

Abstract
Mass-independent fractionation of sulfur isotopes (S MIF) in Archean and Paleoproterozoic rocks provides strong evidence for an anoxic atmosphere before ∼2,400 Ma. However, the origin of this isotopic anomaly remains unclear, as does the identity of the molecules that carried it from the atmosphere to Earth’s surface. Irrespective of the origin of S MIF, processes in the biogeochemical sulfur cycle modify the primary signal and strongly influence the S MIF preserved and observed in the geological record. Here, a detailed model of the marine sulfur cycle is used to propagate and distribute atmospherically derived S MIF from its delivery to the ocean to its preservation in the sediment. Bulk pyrite in most sediments carries weak S MIF because of microbial reduction of most sulfur compounds to form isotopically homogeneous sulfide. Locally, differential incorporation of sulfur compounds into pyrite leads to preservation of S MIF, which is predicted to be most highly variable in nonmarine and shallow-water settings. The Archean ocean is efficient in diluting primary atmospheric S MIF in the marine pools of sulfate and elemental sulfur with inputs from SO2 and H2S, respectively. Preservation of S MIF with the observed range of magnitudes requires the S MIF production mechanism to be moderately fractionating ( 20–40‰). Constraints from the marine sulfur cycle allow that either elemental sulfur or organosulfur compounds (or both) carried S MIF to the surface, with opposite sign to S MIF in SO2 and H2SO4. Optimal progress requires observations from nonmarine and shallow-water environments and experimental constraints on the reaction of photoexcited SO2 with atmospheric hydrocarbons.
20–40‰). Constraints from the marine sulfur cycle allow that either elemental sulfur or organosulfur compounds (or both) carried S MIF to the surface, with opposite sign to S MIF in SO2 and H2SO4. Optimal progress requires observations from nonmarine and shallow-water environments and experimental constraints on the reaction of photoexcited SO2 with atmospheric hydrocarbons.
With few exceptions, the enrichment or depletion of the rare, stable isotopes of sulfur (33S, 34S, and 36S) relative to the abundant isotope (32S) scale as the mass difference between the isotopes (1). Sulfur isotope mass-independent fractionation (S MIF) is defined as a departure from these theoretically derived and empirically observed mass laws, and denoted 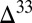 S and
S and  S. S MIF is observed in modern atmospheric sulfate aerosols, as well as in sulfate-bearing layers hosted in glacial ice (2, 3), but is conspicuously absent from the sedimentary record of the last 2,400 My. In contrast, older rocks of the Archean and early Paleoproterozoic eons preserve large and variable S MIF (4, 5). On the basis of experimental SO2 photolysis and atmospheric chemistry models, the prevailing hypothesis to explain this observation is that the absence of atmospheric oxygen before ∼2,400 Ma allowed both the photochemical production of S MIF and its delivery to the surface in the reduced and oxidized products of sulfur photochemistry (4–7). Thus, S MIF is considered strong geochemical evidence for an anoxic atmosphere before ∼2,400 Ma.
S. S MIF is observed in modern atmospheric sulfate aerosols, as well as in sulfate-bearing layers hosted in glacial ice (2, 3), but is conspicuously absent from the sedimentary record of the last 2,400 My. In contrast, older rocks of the Archean and early Paleoproterozoic eons preserve large and variable S MIF (4, 5). On the basis of experimental SO2 photolysis and atmospheric chemistry models, the prevailing hypothesis to explain this observation is that the absence of atmospheric oxygen before ∼2,400 Ma allowed both the photochemical production of S MIF and its delivery to the surface in the reduced and oxidized products of sulfur photochemistry (4–7). Thus, S MIF is considered strong geochemical evidence for an anoxic atmosphere before ∼2,400 Ma.
Although research converges on atmospheric processes as the source of S MIF, its production mechanism and the identity of its vectors to the surface remain unclear. A focus on photolysis experiments and measurements of SO2 isotopologue (molecules of 3xSO2 with x = 2, 3, 4, 6) absorption cross-sections has led to significant progress in understanding the origin of S MIF (8–12). However, attempts to relate experimental results to geologic observations, or to use the observations to inform the experimental search for a mechanism, are unavoidably compounded by the myriad of oceanographic and sedimentologic processes that operate in the sulfur cycle (13). These processes control the degree of postatmospheric mixing among the various S MIF carriers and ultimately the magnitude and spatial distribution of the observable signal. Many of these processes may be quantified using independent knowledge of the sulfur cycle and low-temperature geochemistry. This allows a more complete and more refined use of the information encoded in the observable record, with the objective of better understanding the nature of the atmospheric signal. In this paper, a detailed, spatially resolved model for the coupled biogeochemical cycles of iron and sulfur is developed and used to constrain processes in the Archean sulfur cycle, the identity of atmospheric S MIF carriers, and the sign and magnitude of 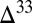 S in these carriers.
S in these carriers.
