

Significance
GabR is a member of the MocR/GabR subfamily of the GntR family of bacterial transcription regulators. It regulates the metabolism of γ-aminobutyric acid, an important nitrogen and carbon source in many bacteria. The crystal structures reported here show that this protein has evolved from the fusion of a type I aminotransferase and a winged helix-turn-helix DNA-binding protein to form a chimeric protein that adopts a dimeric head-to-tail configuration. The pyridoxal 5′-phosphate–binding regulatory domain of GabR is therefore an example of a coenzyme playing a role in transcription regulation rather than in enzymatic catalysis. Our structural and biochemical studies lay the mechanistic foundation for understanding the regulatory functions of the MocR/GabR subfamily of transcription regulators.
Abstract
Bacillus subtilis GabR is a transcription factor that regulates gamma-aminobutyric acid (GABA) metabolism. GabR is a member of the understudied MocR/GabR subfamily of the GntR family of transcription regulators. A typical MocR/GabR-type regulator is a chimeric protein containing a short N-terminal helix-turn-helix DNA-binding domain and a long C-terminal pyridoxal 5′-phosphate (PLP)-binding putative aminotransferase domain. In the presence of PLP and GABA, GabR activates the gabTD operon, which allows the bacterium to use GABA as nitrogen and carbon sources. GabR binds to its own promoter and represses gabR transcription in the absence of GABA. Here, we report two crystal structures of full-length GabR from B. subtilis: a 2.7-Å structure of GabR with PLP bound and the 2.55-Å apo structure of GabR without PLP. The quaternary structure of GabR is a head-to-tail domain-swap homodimer. Each monomer comprises two domains: an N-terminal winged-helix DNA-binding domain and a C-terminal PLP-binding type I aminotransferase-like domain. The winged-helix domain contains putative DNA-binding residues conserved in other GntR-type regulators. Together with sedimentation velocity and fluorescence polarization assays, the crystal structure of GabR provides insights into DNA binding by GabR at the gabR and gabT promoters. The absence of GabR-mediated aminotransferase activity in the presence of GABA and PLP, and the presence of an active site configuration that is incompatible with stabilization of the GABA external aldimine suggest that a GabR aminotransferase-like activity involving GABA and PLP is not essential to its primary function as a transcription regulator.
The MocR/GabR subfamily of chimeric proteins is widespread in bacteria and regulates a variety of biological processes. All members of this subfamily are typically composed of a 60- to 120-residue N-terminal winged-helix DNA-binding domain resembling other bacterial transcriptional regulators from the GntR family (1), and a large C-terminal putative aminotransferase domain, averaging ∼300 aa in size and capable of binding to the vitamin B6 coenzyme, pyridoxal 5′-phosphate (PLP) (2). The PLP-binding putative aminotransferase domains are closely related by amino acid sequence to homodimeric type I aminotransferases (3–5).
GABA is an important nitrogen and carbon source in many bacteria. The GABA-inducible gabTD pathway in Bacillus subtilis allows utilization of extracellular GABA as nitrogen and carbon sources. The gabR gene is located upstream of the gabTD operon and is transcribed divergently. GabR functions as the PLP- and GABA-dependent transcriptional activator of the gabTD operon and as a GABA-independent negative autoregulator (Fig. 1) (2, 6). Mutations in the conserved residues of the putative aminotransferase domain presumed to be crucial for the folding of the domain and PLP-binding abolished the ability of GabR to activate the gabT promoter in vivo but did not affect its activity as an autorepressor (2).
Fig. 1.

Schematic of GabR binding sites at the gabR and gabTD promoters. A schematic of the gabR and gabTD gene loci, as previously elucidated (2), is shown here. The 47-bp GabR binding site (−63 to −17) within the gabR−gabT regulatory region is shown in detail, and the two direct repeats (ATACCA) are indicated. The arrows indicate the transcription start sites and directions for the gabR and gabT genes.
The gabT and gabD genes encode two enzymes for an alternative route of glutamate biosynthesis using GABA: GABA aminotransferase (GabT) and succinic semialdehyde dehydrogenase (GabD), respectively (2). GabT is a type I aminotransferase (5, 7) that catalyzes the ping-pong transamination reaction using GABA as an amino group donor, α-ketoglutarate as an amino group acceptor and PLP as coenzyme to form the products, succinic semialdehyde (SS) and glutamate. During the first half-reaction, GABA donates its amino group to PLP, thereby releasing SS and converting PLP to pyridoxamine 5′-phosphate (PMP) (Fig. S1). In the second half-reaction, α-ketoglutarate accepts the amino group to become glutamate and regenerate the PLP form of the enzyme (Fig. S1). In the downstream reaction, the NAD-dependent dehydrogenase, GabD converts SS to succinate. Null mutants of gabR, gabT, and gabD in B. subtilis were reported to be incapable of using GABA as the sole nitrogen source (2).
