.1126/science.213.4504.214. [DOI] [PubMed] [Google Scholar]
- 12.Vasconcelos C, McKenzie JA, Bernasconi S, Grujic D, Tien AJ. Microbial mediation as a possible mechanism for natural dolomite formation at low temperatures. Nature. 1995;377:220–222. [Google Scholar]
- 13.Land LS. Failure to precipitate dolomite at 25 degrees C from dilute solution despite 1000-fold oversaturation after 32 years. Aquat Geochem. 1998;4(3-4):361–368. [Google Scholar]
- 14.Arvidson RS, Mackenzie FT. The dolomite problem: Control of precipitation kinetics by temperature and saturation state. Am J Sci. 1999;299:257–288. [Google Scholar]
- 15.Deelman JC. Breaking Ostwald’s rule. Chem Erde-Geochem. 2001;61(3):224–235. [Google Scholar]
- 16.Kelleher IJ, Redfern SAT. Hydrous calcium magnesium carbonate, a possible precursor to the formation of sedimentary dolomite. Mol Simul. 2002;28:557–572. [Google Scholar]
- 17. Zenger DH, Dunham JB, Ethington RL (1980) Concepts and Models of Dolomitization, Special Publication No. 28 (Society of Economic Paleontologists and Mineralogists, Tulsa, OK), 320 pp.
- 18.Benson SM, Cole DR. CO2 sequestration in deep sedimentary formations. Elements. 2008;4:325–331. [Google Scholar]
- 19.Radha, et al. Energetic and structural studies of amorphous Ca1-xMgxCO3⋅nH2O (0 ≤ x ≤ 1) Geochim Cosmochim Acta. 2012;90:83–95. [Google Scholar]
- 20.Loste E, Wilson RM, Seshadri R, Meldrum FC. The role of magnesium in stabilizing amorphous calcium carbonate and controlling calcite morphologies. J Cryst Growth. 2003;254:206–218. [Google Scholar]
- 21.Politi Y, et al. Transformation mechanism of amorphous calcium carbonate into calcite in the sea urchin larval spicule. Proc Natl Acad Sci USA. 2008;105(45):17362–17366. doi: 10.1073/pnas.0806604105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamm LM, Wallace AF, Dove PM. Molecular dynamics of ion hydration in the presence of small carboxylated molecules and implications for calcification. J Phys Chem B. 2010;114(32):10488–10495. doi: 10.1021/jp9108893. [DOI] [PubMed] [Google Scholar]
- 23.Konigsberger E, Konigsberger LC, Gamsjager H. Low-temperature thermodynamic model for the system Na2CO3-MgCO3-CaCO3-H2O. Geochim Cosmochim Acta. 1999;63:3105–3119. [Google Scholar]
- 24.Haechen M, Prigiobbe V, Baciocchi R, Mazzotti M. Precipitation in the Mg-carbonate system - effects of temperature and CO2 pressure. Chem Eng Sci. 2008;63:1012–1028. [Google Scholar]
- 25.Ming DW, Franklin WT. Synthesis and characterization of lansfordite and nesquehonite. Soil Sci Soc Am J. 1985;49:1303–1308. [Google Scholar]
- 26.Kloprogge JT, Martens WN, Nothdurft L. Low temperature synthesis and characterization of nesquehonite. J Mater Sci Lett. 2003;22:825–829. [Google Scholar]
- 27.Zhao L, Sang L, Chen J, Ji J, Teng HH. Aqueous carbonation of natural brucite: Relevance to CO2 sequestration. Environ Sci Technol. 2010;44(1):406–411. doi: 10.1021/es9017656. [DOI] [PubMed] [Google Scholar]
- 28.Davies PJ, Bubela B. Transformation of nesquehonite into hydromagnesite. Chem Geol. 1973;12:289–300. [Google Scholar]
- 29.Fernandez AI, Chimenos JM, Segarra M. Procedure to obtain hydromagnesite from MgO-containing residues – A kinetic study. Ind Eng Chem Res. 2000;39:3653–3658. [Google Scholar]
- 30.Zhang Z, et al. Temperature- and pH-dependent morphology and FT-IR analysis of magnesium carbonate hydrates. J Phys Chem B. 2006;110(26):12969–12973. doi: 10.1021/jp061261j. [DOI] [PubMed] [Google Scholar]
- 31.Sayles FL, Fyfe WS. Crystallization of magnesite from aqueous solution. Geochim Cosmochim Acta. 1973;37:87–99. [Google Scholar]
- 32.Deelman JC. Low-temperature nucleation of magnesite and dolomite. Neues Jb Miner Monat. 1999;7:289–302. [Google Scholar]
- 33.Wolf GH, Chizmeshya AVG, Diefenbacher J, McKelvy MJ. In situ observation of CO2 sequestration reactions using a novel microreaction system. Environ Sci Technol. 2004;38(3):932–936. doi: 10.1021/es0346375. [DOI] [PubMed] [Google Scholar]
- 34.Giammar DE, Bruant RG, Peters CA. Forsterite dissolution and magnesite precipitation at conditions relevant for deep saline aquifer storage and sequestration of carbon dioxide. Chem Geol. 2005;217:257–276. [Google Scholar]
- 35. Lippmann F (1973) Sedimentary carbonate minerals. Minerals, Rocks and Inorganic Materials, Monograph Series of Theoretical and Experimental Studies (Springer, Berlin), Vol 6, 228 pp.