Existing Constraints
Systematic sampling of the Archean and Proterozoic rock record has shown that 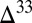 S in pyrite spans a range of approximately
S in pyrite spans a range of approximately 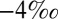 to
to 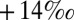 (4, 14–16), that barite (BaSO4 and, by inference, marine sulfate) carries
(4, 14–16), that barite (BaSO4 and, by inference, marine sulfate) carries 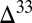 S values around
S values around 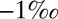 (4, 16, 17), and that the data form a loose array of
(4, 16, 17), and that the data form a loose array of 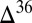 S against
S against 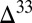 S with a slope of approximately
S with a slope of approximately 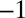 (14). Sulfide minerals with low
(14). Sulfide minerals with low 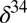 S and some in Archean volcanogenic massive sulfide deposits also carry weak
S and some in Archean volcanogenic massive sulfide deposits also carry weak 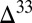 S
S  0 (7, 18, 19). Both are interpreted to have originated from seawater sulfate, the former through microbial reduction and the latter through mixing with volcanogenic sulfur, supporting
0 (7, 18, 19). Both are interpreted to have originated from seawater sulfate, the former through microbial reduction and the latter through mixing with volcanogenic sulfur, supporting 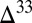 S
S  0 in seawater sulfate. Sulfur isotope analyses of carbonate-associated sulfate (CAS) suggest instead that marine sulfate carried
0 in seawater sulfate. Sulfur isotope analyses of carbonate-associated sulfate (CAS) suggest instead that marine sulfate carried  S
S  0 (20). However, carbonates that precipitated out of the sulfate-poor Archean ocean contain little sulfate, requiring dissolution of large volumes of rock for analysis and making it difficult to rule out contamination by trace pyrite or organic sulfur. Improvements in small-sample analysis promise to overcome these difficulties (21).
0 (20). However, carbonates that precipitated out of the sulfate-poor Archean ocean contain little sulfate, requiring dissolution of large volumes of rock for analysis and making it difficult to rule out contamination by trace pyrite or organic sulfur. Improvements in small-sample analysis promise to overcome these difficulties (21).
Experimental UV photolysis of SO2 was found to produce large S MIF in H2SO4, in cyclic octa-atomic elemental sulfur (S8), and in the residual SO2 (4, 8–10). The magnitudes of 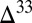 S and
S and 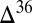 S, as well as the
S, as well as the 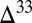 S–
S–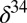 S and
S and  S–
S–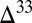 S relationships vary with wavelength, with the photolysis column optical thickness, and with bath gas composition and pressure. Once produced, atmospheric models indicate that S MIF would reach the surface in the oxidized and reduced products of sulfur photochemistry only if atmospheric oxygen was below
S relationships vary with wavelength, with the photolysis column optical thickness, and with bath gas composition and pressure. Once produced, atmospheric models indicate that S MIF would reach the surface in the oxidized and reduced products of sulfur photochemistry only if atmospheric oxygen was below 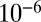 to
to 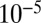 present atmospheric levels, depending on the trace gas composition of the atmosphere (6, 7, 22, 23). Thermal sulfate reduction has been suggested as the source of Archean S MIF (24), but recent experiments yield only anomalous fractionation of 33S (
present atmospheric levels, depending on the trace gas composition of the atmosphere (6, 7, 22, 23). Thermal sulfate reduction has been suggested as the source of Archean S MIF (24), but recent experiments yield only anomalous fractionation of 33S (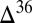 S
S  0), suggesting a magnetic isotope effect and in contrast to Archean S MIF (25).
0), suggesting a magnetic isotope effect and in contrast to Archean S MIF (25).
The emerging picture is of photochemical pathways, which persisted over hundreds of millions of years, generating 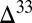 S
S  0 (
0 (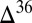 S
S  0) in atmospheric SO2 and H2SO4 and
0) in atmospheric SO2 and H2SO4 and  S
S  0 (
0 (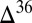 S
S  0) in other products of sulfur photochemistry, taken to be S8. Characteristic
0) in other products of sulfur photochemistry, taken to be S8. Characteristic 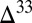 S–
S–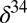 S and
S and 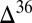 S–
S–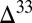 S relationships are thought to provide a fingerprint of these photochemical pathways, with modification of the former, but not the latter, by subsequent biogeochemical sulfur cycling (7).
S relationships are thought to provide a fingerprint of these photochemical pathways, with modification of the former, but not the latter, by subsequent biogeochemical sulfur cycling (7).
S MIF Production Mechanism
The mechanism by which S MIF was generated remains unclear. Explanations divide broadly into two categories: effects related to different photoexcitation probabilities of the SO2 isotopologues, and kinetic isotope effects associated with the relaxation or ultimate dissociation of photoexcited SO2. Effects in the latter category have yet to be discussed in the literature for SO2, but may be analogous to isotope-dependent relaxation rates of excited ozone or of state-to-state transformations of carbon monoxide, which lead to mass-independent oxygen isotope effects (26). Effects in the former category stem from subtle differences in the absorption cross-sections of the SO2 isotopologues. Self-shielding occurs when the atmosphere is optically thick in one or more of the isotopologues. The radiation suitable for the photolysis of the abundant isotopologue is absorbed at altitude, whereas the radiation suitable for photolysis of the rare isotopologues penetrates deeper into the atmosphere. The proportions of sulfur isotopes in the photoproducts and residual SO2 then deviate from mass dependence (27, 28). Calculations with absorption cross-sections at the highest currently available spectral resolution indicate that a significant component of S MIF generated in this way is inconsistent with geological observations (12). A second effect related to differences in absorption spectra occurs at any optical density of SO2. Here, the subtle differences among the cross-sections lead to higher (lower) rates of photo-excitation of one or more of the isotopologues, depending on the spectrum of incident radiation (11–13, 29). The photoproducts then are mass-independently enriched (depleted) in this isotopologue relative to the residual SO2.