GabR binds specifically to an extended region of DNA that overlaps the −35 and −10 elements of the gabT and gabR promoters, respectively (6) (Fig. 1). Using DNase I footprinting experiments and native gel-shift experiments with purified GabR, a 47-bp region of DNA (−63 to −17 relative to the gabT transcription start site) was identified as the GabR binding site. The 47-bp minimal fragment contains two copies of the direct repeat, ATACCA, which may be essential for GabR binding. Addition of PLP and GABA did not noticeably alter the DNase I footprints, indicating that GabR is capable of binding DNA in the absence of these molecules. Based on the extended 47-bp footprint, it was proposed that GabR binds to DNA either as a dimer or tetramer (6).
It has been reported that a GabR-catalyzed “partial” aminotransferase-like reaction involving GABA and PLP may be essential for its transcriptional activator function (6). Interestingly, no “full” two-step ping-pong–type GABA aminotransferase activity for GabR could be detected in vivo, and overexpression of GabR was unable to restore GABA aminotransferase activity in strains lacking gabT (6).
Here, we report two crystal structures of the PLP-bound and PLP-free forms of full-length B. subtilis GabR determined to be 2.7 and 2.55 Å, respectively. The crystal structure of GabR reveals a two-domain protein organized as a distinct head-to-tail domain-swap homodimer. A long linker of 29 aa separates the N-terminal winged-helix domain from the C-terminal domain, which has the same overall fold as type I aminotransferases and is hereby called AT-fold domain. Together with sedimentation velocity and fluorescence polarization data, the GabR structure provides a model for DNA binding at the gabR-gabT locus by one or more GabR homodimers. Key amino acid sequence changes in the putative substrate-binding pocket of GabR relative to those of well-characterized type I aminotransferases suggest that a GabR catalytic activity involving GABA and PLP is either absent or very poor under physiological conditions. Our results strongly suggest that the AT-fold domain in B. subtilis GabR, in a unique combination with the winged-helix domain, is primarily relegated to a regulatory function at the gabR and gabT promoters.
Results and Discussion
GabR Is a Head-to-Tail Domain-Swap Homodimer.
Sequence alignments of members of the MocR/GabR subfamily show that these proteins comprise a small N-terminal winged-helix domain and a large C-terminal putative type I aminotransferase domain (1, 2). The crystal structure of GabR described here is consistent with these reports and shows that a GabR monomer folds into two domains (Fig. 2): an N-terminal helix-turn-helix (HTH) DNA-binding domain (residues 3–80) of the winged-helix subtype (8) and a C-terminal AT-fold domain (residues 110–470) characteristic of the type I aminotransferase family (9, 10) and containing the signature long bent α-helix (residues 345–386) connecting two subdomains. A long 29-residue linker (residues 81–109) connects the winged-helix domain to the AT-fold domain (Fig. 2 and Fig. S2). GabR crystallizes as a homodimer with the two monomers organized in a head-to-tail domain-swap arrangement. This arrangement creates two distinct dimer interfaces. The larger interface is composed of two AT-fold domains, which are also arranged in a head-to-tail manner to form the characteristic homodimeric type I aminotransferase fold (5, 9). The interface contains a buried area of 1,695 Å2 with a binding energy of −11.4 kcal/mol, suggesting that this interface stabilizes the GabR homodimer and is physiological. This interface of GabR, as seen in all type I aminotransferases (9), contains two PLP-binding pockets bound by one molecule of PLP in each pocket.
Fig. 2.
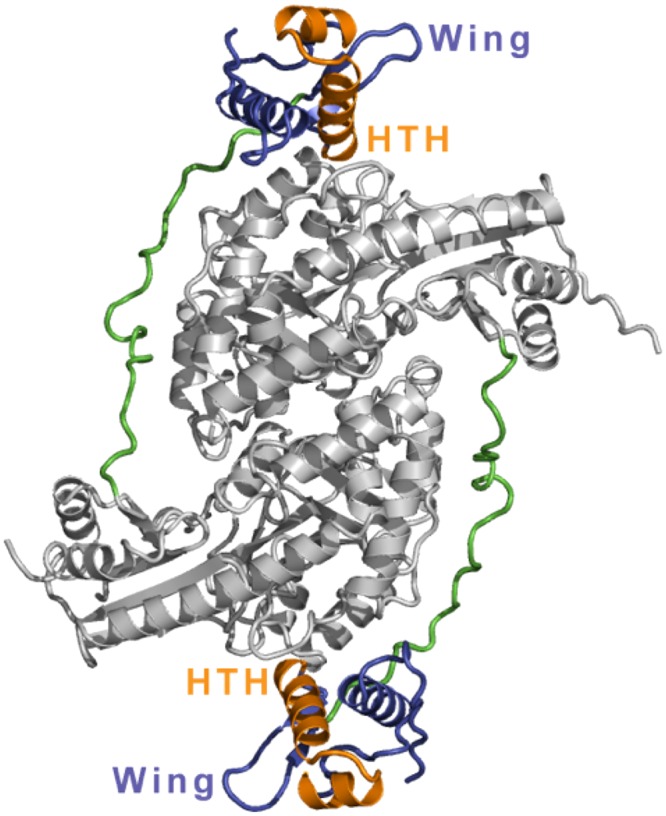
Quaternary structure of GabR. GabR is a head-to-tail domain-swap homodimer. The AT-fold domains are shown in light gray. The long linkers connecting the N-terminal winged-helix domains to the C-terminal AT-fold domains are shown in green. The winged-helix domains are shown in blue-violet. The HTH motifs (orange) and the wing motifs are labeled. An orthogonal view of the structure is shown in Fig. S2.