- 36.De Leeuw NH, Parker SC. Surface-water interactions in the dolomite problem. Phys Chem Chem Phys. 2001;3:3217–3221. [Google Scholar]
- 37.Jiao D, King C, Grossfield A, Darden TA, Ren P. Simulation of Ca2+ and Mg2+ solvation using polarizable atomic multipole potential. J Phys Chem B. 2006;110(37):18553–18559. doi: 10.1021/jp062230r. [DOI] [PubMed] [Google Scholar]
- 38.Di Tommaso D, de Leeuw NH. Structure and dynamics of the hydrated magnesium ion and of the solvated magnesium carbonates: Insights from first principles simulations. Phys Chem Chem Phys. 2010;12(4):894–901. doi: 10.1039/b915329b. [DOI] [PubMed] [Google Scholar]
- 39.Yang Y, Sahai N, Romanek CS, Chakraborty S. A computational study of Mg2+ dehydration in aqueous solution in the presence of HS- and other monovalent anions – Insights to dolomite formation. Geochim Cosmochim Acta. 2012;88:77–87. [Google Scholar]
- 40.Pavlov M, Siegbahn PEM, Sandstro M. Hydration of beryllium, magnesium, calcium, and zinc ions using density functional theory. J Phys Chem A. 1998;102:219–228. [Google Scholar]
- 41.Markham GD, Glusker JP, Bock CW. The arrangement of first- and second-sphere water molecules in divalent magnesium complexes: Results from molecular orbital and density functional theory and from structural crystallography. J Phys Chem B. 2002;106:5118–5134. doi: 10.1021/ic020602e. [DOI] [PubMed] [Google Scholar]
- 42.Bock CW, Kaufman A, Glusker JP. Coordination of water to magnesium cations. Inorg Chem. 1994;33:419–427. [Google Scholar]
- 43.Berg, et al. Stability and reactivity of hydrated magnesium cations. Chem Phys. 1998;239:379–392. [Google Scholar]
- 44.Barran PE, Walker NR, Stace AJ. Competitive charge transfer reactions in small [Mg(H2O)N]2+ clusters. J Chem Phys. 2000;112:6173–6177. [Google Scholar]
- 45.Lightstone FC, Schwegler E, Hood RQ, Gygi F, Galli G. A first principles molecular dynamics simulation of the hydrated magnesium ion. Chem Phys Lett. 2001;343:549–555. [Google Scholar]
- 46.Pullman A, Berthod H, Giessnerprettre C, Hinton JF, Harpool D. Hydrogen bonding in pure and aqueous formamide. J Am Chem Soc. 1978;100:3991–3994. [Google Scholar]
- 47.Zhang F, Xu H, Konishi H, Roden EE. A relationship between d(104) value and composition in the calcite-disordered dolomite solid-solution series. Am Mineral. 2010;95:1650–1656. [Google Scholar]
- 48.Davies KJ, Dove PM, Wasylenki LE, De Yoreo JJ. Morphological consequences of differential Mg2+ incorporation at structurally distinct steps on calcite. Am Mineral. 2004;89:714–720. [Google Scholar]
- 49.Mucci A, Morse JW. The incorporation of Mg2+ and Sr2+ into calcite overgrowths-Influence of growth rate and solution composition. Geochim Cosmochim Acta. 1983;47:217–233. [Google Scholar]
- 50.Mucci A. Growth-kinetics and composition of magnesian calcite overgrowths precipitated from seawater – quantitative influence of ortho-phosphate ions. Geochim Cosmochim Acta. 1986;50:2255–2265. [Google Scholar]
- 51.Hartley G, Mucci A. The influence of PCO2 on the partitioning of magnesium in calcite overgrowths precipitated from artificial seawater at 25° and 1-atm total pressure. Geochim Cosmochim Acta. 1996;60:315–324. [Google Scholar]
- 52.Huang YM, Fairchild IJ. Partitioning of Sr2+ and Mg2+ into calcite under karst-analogue experimental conditions. Geochim Cosmochim Acta. 2001;65:47–62. [Google Scholar]
- 53.Aizenberg J, Lambert G, Weiner S, Addadi L. Factors involved in the formation of amorphous and crystalline calcium carbonate: A study of an ascidian skeleton. J Am Chem Soc. 2002;124(1):32–39. doi: 10.1021/ja016990l. [DOI] [PubMed] [Google Scholar]
- 54.Raz S. The transient phase of amorphous calcium carbonate in sea urchin larval spicules: The involvement of proteins and magnesium ions in its formation and stabilization. Adv Funct Mater. 2003;13:480–486. [Google Scholar]