The identity of the S MIF carriers other than SO2 and H2SO4 also is uncertain. Photolysis experiments that produced S8 involved high concentrations of SO2, which favor oligomerization of elemental sulfur to form S8 rings. Recent experiments with realistic abundances of SO2 do not produce S8, but in the presence of realistic methane concentrations, do generate methanesulfonic acid (MSA; CH3SO3H) (30). Experiments with other atmospherically relevant hydrocarbons generate additional organosulfur molecules (31). Unlike S8, MSA is generated not from the UV dissociation of SO2, but from the reaction of photoexcited SO2 with methane. Calculations convolving SO2 isotopologue absorption cross-sections with the solar spectrum at wavelengths that cause photoexcitation (λ
 240 nm) yield S MIF with a
240 nm) yield S MIF with a 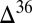 S–
S–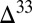 S relationship consistent with the Archean record, but with
S relationship consistent with the Archean record, but with 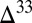 S
S  0 in the residual SO2 and
0 in the residual SO2 and 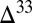 S
S  0 in the photoexcitation products (32). A third possibility is that H2SO4 and SO2 carried opposite signs of S MIF. Atmospheric H2SO4 formed from direct oxidation of SO2 would carry the same sign S MIF as the SO2, but H2SO4 formed from photoexcited SO2 would carry S MIF of the opposite sign.
0 in the photoexcitation products (32). A third possibility is that H2SO4 and SO2 carried opposite signs of S MIF. Atmospheric H2SO4 formed from direct oxidation of SO2 would carry the same sign S MIF as the SO2, but H2SO4 formed from photoexcited SO2 would carry S MIF of the opposite sign.
Model of the Archean Sulfur Cycle
Independent constraints on the carriers of S MIF and on the characteristics of S MIF in those carriers might valuably inform theoretical and experimental work. Given current uncertainty, the approach here is not to directly calculate S MIF generation, but to prescribe S MIF-bearing fluxes from the atmosphere to the ocean and to model in detail the marine processes that distribute, mix, and ultimately bury the isotopic anomaly in seafloor sediments. The premise of the approach is that with the marine biogeochemistry of the various S MIF carriers accounted for in the model, prescribed atmospheric fluxes of these carriers will have diagnostic outcomes in the preserved magnitude, sign, and spatial distribution of S MIF.
The model is described in detail in the Materials and Methods, SI Materials and Methods, and Tables S1–S5. The major ocean boxes (Fig. 1) were chosen to capture large-scale effects of global, density-driven circulation on ocean chemistry. Additional boxes represent specific physical environments, which are sampled by observations (e.g., continental shelf sediments, hydrothermal mounds). S MIF in these environments is expected to reflect the local sources of sulfur and the locally dominant biogeochemical processes. The model tracks the concentrations of seven sulfur species: sulfate 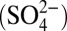 , sulfite
, sulfite  , polythionates
, polythionates 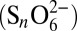 , thiosulfate
, thiosulfate 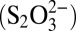 , elemental sulfur particles (S8), sulfide (
, elemental sulfur particles (S8), sulfide (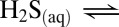
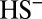 ), and organic sulfur (MSA and similar molecules); and three iron species: dissolved
), and organic sulfur (MSA and similar molecules); and three iron species: dissolved 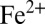 and
and 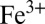 (and their complexes in seawater) and particulate Fe(OH)3. The concentrations of polysulfides
(and their complexes in seawater) and particulate Fe(OH)3. The concentrations of polysulfides 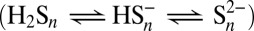 are not tracked dynamically but calculated in equilibrium with sulfide and S8. Coupled, nonlinear mass-balance equations for the 10 model species in each box include natural source terms (rivers, atmospheric deposition, hydrothermal activity, etc.), ultimate sinks (mineral precipitation, loss to sediments and hydrothermal circulation), transport between model boxes (advection, particle settling, etc.), and biogeochemical transfer of material among the various species (chemical reaction, biological utilization, etc.). The preservation of S MIF suggests that water-column biological activity did not cycle sulfur between its oxidation states enough to strongly attenuate atmospherically generated S MIF (13). Therefore, here biological sulfur cycling is taken to occur within the sediments.
are not tracked dynamically but calculated in equilibrium with sulfide and S8. Coupled, nonlinear mass-balance equations for the 10 model species in each box include natural source terms (rivers, atmospheric deposition, hydrothermal activity, etc.), ultimate sinks (mineral precipitation, loss to sediments and hydrothermal circulation), transport between model boxes (advection, particle settling, etc.), and biogeochemical transfer of material among the various species (chemical reaction, biological utilization, etc.). The preservation of S MIF suggests that water-column biological activity did not cycle sulfur between its oxidation states enough to strongly attenuate atmospherically generated S MIF (13). Therefore, here biological sulfur cycling is taken to occur within the sediments.
Fig. 1.

Model geometry. Seawater boxes are white and sediment boxes are gray. Dotted black arrows denote atmospheric fluxes. Thin black bidirectional arrows denote diffuse exchange between model boxes. Thicker colored arrows denote directional flow (blue, thermohaline circulation; red, coastal upwelling; orange, riverine influx and evaporative outflux). Values of geometric and transport parameters are in Tables S1 and S2.