The second interface is formed between the winged-helix domain of one monomer and the AT-fold domain of the other monomer. This interface has a buried area of 422 Å2 with a relatively low free energy of −3.8 kcal/mol, suggesting that the overall quaternary structure of GabR may be dynamic in solution and that the winged-helix domain may undergo large-scale domain movements, especially when bound by DNA. The large dimer interface between the two AT-domains further suggests that any such domain movements by the winged-helix domain do not require restructuring of the AT−AT interface. Interestingly, we note that the extended N-terminal arms of the two monomers of aspartate aminotransferase (Asp-AT) also reach over and interact with the other subunit in that homodimer (10). Because Asp-AT does not have an N-terminal winged-helix domain, the significance of this “crossing over” of the N-terminal arm to the other subunit in Asp-AT is not clear. Although there is reasonable electron density for the long extended linker that connects the winged-helix domain to the AT-fold domain, the linker does not form any significant contacts with the rest of the protein, suggesting that it may be flexible in solution and may thereby facilitate the proposed winged-helix domain movements upon DNA binding.
We also crystallized GabR without exogenous PLP. In the resulting apo-GabR structure, PLP is no longer bound in the PLP-binding pocket, and instead, a molecule of imidazole is bound in the same pocket (Fig. S3). The purified protein is pale yellow in color, suggesting that endogenous PLP from Escherichia coli is still bound to GabR immediately after purification. Because imidazole is present in high concentrations in the purification and crystallization buffers, the replacement of PLP by imidazole is most likely a crystallization artifact. The overall similarity between the GabR−imidazole and GabR−PLP structures suggests that the AT-fold domain maintains its overall structural integrity even when PLP is no longer bound.
GabR Is a Dimer in Solution.
We performed sedimentation velocity analysis to investigate the oligomeric state of GabR−PLP in solution. In the absence of GABA, the sedimentation velocity plot shows a predominant peak at ∼100 kDa, suggesting a dimer of GabR in solution even at very low protein concentrations (Fig. S4A; GabR molecular weight, 56 kDa). The plot also shows two minor peaks at about 50 and 10 kDa, which may represent a small proportion of monomer and degradation products, respectively. The sedimentation velocity plot of GabR−PLP in the presence of GABA similarly shows a predominant peak at ∼100 kDa and two low–molecular-weight peaks (Fig. S4B), again indicating a dimer of GabR in solution. Also, during protein purification, GabR elutes as one major peak on a Superdex 200 gel-filtration column with an apparent molecular weight of a dimer (Fig. S5). Based on the crystal structure, our expectation is that the AT−AT dimer interface in the solution state of GabR may be similar to that found in the crystal structure. Our sedimentation velocity data do not reveal tetramers and higher oligomers in solution; however, further oligomerization of GabR may still occur when GabR binds to DNA.
Conserved Putative DNA-Binding Residues in the N-Terminal Winged-Helix Domain.
The GabR N-terminal domain (residues 3–80) contains a canonical winged-helix domain (α1-β1-α2-T-α3-β2-W-β3), where α2-T-α3 (residues 42–66) form the HTH motif and W (residues 71–76) is the wing (Fig. S6). In the case of GabR, a second wing is absent. Although many winged-helix proteins contain a second wing, there are several examples of winged-helix proteins that bind DNA and in which the second wing is absent (8). X-ray and NMR structures of winged-helix proteins in complex with DNA show that the winged-helix domain uses diverse modes of DNA binding, with examples of this domain binding DNA as a monomer, homodimer, or heterodimer (8). In general, the most basic or positively charged surface of the winged-helix domain binds to the negatively charged phosphate backbone or nucleotide side-chain groups (8).
Because the putative DNA-binding domain is conserved among GntR-type transcriptional regulators, we searched the Conserved Domain Database (11) using the GabR winged-helix domain sequence as a query. We found several hits, including E. coli acyl-CoA–responsive transcription factor, FadR, and identified three putative DNA-binding residues in GabR (R43, S52, and K75) based on sequence conservation in FadR and other GntR family members. Because these three residues (R43 and S52 in the HTH; K75 in the wing) also map to the most basic surface on the GabR winged-helix domain (Fig. S7) and are fully exposed in the GabR homodimer (Fig. S8), we predict that they may constitute a major DNA-binding surface. To examine the DNA-binding roles of these conserved residues, we generated a GabR−DNA model by superimposing the structural coordinates of the GabR winged-helix domain onto those of the FadR−DNA complex [Protein Data Bank (PDB) ID code 1H9T (12)]. In the FadR−DNA complex (Fig. 3A), two winged-helix domains of a FadR homodimer bind to a 19-bp operator sequence containing two copies of an inverted repeat, TCTGGT, separated only by 3 bp (12). In contrast, the 47-bp GabR binding site contains two copies of the direct repeat ATACCA separated by 34 bp of DNA. Because we do not know exactly how GabR binds DNA, we only used one copy of the GabR winged-helix domain for the structural superposition (Fig. 3B). The resulting GabR−DNA model (Fig. 3B) is identical no matter which FadR monomer we use for the superposition and has several salient features.