- 55.Faatz M, Grohn F, Wegner G. Amorphous calcium carbonate: Synthesis and potential intermediate in biomineralization. Adv Mater. 2004;16:996–1000. [Google Scholar]
- 56.Oh KD, Morikawa H, Iwai S, Aoki H. The crystal structure of magnesite. Am Mineral. 1973;58:1029–1033. [Google Scholar]
- 57.Markgraf SA, Reeder RJ. High-temperature structure refinements of calcite and magnesite. Am Mineral. 1985;70:590–600. [Google Scholar]
- 58.Gottlicher S, Vegas A. Electron-density distribution in magnesite (MgCO3) Acta Crystallogr B. 1988;44:362–367. [Google Scholar]
- 59.Maslen EN, Streltsov VA, Streltsova NR. X-ray study of the electron density in calcite, CaCO3. Acta Crystallogr. 1993;49:636–641. [Google Scholar]
- 60.Skinner AJ, LaFemina JP, Jansen HJF. Structure and bonding of calcite: A theoretical study. Am Mineral. 1994;79:205–214. [Google Scholar]
- 61.Catti M, Pavese A. Theoretical structure factors and electron density of magnesite (MgCO3) Acta Crystallogr A. 1996;52:413–418. [Google Scholar]
- 62.Peterson, et al. Electron density study of calcite. EOS. 1979;60:415–416. [Google Scholar]
- 63.Reeder RJ, Wenk HR. Structure refinements of some thermally disordered dolomite. Am Mineral. 1983;68:769–776. [Google Scholar]
- 64.Williams Q, Collerson B, Knittle E. Vibrational spectra of magnesite (MgCO3) and calcite-III at high pressures. Am Mineral. 1992;77:1158–1165. [Google Scholar]
- 65.Gillet P, Biellmann C, Reynard B, McMillan P. Raman spectroscopic studies of carbonates. Part I: High-pressure and high-temperature behavior of calcite, magnesite, dolomite and aragonite. Phys Chem Miner. 1993;20:1–18. [Google Scholar]
- 66.Grzechnik A, Simon P, Gillet P, McMillan P. An infrared study of MgCO3 at high pressure. Physica B. 1999;262:67–73. [Google Scholar]
- 67.Santillan J, Catalli K, Williams Q. An infrared study of carbon-oxygen bonding in magnesite to 60 GPa. Am Mineral. 2005;90:1669–1673. [Google Scholar]
- 68.Fiquet, et al. Structural refinements of magnesite at very high pressure. Am Mineral. 2002;87:1261–1265. [Google Scholar]
- 69.Reddy MM, Nancollas GH. The crystallization of calcium carbonate. (IV) The effect of magnesium, strontium and sulfate ions. J Cryst Growth. 1976;35:33–38. [Google Scholar]
- 70.Ma Y, et al. The grinding tip of the sea urchin tooth exhibits exquisite control over calcite crystal orientation and Mg distribution. Proc Natl Acad Sci USA. 2009;106(15):6048–6053. doi: 10.1073/pnas.0810300106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zolotoyabko, et al. Differences between bond lengths in biogenic and geological calcite. Cryst Growth Des. 2010;10:1207–1214. [Google Scholar]
- 72.Becker A, Ziegler A, Epple M. The mineral phase in the cuticles of two species of Crustacea consists of magnesium calcite, amorphous calcium carbonate, and amorphous calcium phosphate. Dalton Trans. 2005;10(10):1814–1820. doi: 10.1039/b412062k. [DOI] [PubMed] [Google Scholar]
- 73.Elstnerová P, et al. Ab initio study of thermodynamic, structural, and elastic properties of Mg-substituted crystalline calcite. Acta Biomater. 2010;6(12):4506–4512. doi: 10.1016/j.actbio.2010.07.015. [DOI] [PubMed] [Google Scholar]
- 74.Effenberger H, Mereiter K, Zemann J. Crystal structure refinements of magnesite, calcite, rhodochrosite, siderite, smithonite, and dolomite, with discussion of some aspects of the stereochemistry of calcite type carbonates. Z Kristallogr. 1981;156:233–243. [Google Scholar]
- 75.Burton B, Kikuchi R. Thermodynamic analysis of the system CaCO3-MgCO3 in the tetrahedron approximation of the cluster variation method. Am Mineral. 1984;69:165–175. [Google Scholar]
- 76.Steinfink H, Sans FJ. Refinement of the crystal structure of dolomite. Am Mineral. 1959;44:679–682. [Google Scholar]