The atmospheric influxes to the surface boxes are based on the results of photochemical modeling studies (6, 7, 22). Proportions of atmospherically deposited species vary among the models (Table 1) as a result of differences in the trace-gas composition of the atmosphere, the oxidation state of volcanic emissions, and the included chemical reactions and their rates. As discussed below, this leads to variability both in the concentrations of marine sulfur-bearing compounds and in their S MIF composition. Using atmospheric fluxes from the photochemical models and the dynamics described above, the equations are solved numerically for the steady-state concentrations of the model species. The steady-state concentrations and fluxes, together with specified values of 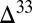 S in the atmospheric influxes, then are used to solve for the S MIF composition of the marine and sedimentary reservoirs. Empirical knowledge and isotopic mass balance guide the values of
S in the atmospheric influxes, then are used to solve for the S MIF composition of the marine and sedimentary reservoirs. Empirical knowledge and isotopic mass balance guide the values of 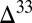 S carried in the atmospheric vectors (Fig. 2 and Materials and Methods).
S carried in the atmospheric vectors (Fig. 2 and Materials and Methods).
Table 1.
Approximate proportions of atmospheric species in total flux to the surface
| Model | H2SO4 | SO2 | S8 | H2S* | Sorg |
| (A) Pavlov and Kasting (6) | 0.07 | 0.24 | 0.33 | 0.36 | 0.00 |
| (B) Ono et al. (7) | 0.16 | 0.11 | 0.63 | 0.10 | 0.00 |
| (C) Zahnle et al. (22) | 0.56 | 0.18 | 0.26 | 0.00 | 0.00 |
| (D) Sorg | 0.05 | 0.45 | 0.00 | 0.45 | 0.05 |
*Including HS.
Fig. 2.

Conceptual model for distribution of S MIF in products of sulfur photochemistry. Arrows may represent complex, multistep reaction pathways rather than single reactions. S MIF in the photoproducts and in the residual SO2 are shown in red. At wavelengths less than ∼220 nm, SO2 dissociates and photoproducts are fractionated by a factor, ED. Photoexcitation at wavelengths greater than ∼260 nm and subsequent reaction of the excited SO2 may lead to a variety of final products. These photoproducts are fractionated by a different factor, E*. SO3 (and subsequently H2SO4) may form by direct oxidation of SO2, in which case the oxidation products will carry S MIF similar to the SO2. The fractions of H2SO4 and S8 formed by reaction of photoexcited SO2 are denoted 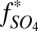 and
and 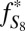 , respectively.
, respectively.
Results and Discussion
Steady-state concentrations (Fig. 3) and isotopic compositions (Fig. 4) of the marine species are presented for four cases. In cases A through C, inorganic sulfur compounds (S8 or H2SO4) are the S MIF carrier (in addition to SO2). In case D, organic sulfur compounds (Sorg) carry S MIF, and this is the only case in which marine concentrations of Sorg are nonzero. Cases A through C cover a range of atmospheric deposition fluxes from photochemical models (Table 1).
Fig. 3.

Sulfur species concentrations (bars) and their proportions in fluxes to the sediment (pie charts). Sulfite, polythionates, and thiosulfate, which derive almost exclusively from deposited atmospheric SO2 and exchange sulfur rapidly with one another, carry identical S MIF and therefore are plotted together. Their combined concentration is dominated by thiosulfate, with the other two species at least four orders of magnitude less abundant. The vertical extent of the bars reflects the range of concentrations obtained with different atmospheric deposition fluxes (Table 1, cases A–D) and with variable values for uncertain reaction rate constants (see text). Labels on the leftmost pie charts correspond to the model cases (B, fluxes from ref. 7; C, fluxes from ref. 22; D,  carrier with fluxes in Table 1).
carrier with fluxes in Table 1).
Fig. 4.

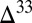 S in marine sulfur species for five scenarios (S8 carrier in cases B and C, H2SO4 carrier in cases B and C, and
S in marine sulfur species for five scenarios (S8 carrier in cases B and C, H2SO4 carrier in cases B and C, and 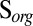 carrier in case D) in the deep-ocean and estuarine boxes. Values of
carrier in case D) in the deep-ocean and estuarine boxes. Values of 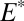 ,
,  ,
, 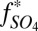 , and
, and 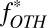 are below each of the scenario
are below each of the scenario 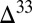 S results. The shaded region brackets the observed range of Archean
S results. The shaded region brackets the observed range of Archean 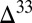 S.
S.
Sulfur Chemistry in the Archean Ocean.
Species’ concentrations are high near their sources and low near their sinks. For example, sulfide concentrations are higher in the sediments, where sulfide is produced by microbial reduction of more oxidized forms of sulfur, and lower in surface boxes, where sulfide is scavenged by photooxidation. Sulfur exits the ocean primarily as pyrite, with sulfate evaporite and barite formation accounting for 5–9% of outfluxes, consistent with suggestions that pyrite burial accounted for essentially all the sulfur exiting the oceans over much of the Precambrian (33).