Fig. 3.

Conserved putative DNA-binding residues in GabR. (A) The crystal structure of the FadR−DNA complex is shown (PDB ID code 1H9T) (12). FadR binds DNA as a homodimer. The protein and DNA are shown in cartoon and stick representation, respectively. Side chains of some of the DNA-binding residues are shown as sticks and labeled. The corresponding phosphate groups and nucleotides contacted by the protein side chains are colored in orange. The central base pair in the 19-mer DNA is shown in pink. Only the winged-helix domains of FadR are shown; the acyl-CoA–binding domains are hidden for clarity. (B) A model of the GabR winged-helix domain bound to the 19-bp FadR DNA sequence. Again, the central base pair in the 19-mer is colored in pink, although only 12 of the 19 bp are shown for clarity. The phosphate groups and nucleotides putatively contacted by GabR protein side chains are colored in orange. The GabR protein side chains predicted to be involved in DNA binding are shown as sticks and are labeled.
First, the HTH motif in FadR binds in the major groove of DNA, whereas the wing invades the minor groove (Fig. 3A). Residue R35 in helix α2 of the FadR HTH motif makes two H-bonds with the first guanine in the TCTGGT repeat (Fig. 3A). The conserved arginine residue R43 in GabR (Fig. 3B) may form similar sequence-specific contacts with one of the guanines in its direct repeat (double-stranded sequence, ATACCA•TATGGT). In the turn of the HTH motif, FadR residue T44 (S52 in GabR) makes H bonds with the phosphate backbone and provides a hydrophobic contact to the central C−G base pair in the 19-mer, whereas residues in the α3 recognition helix, which are typically known to bind the major groove of DNA (8), either contact the central C−G base pair in the 19-mer (T46 in FadR; N54 in GabR) or a phosphate group (T47 in FadR; S55 in GabR) in the major groove. In the FadR wing, residue K67 points between two phosphate groups (Fig. 3A). The GabR residue K75 (Fig. 3B) could similarly point between two phosphate groups. Because many residues are conserved between FadR and GabR (Fig. S9) and because the FadR and GabR DNA repeats have similar sequences, we predict that some of the protein−DNA interactions will be similar between the FadR−DNA and GabR−DNA complexes. However, because the GabR and FadR repeat sequences are dissimilar in type, direction, and spacing, a crystal structure of the GabR−DNA complex is required to eludicate all of the individual pairwise protein−DNA interactions.
Second, because the two winged-helix domains of FadR interact with each other on DNA (Fig. 3A) (12), we examined whether such an interface is possible in GabR. The GabR α3 helix is surface exposed (Fig. 2) and has a sequence that is capable of forming an α3−α3 interface similar to that of FadR (Figs. S9 and S10). Thus, two copies of the GabR winged-helix domain may be able to interact with each other on DNA under specific circumstances, for instance, when the direct repeats that are 34 bp apart are brought into close proximity.
Third, mutating the CC doublets in either of the two GabR direct repeats to GG led to a 10-fold decrease in DNA-binding affinity. Furthermore, mutating both CC doublets to GG completely abolished GabR binding (6). These observations are consistent with our GabR−DNA model, which suggests that residue R43 of GabR may form sequence-specific contacts with one of the guanines in its direct repeat. As is true for FadR (13), our GabR−DNA model predicts that mutating conserved residues like R43 and K75 may abrogate or interfere with GabR−DNA interactions.
GabR Binds DNA with High Affinity.
The binding affinity of GabR for a 49-bp DNA containing the 47-bp GabR binding site was determined by fluorescence polarization and competitive binding assays. Previous studies estimated the apparent dissociation constant of a GabR−DNA complex containing a 250-bp gabR−gabT fragment to be around 1 nM (6). When a range of GabR protein concentrations were titrated against the 47-bp carboxytetramethylrhodamine (TAMRA)-labeled DNA, we observed a dose-dependent increase in polarization (Fig. 4A), indicative of the binding of GabR protein to DNA. The curve was fitted to the modified Hill equation (Eq. S1 in SI Materials and Methods) and the dissociation constant of the GabR−DNA complex was determined to be 70.2 ± 3.7 nM. A Hill coefficient of 2.2 ± 0.2 was obtained, indicating positive cooperativity in the binding process. To establish specificity of binding and rule out nonspecific effects from the TAMRA label, a competitive binding assay was performed with unlabeled oligonucleotides containing the same sequence as the TAMRA-labeled DNA (Fig. 4B). A preformed mixture of equimolar concentrations of TAMRA-labeled DNA and GabR was titrated with increasing concentrations of unlabeled DNA. The fluorescence polarization decreased with increasing amounts of unlabeled DNA (Fig. 4B). The curve was fitted to the Hill equation (Eq. S2) and the IE50 of unlabeled DNA for GabR was calculated to be 98.1 ± 7.8 nM, which is similar to the dissociation constant of the TAMRA-labeled DNA−GabR complex (Fig. 4A). A Hill coefficient of 1.7 ± 0.4 was obtained, supporting the same type of positive cooperativity as shown in Fig. 4A. Although the precise binding stoichiometry of GabR for DNA is still unknown, the dissociation constants that we determined suggest a high-affinity GabR−DNA complex. The Hill coefficients in both titrations indicate positive cooperativity in the binding of GabR dimers to DNA.