A qualitative difference between the seawater and sediment boxes arises as the result of microbial activity within the sediments. In seawater boxes, several species reach high concentrations, notably sulfate (∼20–60 μM), thiosulfate (∼2–10 μM), and Sorg in case D (∼20 μM). These concentrations are in agreement with recent estimates of Archean seawater sulfate concentrations from multiple sulfur isotopes in volcanogenic massive sulfide deposits (19). By contrast, concentrations of sulfite, polythionates, sulfide, and S8 are subnanomolar. Despite low concentrations, some of these species react rapidly with the more abundant species, facilitating isotopic exchange between sulfur compounds of different oxidation states (e.g., polythionate lengthening and shortening reactions involve exchange of sulfur among polythionates, thiosulfate, and sulfite). Although atmospheric fluxes to the ocean contain large proportions of SO2, S8, and H2S, the total concentration of sulfur species in the ocean is heavily dominated by sulfate and thiosulfate. The reason for this is that the atmospherically deposited species react or settle rapidly once in the ocean. Sulfate and thiosulfate, on the other hand, are produced rapidly from less stable species, but are only slowly removed by microbial reduction within the sediments (SI Materials and Methods). This concentration of sulfoxy anions (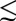 70 μM) is high enough that isotopic fractionation associated with microbial reduction should be large, as suggested by sulfur isotope ratios in Lake Matano, where sulfate concentrations in surface waters are
70 μM) is high enough that isotopic fractionation associated with microbial reduction should be large, as suggested by sulfur isotope ratios in Lake Matano, where sulfate concentrations in surface waters are  25 μM yet large fractionations are observed (21). However, the geologic record of
25 μM yet large fractionations are observed (21). However, the geologic record of 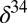 S in bulk pyrite shows clear evidence for strong fractionation of sulfur isotopes between sulfate and sulfide starting only ∼2,500 Ma (14). The model suggests that this is not because low sulfate levels limited isotopic fractionation (34), but because quantitative reduction of oxidized sulfur species followed by rapid pyrite formation prevented influence of microbial reduction on the isotopic composition of seawater sulfate. This is consistent with large variability in
S in bulk pyrite shows clear evidence for strong fractionation of sulfur isotopes between sulfate and sulfide starting only ∼2,500 Ma (14). The model suggests that this is not because low sulfate levels limited isotopic fractionation (34), but because quantitative reduction of oxidized sulfur species followed by rapid pyrite formation prevented influence of microbial reduction on the isotopic composition of seawater sulfate. This is consistent with large variability in 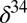 S within single samples, indicating that microbial reduction resulted in strong fractionation as early as 3,450 Ma (35).
S within single samples, indicating that microbial reduction resulted in strong fractionation as early as 3,450 Ma (35).
Depending on the proportion of H2SO4 in atmospheric fluxes to the ocean, up to 80% of the sulfate in the ocean is produced by oxidation and disproportionation of aqueous sulfite species, and essentially all the thiosulfate is produced by sulfite disproportionation. The model, therefore, is sensitive to the rate of this reaction, which is unknown at environmental temperatures, as discussed below. When H2S proportions in fluxes from the atmosphere are high, another reaction of importance is aqueous-phase photooxidation of H2S to form S8, which may replace atmospheric deposition as the main source of marine S8. In this case, the S8 concentration is governed by a balance between H2S photooxidation and settling of S8 particles. Importantly, S MIF in the marine S8 pool is variably diluted relative to S MIF in atmospheric S8 by contributions from H2S, which carries no S MIF.
Fluxes of sulfur to the sediment are dominated by sulfate, thiosulfate, and particles of S8 (Fig. 3 pie charts). The proportions of these species in the flux to the sediment show a dependence on their proportions in atmospheric deposition fluxes, but are variably altered by their production and consumption in the ocean. For example, the proportion of S8 in fluxes to the sediment is high even when its atmospheric deposition is modest as the result of S8 production in the water column. In the sediment boxes, microbial reduction generates sulfide (∼10–60 μM) and depresses the concentration of sulfur oxyanions and Sorg. Relatively high concentrations of S8 (∼0.01–5 μM) are maintained in the sediments by rapid settling of particles and by partial reoxidation of sulfide by  particles. Rapid reaction between H2S and dissolved S8 generates polysulfides, which promote isotopic mixing between these sulfur pools, although mixing with S8 may be limited by the kinetics of S8 particle dissolution. The confluence of sulfur from all oxidation states into porewater sulfide affects the minor isotopic composition of pyrite but not of the ocean, because pyrite formation rapidly scavenges most of the reduced sulfur.
particles. Rapid reaction between H2S and dissolved S8 generates polysulfides, which promote isotopic mixing between these sulfur pools, although mixing with S8 may be limited by the kinetics of S8 particle dissolution. The confluence of sulfur from all oxidation states into porewater sulfide affects the minor isotopic composition of pyrite but not of the ocean, because pyrite formation rapidly scavenges most of the reduced sulfur.
Controls on Preserved S MIF.
Microbial reduction of all sulfur species to sulfide in the sediments leads to near-zero 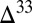 S in bulk pyrite. However, the species that together contribute to this value each carry appreciable S MIF (Fig. 4). If pyrite precipitation rapidly follows microbial production of sulfide, then S MIF in the different sulfur species may avoid homogenization and get preserved. This is in agreement with observations of highly variable
S in bulk pyrite. However, the species that together contribute to this value each carry appreciable S MIF (Fig. 4). If pyrite precipitation rapidly follows microbial production of sulfide, then S MIF in the different sulfur species may avoid homogenization and get preserved. This is in agreement with observations of highly variable 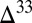 S in finely disseminated pyrite and relatively constant
S in finely disseminated pyrite and relatively constant 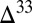 S in co-occurring nodular pyrite (18), with the former possibly representing a small-scale contribution from single sulfur species and the latter representing the bulk sulfide produced by microbial reduction. Correlation between iron content and
S in co-occurring nodular pyrite (18), with the former possibly representing a small-scale contribution from single sulfur species and the latter representing the bulk sulfide produced by microbial reduction. Correlation between iron content and  S magnitude in some rocks (36) also supports the idea that rapid pyrite formation relative to microbial sulfide production favors the preservation of a range of S MIF values. Locally within the sediment, therefore, the full range of S MIF compositions in the species delivered from the water column may get preserved and may account for the large S MIF variability in Archean sulfide minerals. Furthermore, observations of low
S magnitude in some rocks (36) also supports the idea that rapid pyrite formation relative to microbial sulfide production favors the preservation of a range of S MIF values. Locally within the sediment, therefore, the full range of S MIF compositions in the species delivered from the water column may get preserved and may account for the large S MIF variability in Archean sulfide minerals. Furthermore, observations of low 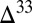 S magnitudes in Mesoarchean rocks (37) may be the result of local homogenization of S MIF in the sampled paleoenvironments rather than decreased S MIF production, as also suggested by recent observations, which significantly stretch the envelope of Mesoarchean S MIF (16).
S magnitudes in Mesoarchean rocks (37) may be the result of local homogenization of S MIF in the sampled paleoenvironments rather than decreased S MIF production, as also suggested by recent observations, which significantly stretch the envelope of Mesoarchean S MIF (16).
The model predicts differences in sulfur species fluxes to the various sedimentary environments (Fig. 3, pie charts), with the implication that the sign and range of 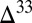 S preserved in sedimentary pyrite should vary among these environments. For example, S8 particles comprise more than 90% of the sulfur delivered to deep ocean sediments. Pyrite formed in deep-water sediments therefore likely preserves a relatively narrow range of
S preserved in sedimentary pyrite should vary among these environments. For example, S8 particles comprise more than 90% of the sulfur delivered to deep ocean sediments. Pyrite formed in deep-water sediments therefore likely preserves a relatively narrow range of 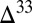 S values, similar to those in marine S8. This is consistent with
S values, similar to those in marine S8. This is consistent with 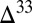 S
S  0 in fluvial and proximal marine facies and
0 in fluvial and proximal marine facies and 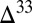 S
S  0 in distal marine facies of a Mesoarchean foreland basin (Witwatersrand Supergroup, South Africa) (15). The model prediction, that S8 should be the predominant component of the flux to the sediment in the deep ocean, is consistent with the view that S8 carried positive
0 in distal marine facies of a Mesoarchean foreland basin (Witwatersrand Supergroup, South Africa) (15). The model prediction, that S8 should be the predominant component of the flux to the sediment in the deep ocean, is consistent with the view that S8 carried positive 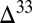 S to these distal settings. In contrast, in shallow-water settings (continental shelves and estuaries), the fractions of sulfate, thiosulfate, S8, and Sorg in fluxes to the sediment are of comparable magnitude, and the potential exists for local preservation of a larger range of
S to these distal settings. In contrast, in shallow-water settings (continental shelves and estuaries), the fractions of sulfate, thiosulfate, S8, and Sorg in fluxes to the sediment are of comparable magnitude, and the potential exists for local preservation of a larger range of 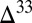 S values. This is consistent with the largest observed range of S MIF in shallow-water volcaniclastic units of the Paleoarchean Fig Tree Group (16), and with observations of larger
S values. This is consistent with the largest observed range of S MIF in shallow-water volcaniclastic units of the Paleoarchean Fig Tree Group (16), and with observations of larger 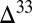 S magnitude and variability in the more proximal of two drill cores into the Neoarchean Transvaal Supergroup in South Africa (36).
S magnitude and variability in the more proximal of two drill cores into the Neoarchean Transvaal Supergroup in South Africa (36).
Sulfate minerals, in contrast to pyrite, form exclusively from the marine sulfate pool, which the model suggests is large enough to be chemically and isotopically well mixed throughout the ocean (Fig. 3). This may be the reason for the relatively homogeneous S MIF in Archean barite (17). However, S MIF in barite or CAS should not be interpreted as directly representing atmospheric H2SO4, because marine sulfate carries a mixture of S MIF signatures from the atmospheric fluxes of both H2SO4 and SO2 (through aqueous oxidation and disproportionation of sulfite). Depending on the S MIF production mechanism (Fig. 2), these two signatures may or may not be similar. This uncertainty may be resolved by future observations of S MIF preserved in nonmarine environments, which more likely represent unaltered atmospheric signatures.