Fig. 4.

Fluorescence polarization assays to measure DNA binding by GabR. Each data point was measured in triplicate. The average values and errors were plotted and fitted to the Hill equation. The abbreviations used in the figure are as follows: FPFTD, fluorescence polarization signal of free labeled DNA; FPGTD, fluorescence polarization signal of the GabR−DNA complex; KD, the dissociation constant; n, the Hill coefficient; and IE50, the concentration of unlabeled DNA required to bind 50% of protein. (A) Titration of GabR against 50 nM TAMRA-DNA. The GabR concentrations were varied from 0 to 1,000 nM. Error bars are too small to be displayed if the errors are less than 1/1,000th of the measured value. (B) Competitive binding assay. Titration of unlabeled DNA (0–1,000 nM) against a fixed concentration of preformed GabR-labeled DNA complex (100 nM each of GabR and TAMRA-DNA).
PLP Is Bound in the C-Terminal AT-Fold Domain.
Like in other type I aminotransferases (9, 10), the AT-fold domain of GabR has two subdomains, the large subdomain containing the invariant lysine (K312) that forms the Schiff base with PLP and the small subdomain that typically interacts with the α-carboxylate group of the substrate. One molecule of PLP is bound per GabR monomer as an internal aldimine at K312. We next compared the PLP-binding pocket of B. subtilis GabR (Fig. S11) with those of E. coli Asp-AT (PDB ID code 2AAT), the best-characterized member of the type I aminotransferase family; and pig liver GABA-AT bound to the substrate analog, γ-ethynyl GABA (GEG) (PDB ID code 1OHY) (10, 14). The active site residues that facilitate PLP binding are conserved in all three cases. For example, the phosphate group of PLP is situated very close to the N terminus of a nearby helix in all cases and is stabilized by hydrogen bonds from polar residues (T181, T309, and S311 in the case of GabR) to the phosphate oxygens. The salt bridge between an arginine residue (residue R319) and the phosphate group is also conserved in GabR, although the phosphate group has a different orientation compared with those in Asp-AT and GABA-AT.
The primary function of PLP is to stabilize the negative charge generated at the Cα atom of the transition state or external aldimine (Schiff base between an amino acid substrate and PLP) by delocalizing the negative charge through the π-electron system of the cofactor. A conserved aspartate residue [D279 in GabR, D222 in E. coli Asp-AT, D223 in chicken mitochondrial Asp-AT (4), and D298 in GABA-AT; Figs. 5A and 6A] promotes this electron sink nature of PLP by maintaining PLP in the protonated state through interaction with the pyridinium nitrogen.
Fig. 5.

PLP binding in aminotransferases and the arginine switch. (A) A schematic of the interactions of the phenolic ring of PLP with surrounding protein side chains. The corresponding residue numbers for Asp-AT (D223, N195, K258) and GABA-AT (D298, Q301, K329) are indicated. The precise locations of the hydrogen atoms have not been observed or measured so far. The H-atom positions indicated in the schematic are therefore only inferred from reaction chemistry. (B) A schematic model for the arginine switch in GABA-AT. The model is inferred from the crystal structure of pig liver GABA-AT−GEG complex and adapted from refs. 14 and 9. The positions of the two conserved arginine residues in the active site of GABA-AT and their interactions with the α- and γ-carboxylate groups of glutamate are indicated.
Fig. 6.

PLP is bound in the AT-fold domain of GabR. (A) The substrate-binding pocket in the GABA-AT−GEG structure. The secondary structure elements are shown in cartoon form. Side chains of the key residues that interact with PLP and substrate analog, GEG, are shown in stick representation and labeled. Hydrogen bonds are shown as dotted green lines. (B) The putative substrate-binding pocket in the crystal structure of GabR. The figure is drawn as described in A. The 2Fo − Fc map (blue mesh) around PLP is shown at 1.2σ level.
Is GabR an Enzyme?
We were currently unable to obtain crystals of a GabR−PLP−GABA complex, and therefore, we examined the putative GABA-binding site in GabR−PLP to understand whether GabR might catalyze an aminotransferase reaction involving GABA. Transamination reactions are ping-pong reactions that require two different amino acids to be able to bind in the active site (GABA and glutamate, if GabR were an α-ketoglutarate–dependent GABA aminotransferase). Most aminotransferases use either two conserved arginines or an extended hydrogen-bonding network to recognize and bind two amino acids with different shapes and properties (9). We superimposed the crystal structures of B. subtilis GabR, pig liver GABA-AT−GEG (14), and E. coli Asp-AT (10) to elucidate how different amino acids might bind in the active site of GabR. Even though GabR, GABA-AT, and Asp-AT share the overall type I aminotranferase fold, their polypeptide sequences are highly divergent. Also, the lengths of their secondary structural elements vary significantly and contain insertions of α-helices and β-strands in different parts of the structures, making it difficult to obtain secondary structure-based superposition. Therefore, we manually superimposed the pyridine ring of PLP to compare the putative substrate-binding pockets in all cases.