A small number of aqueous reactions turn out to govern the concentrations and 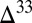 S of the sulfur species, but some of the rates of these reactions are poorly constrained. Notable among these is sulfite disproportionation to form sulfate and thiosulfate (38, 39). The rate of this reaction is not well known at seawater temperatures but is critical in determining the concentration of marine sulfoxy anions and in delivering S MIF from atmospheric SO2 to these sulfur species. A second example is the rate of thiosulfate acid decomposition to form sulfite and S8. The pH-dependent reaction rate has been determined experimentally around a pH of 2 (40), much lower than that expected in the Archean ocean (∼7; SI Materials and Methods). If the pH dependence determined under acidic conditions is valid at neutral pH, then this reaction provides an avenue for spreading S MIF from atmospheric SO2 to thiosulfate and from it to S8, thereby mixing
S of the sulfur species, but some of the rates of these reactions are poorly constrained. Notable among these is sulfite disproportionation to form sulfate and thiosulfate (38, 39). The rate of this reaction is not well known at seawater temperatures but is critical in determining the concentration of marine sulfoxy anions and in delivering S MIF from atmospheric SO2 to these sulfur species. A second example is the rate of thiosulfate acid decomposition to form sulfite and S8. The pH-dependent reaction rate has been determined experimentally around a pH of 2 (40), much lower than that expected in the Archean ocean (∼7; SI Materials and Methods). If the pH dependence determined under acidic conditions is valid at neutral pH, then this reaction provides an avenue for spreading S MIF from atmospheric SO2 to thiosulfate and from it to S8, thereby mixing 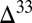 S of opposite signs.
S of opposite signs.
Identity of S MIF Carriers.
With variable values of the S MIF factors (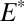 and
and 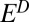 ) and the fractions of atmospheric H2SO4 and Sorg generated from photoexcited SO2 (
) and the fractions of atmospheric H2SO4 and Sorg generated from photoexcited SO2 (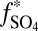 and
and 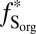 ), the model can preserve S MIF with the full range of
), the model can preserve S MIF with the full range of 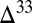 S observed in Archean pyrite. This is possible when S MIF in SO2 is balanced primarily by S MIF of opposite sign in H2SO4, S8, or Sorg. Several successful parameter combinations are shown in Fig. 4 for cases B through D. All these scenarios are plausible in terms of the required values of
S observed in Archean pyrite. This is possible when S MIF in SO2 is balanced primarily by S MIF of opposite sign in H2SO4, S8, or Sorg. Several successful parameter combinations are shown in Fig. 4 for cases B through D. All these scenarios are plausible in terms of the required values of 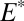 and
and 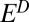 (
(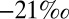 to
to 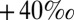 ), which are similar in approximate absolute magnitude to values obtained in experiments of broadband UV SO2 photolysis (9–11). Their plausibility as explanations for the Archean record of S MIF may be tested further by the values of
), which are similar in approximate absolute magnitude to values obtained in experiments of broadband UV SO2 photolysis (9–11). Their plausibility as explanations for the Archean record of S MIF may be tested further by the values of 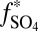 they require and by the sign of the
they require and by the sign of the 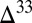 S in the marine reservoirs.
S in the marine reservoirs.
First, a possibility not previously suggested is that H2SO4 and SO2 were the only major S MIF carriers. Sulfate derived exclusively from oxidation of atmospheric and marine SO2 (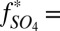 0) will have
0) will have 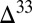 S similar in sign and magnitude to the SO2. In contrast, when atmospheric sulfate comes from reaction of photoexcited SO2 (
S similar in sign and magnitude to the SO2. In contrast, when atmospheric sulfate comes from reaction of photoexcited SO2 (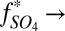 1),
1), 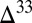 S in sulfate and SO2 diverge and may have opposite sign (e.g., Fig. 4, cases B and C with an H2SO4 carrier). In the ocean, sulfite oxidation and disproportionation deliver S MIF from SO2 to marine sulfate, but delivery from sulfate to sulfite is negligibly small. Thus, two distinct fluxes of S MIF of opposite sign may reach the sediment without the requirement for atmospheric production of S8 or Sorg. This possibility appears unlikely for two reasons. First, it requires high values of
S in sulfate and SO2 diverge and may have opposite sign (e.g., Fig. 4, cases B and C with an H2SO4 carrier). In the ocean, sulfite oxidation and disproportionation deliver S MIF from SO2 to marine sulfate, but delivery from sulfate to sulfite is negligibly small. Thus, two distinct fluxes of S MIF of opposite sign may reach the sediment without the requirement for atmospheric production of S8 or Sorg. This possibility appears unlikely for two reasons. First, it requires high values of 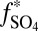 , which are unlikely because most H2SO4 in the Archean atmosphere would have formed by direct oxidation of SO2. Second, microbial reduction of both sulfate and sulfite–polythionate–thiosulfate within the sediments tend to homogenize and erase the isotopic anomaly.
, which are unlikely because most H2SO4 in the Archean atmosphere would have formed by direct oxidation of SO2. Second, microbial reduction of both sulfate and sulfite–polythionate–thiosulfate within the sediments tend to homogenize and erase the isotopic anomaly.