If GabR were a bona fide aminotransferase, then the active site must be able to interact with a keto acid such as α-ketoglutarate. In the active sites of Asp-AT and GABA-AT, two conserved arginines stabilize such a keto acid to subsequently form the related amino acid (Fig. 5B) (9). However, we found two key differences in the “active site” of GabR, compared with these aminotransferases, which may compromise its ability to transaminate GABA. First, the GabR active site may contain only one structurally conserved arginine (R207) that is, however, not conserved by sequence. In Asp-AT and GABA-AT, the equivalent arginine residue (R292* in the other subunit of the dimer in Asp-AT and R192 in GABA-AT; Fig. 5B) is known to form a bidentate hydrogen bond/ion pair with the γ-carboxylate of a dicarboxylate substrate such as glutamate, whereas the second arginine (R386 in Asp-AT and R445 in GABA-AT; Fig. 5B) makes a similar direct contact with the α-carboxylate of the glutamate. Furthermore, GABA-AT employs an arginine switch to distinguish between GABA and glutamate (9, 14). When GABA binds, R192 stays in place to contact the carboxylate of GABA (Fig. 6A), which is located in the same position as the γ-carboxylate of glutamate, while R445 moves away to accommodate GABA and engages instead in a salt bridge with a nearby glutamate residue (E270; Fig. 6A). Because only the first arginine residue in GabR may be structurally conserved (R207; Fig. 6B) while the second arginine is not, this suggests that GabR may bind GABA but not glutamate or other dicarboxylate substrates. In the absence of conserved arginines, aromatic amino acid aminotransferases and branched-chain aminotransferases use large-scale rearrangements of the active site hydrogen-bond network or hydrophobic cavities lined with polar residues to accommodate two different amino acids in the same site (refs. 9 and 15, and references therein). However, because these features are not preserved in GabR, these alternative substrate-binding mechanisms are unlikely for GabR.
The second key difference in the active site of GabR is related to its ability to stabilize the GABA external aldimine. When the amino acid substrate forms an external aldimine, the imine N atom of the amino acid and phenolic O atom of PLP could share an H-atom. Therefore, the phenolic O must be optimally oriented to accept the hydrogen from the imine N during reprotonation and hydrolysis to form PMP and release the deaminated substrate (Fig. 5A). In Asp-AT, an asparagine (N195; Fig. 5A) interacts with the phenolic O to hold it in place, and in GABA-AT, a glutamine residue (Q301; Fig. 5A) and a water molecule perform the same function. In GabR, there is no such interaction with the phenolic O, implying that the GABA external aldimine might not be optimally oriented for catalysis.
A sequence alignment of 10 MocR/GabR protein sequences from different strains of B. subtilis, E. coli, Pseudomonas aeruginosa, Salmonella enterica, and Streptococcus pneumoniae revealed an overall lack of evolutionary conservation for the arginine switch. Although the residues involved in PLP-binding (equivalent of B. subtilis GabR residues, D279, R319, and K312) are conserved in the examined homologs, we found the residue corresponding to B. subtilis GabR (R207) to be highly variable. In contrast to GabR, all of the PLP-interacting residues and the two substrate-interacting arginines are identical between B. subtilis GabT, a known GABA aminotransferase, and pig liver GABA-AT (Fig. S12). Such a contrast indicates the functional divergence of GabR from its evolutionary relatives.
Together, these critical differences in the active site architecture of GabR strongly suggest that GabR may bind GABA in a different manner than a true transaminase and may only catalyze the “full” transamination reaction with poor efficiency, if at all. Without the ability to accommodate glutamate effectively, GabR may not be able to complete the second half-reaction and enzymatically regenerate the PLP form of GabR. Consistent with our crystal structure and with previous reports that GabR does not rescue GabT activity (6), we were unable to measure a GABA-AT activity for GabR in coupled steady-state enzyme assays (described in SI Materials and Methods). Also, UV-visible scanning spectrophotometry experiments of GabR in the presence of PLP and GABA did not reveal any of the spectral shifts typically produced by the PLP or PMP intermediates in a PLP-dependent transamination reaction. Together, these experiments suggest that GabR may have very poor or no GABA aminotransferase activity under physiological conditions.
Although the trifunctional NAD-dependent S. enterica repressor NadR is an example of a cofactor-dependent protein containing an N-terminal DNA-binding domain and two catalytically active domains [a middle adenylyltransferase domain and a C-terminal nicotinamide ribose kinase (16)], there is also precedence for adaptation of enzymes to regulatory roles in other proteins. For instance, PyrR, a UMP-dependent transcription attenuator for pyrimidine biosynthesis in B. subtilis contains a type I phosphoribosyltransferase (PRT) domain without measurable catalytic activity (17). The B. subtilis purine repressor PurR has a two-domain architecture like GabR: an N-terminal winged-helix domain and a C-terminal PRT domain devoid of catalytic activity (18). It is of course possible that GabR recognizes other substrates or possesses a different PLP-dependent enzymatic activity that may or may not be related to its function as a PLP- and GABA-dependent transcriptional activator of gabT. However, so far, no other enzymatic activity for GabR has been reported.
Model for GabR-Mediated Transcriptional Regulation at the gabR and gabT Promoters.
Based on the crystal structure of GabR, the sedimentation velocity data and the model for the GabR−DNA complex, we propose three possible modes for GabR binding and regulation at the gabR and gabT promoters (Fig. 7). Because we do not know the precise GabR:DNA binding ratio, we consider different models with one or more GabR dimers bound to DNA. In model 1, two copies of GabR dimers bind to the 47-bp DNA with only one winged-helix domain from each dimer binding to each direct repeat. That the Hill coefficient for the GabR−DNA complex is close to 2 (Fig. 4) further suggests that GabR may cooperatively bind DNA as a dimer of dimers in the same way that a dimer of λcII dimers binds its DNA operator (19). In the λcII−DNA crystal structure, as in model 1, only one monomer from each λcII dimer binds its DNA sequence comprising two direct repeats. Even though the spacings between the direct repeats in the binding sites of λcII and GabR are different (6 and 34 bp, respectively), the cooperative binding of GabR to DNA as a dimer of dimers is still possible. In model 2, only one copy of the GabR dimer binds to the 47-bp DNA and both winged-helix domains of the dimer are involved in DNA binding. In this model, the two winged-helix domains of one GabR dimer may be rearranged and some bending of DNA may occur. The small buried surface area (422 Å2) between the winged-helix domain and AT-fold domains suggests that the position of the winged-helix domain may be dynamic in solution and that the proposed domain movements are possible using large-scale movements facilitated by the long linker. We recognize that other viable models still exist. For example, the second winged-helix domain of a GabR dimer may bind to unidentified cis binding sites located at larger distances from the gabR−gabT locus to form large DNA loops. Intriguingly, a large basic surface at the AT−AT interface begs the question whether this surface is somehow also involved in DNA binding (Fig. S8).
Fig. 7.

A model for transcriptional regulation at the gabR and gabT promoters. Different models for GabR binding in the gabR–gabT regulatory region are depicted. The GabR homodimer is represented as a cartoon with one monomer shown in white and the other colored gray. The larger oval represents the AT-fold domain and the smaller oval represents the winged-helix domain. Putative conformational changes in the presence of GABA are indicated by symbols with dashed lines.
These models suggest that GABA may bring about conformational changes in GabR or the GabR−DNA complex, and that these changes may be necessary to activate gabT transcription, for instance, by recruiting RNA polymerase to the gabT promoter (Fig. 7). Crystal structures of GabR−DNA complexes with and without GABA may help unravel some of these unknowns.
Biological Implications
Our studies suggest that GabR may not possess “full” GABA aminotranferase activity, although it requires PLP and GABA to activate transcription at the gabT promoter. It is likely that the primary function of GabR in B. subtilis is to regulate transcription at the gabR and gabT promoters and that the AT-fold domain of GabR has adapted to primarily serve as an effector binding domain. How the AT-fold domain of GabR assists the winged-helix domain in transcriptional regulation and how GABA mechanistically affects gabT activation by GabR still remain to be determined.
Materials and Methods
All experimental procedures are included in the Supporting Information. The data and refinement statistics for the final models of GabR-PLP and apo-GabR are listed in Table S1. The coordinates and structure factors for GabR-PLP and apo-GabR have been deposited in the PDB with ID codes 4N0B and 4MGR, respectively.
Supplementary Material
Acknowledgments
We thank Boguslaw Nocek (Structural Biology Center, Sector 19) at Advance Photon Source, Argonne National Laboratory for help with data collection; Zbyszek Dauter, Eleanor Dodson, Frank DiMaio, Nathaniel Echols, Julia Klutkowski, Ruslan Sanishvili, Miguel Ballicora, and Janet Smith for helpful discussions, suggestions, and/or technical help. This work was funded by the start-up funds from Loyola University Chicago (to D.L.), an NSF subaward from Grant CHE-1308672 (to D.L.), NSF Grant MCB-0110651 (to B.R.B.), and a grant from the National Institutes of Health (to D.R. and G.A.P.).
Footnotes
The authors declare no conflict of interest.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org [PDB ID codes 4MGR (apo) and 4N0B (GabR-PLP)].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1315887110/-/DCSupplemental.
References
- 1.Rigali S, Derouaux A, Giannotta F, Dusart J. Subdivision of the helix-turn-helix GntR family of bacterial regulators in the FadR, HutC, MocR, and YtrA subfamilies. J Biol Chem. 2002;277(15):12507–12515. doi: 10.1074/jbc.M110968200. [DOI] [PubMed] [Google Scholar]
- 2.Belitsky BR, Sonenshein AL. GabR, a member of a novel protein family, regulates the utilization of gamma-aminobutyrate in Bacillus subtilis. Mol Microbiol. 2002;45(2):569–583. doi: 10.1046/j.1365-2958.2002.03036.x. [DOI] [PubMed] [Google Scholar]
- 3.Bramucci E, Milano T, Pascarella S. Genomic distribution and heterogeneity of MocR-like transcriptional factors containing a domain belonging to the superfamily of the pyridoxal-5′-phosphate dependent enzymes of fold type I. Biochem Biophys Res Commun. 2011;415(1):88–93. doi: 10.1016/j.bbrc.2011.10.017. [DOI] [PubMed] [Google Scholar]
- 4.Kirsch JF, et al. Mechanism of action of aspartate aminotransferase proposed on the basis of its spatial structure. J Mol Biol. 1984;174(3):497–525. doi: 10.1016/0022-2836(84)90333-4. [DOI] [PubMed] [Google Scholar]
- 5.Schneider G, Käck H, Lindqvist Y. The manifold of vitamin B6 dependent enzymes. Structure. 2000;8(1):R1–R6. doi: 10.1016/s0969-2126(00)00085-x. [DOI] [PubMed] [Google Scholar]
- 6.Belitsky BR. Bacillus subtilis GabR, a protein with DNA-binding and aminotransferase domains, is a PLP-dependent transcriptional regulator. J Mol Biol. 2004;340(4):655–664. doi: 10.1016/j.jmb.2004.05.020. [DOI] [PubMed] [Google Scholar]
- 7.Ouchi T, Tomita T, Miyagawa T, Kuzuyama T, Nishiyama M. Dual roles of a conserved pair, Arg23 and Ser20, in recognition of multiple substrates in alpha-aminoadipate aminotransferase from Thermus thermophilus. Biochem Biophys Res Commun. 2009;388(1):21–27. doi: 10.1016/j.bbrc.2009.07.096. [DOI] [PubMed] [Google Scholar]
- 8.Gajiwala KS, Burley SK. Winged helix proteins. Curr Opin Struct Biol. 2000;10(1):110–116. doi: 10.1016/s0959-440x(99)00057-3. [DOI] [PubMed] [Google Scholar]
- 9.Eliot AC, Kirsch JF. Pyridoxal phosphate enzymes: Mechanistic, structural, and evolutionary considerations. Annu Rev Biochem. 2004;73:383–415. doi: 10.1146/annurev.biochem.73.011303.074021. [DOI] [PubMed] [Google Scholar]
- 10.Smith DL, Almo SC, Toney MD, Ringe D. 2.8-A-resolution crystal structure of an active-site mutant of aspartate aminotransferase from Escherichia coli. Biochemistry. 1989;28(20):8161–8167. doi: 10.1021/bi00446a030. [DOI] [PubMed] [Google Scholar]
- 11.Marchler-Bauer A, et al. CDD: A Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2011;39(Database issue):D225–D229. doi: 10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Aalten DM, DiRusso CC, Knudsen J. The structural basis of acyl coenzyme A-dependent regulation of the transcription factor FadR. EMBO J. 2001;20(8):2041–2050. doi: 10.1093/emboj/20.8.2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raman N, Black PN, DiRusso CC. Characterization of the fatty acid-responsive transcription factor FadR. Biochemical and genetic analyses of the native conformation and functional domains. J Biol Chem. 1997;272(49):30645–30650. doi: 10.1074/jbc.272.49.30645. [DOI] [PubMed] [Google Scholar]
- 14.Storici P, et al. Structures of gamma-aminobutyric acid (GABA) aminotransferase, a pyridoxal 5′-phosphate, and [2Fe-2S] cluster-containing enzyme, complexed with gamma-ethynyl-GABA and with the antiepilepsy drug vigabatrin. J Biol Chem. 2004;279(1):363–373. doi: 10.1074/jbc.M305884200. [DOI] [PubMed] [Google Scholar]
- 15.Hirotsu K, Goto M, Okamoto A, Miyahara I. Dual substrate recognition of aminotransferases. Chem Rec. 2005;5(3):160–172. doi: 10.1002/tcr.20042. [DOI] [PubMed] [Google Scholar]
- 16.Grose JH, Bergthorsson U, Roth JR. Regulation of NAD synthesis by the trifunctional NadR protein of Salmonella enterica. J Bacteriol. 2005;187(8):2774–2782. doi: 10.1128/JB.187.8.2774-2782.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tomchick DR, Turner RJ, Switzer RL, Smith JL. Adaptation of an enzyme to regulatory function: Structure of Bacillus subtilis PyrR, a pyr RNA-binding attenuation protein and uracil phosphoribosyltransferase. Structure. 1998;6(3):337–350. doi: 10.1016/s0969-2126(98)00036-7. [DOI] [PubMed] [Google Scholar]
- 18.Sinha SC, et al. The purine repressor of Bacillus subtilis: A novel combination of domains adapted for transcription regulation. J Bacteriol. 2003;185(14):4087–4098. doi: 10.1128/JB.185.14.4087-4098.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jain D, et al. Crystal structure of bacteriophage lambda cII and its DNA complex. Mol Cell. 2005;19(2):259–269. doi: 10.1016/j.molcel.2005.06.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.