When S8 is the S MIF carrier, it delivers large  S to the sediments only when the proportions of both S8 and H2S in atmospheric deposition fluxes are small. The former arises from the requirement for isotopic mass balance in the chain of reactions that generated S MIF, and the latter arises from the requirement for minimal dilution by H2S photooxidation in the ocean. Case C satisfies both these requirements and displays large
S to the sediments only when the proportions of both S8 and H2S in atmospheric deposition fluxes are small. The former arises from the requirement for isotopic mass balance in the chain of reactions that generated S MIF, and the latter arises from the requirement for minimal dilution by H2S photooxidation in the ocean. Case C satisfies both these requirements and displays large 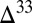 S
S  0 in S8 and small
0 in S8 and small 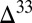 S
S  0 in the combined sulfoxy anion pool, in accordance with the prevailing view of S MIF delivery to the Archean ocean. Cases B and D fail to meet one of the requirements, resulting in the inability of S8 to support large S MIF magnitudes for reasonable values of
0 in the combined sulfoxy anion pool, in accordance with the prevailing view of S MIF delivery to the Archean ocean. Cases B and D fail to meet one of the requirements, resulting in the inability of S8 to support large S MIF magnitudes for reasonable values of 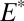 and
and 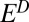 . In case B, the requirement for a small proportion of H2S in atmospheric deposition fluxes is satisfied, but most (63%) of the sulfur is delivered to the ocean as S8. Isotopic mass balance then requires atmospheric S8 to carry smaller
. In case B, the requirement for a small proportion of H2S in atmospheric deposition fluxes is satisfied, but most (63%) of the sulfur is delivered to the ocean as S8. Isotopic mass balance then requires atmospheric S8 to carry smaller 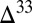 S than the residual SO2. Consequently, although case B displays
S than the residual SO2. Consequently, although case B displays 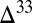 S values bracketing the observed range, large
S values bracketing the observed range, large 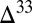 S is found in the combined sulfoxy anion pool, not in S8. In case D, the proportion of H2S in atmospheric deposition fluxes is much higher than that of S8, resulting in dilution of strong S MIF in atmospheric S8 by mass-dependent compositions in S8 generated by aqueous H2S photooxidation. In both cases B and D, if S8 carries
S is found in the combined sulfoxy anion pool, not in S8. In case D, the proportion of H2S in atmospheric deposition fluxes is much higher than that of S8, resulting in dilution of strong S MIF in atmospheric S8 by mass-dependent compositions in S8 generated by aqueous H2S photooxidation. In both cases B and D, if S8 carries 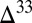 S
S  0, as in the prevailing view, the sense of asymmetry around
0, as in the prevailing view, the sense of asymmetry around  S of zero is reverse to that in the Archean record of S MIF.
S of zero is reverse to that in the Archean record of S MIF.
The sulfoxy anion pool is less susceptible to dilution of the S MIF signal, because production of these species from other aqueous sulfur compounds is negligible at low temperature and in the absence of dissolved oxygen. As such, this pool retains an undiluted 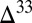 S value very close to the atmospheric input of SO2. In environments in which this pool contributes substantially to the flux to the sediments (e.g., estuaries in Fig. 3), the potential exists for preservation of S MIF very close to the primary atmospheric value. If, as in some atmospheric models (e.g., cases B and C), the fractional deposition of SO2 is small, then this SO2 will carry large magnitudes of
S value very close to the atmospheric input of SO2. In environments in which this pool contributes substantially to the flux to the sediments (e.g., estuaries in Fig. 3), the potential exists for preservation of S MIF very close to the primary atmospheric value. If, as in some atmospheric models (e.g., cases B and C), the fractional deposition of SO2 is small, then this SO2 will carry large magnitudes of 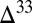 S to the surface, again as a result of the constraints of isotopic mass balance in the S MIF-forming reactions. Conversely, high fractional deposition of SO2 will lead to lower
S to the surface, again as a result of the constraints of isotopic mass balance in the S MIF-forming reactions. Conversely, high fractional deposition of SO2 will lead to lower 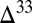 S magnitudes in this SO2. Thus, relatively small and negative
S magnitudes in this SO2. Thus, relatively small and negative 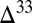 S in atmospheric SO2 (and H2SO4), as in the prevailing view, implies that oxidized sulfur compounds comprised a large fraction of the atmospheric flux to the surface.
S in atmospheric SO2 (and H2SO4), as in the prevailing view, implies that oxidized sulfur compounds comprised a large fraction of the atmospheric flux to the surface.
When organosulfur molecules carry S MIF, the most extreme values of 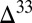 S are in the Sorg pool (Fig. 4, case D). This is because production of mass-dependent MSA by gas-phase and aqueous oxidation of more reduced organosulfur compounds, such as dimethyl sulfide (41), likely was minor in the oxidant-poor Archean ocean atmosphere. As a result, S MIF in MSA remains undiluted, and high positive
S are in the Sorg pool (Fig. 4, case D). This is because production of mass-dependent MSA by gas-phase and aqueous oxidation of more reduced organosulfur compounds, such as dimethyl sulfide (41), likely was minor in the oxidant-poor Archean ocean atmosphere. As a result, S MIF in MSA remains undiluted, and high positive  S, such as that observed in the late Archean, can be reached with modest broadband values of the S MIF factor
S, such as that observed in the late Archean, can be reached with modest broadband values of the S MIF factor 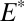 (∼13‰). However, the value of
(∼13‰). However, the value of  required to generate a second reservoir with smaller
required to generate a second reservoir with smaller 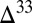 S of opposite sign also depends on the proportions of H2SO4, SO2, and Sorg in atmospheric deposition fluxes, as well as on the fractions of H2SO4 and Sorg generated by reaction of photoexcited SO2 (
S of opposite sign also depends on the proportions of H2SO4, SO2, and Sorg in atmospheric deposition fluxes, as well as on the fractions of H2SO4 and Sorg generated by reaction of photoexcited SO2 ( and
and 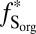 ). For the atmospheric fluxes specified in case D (Table 1), the value of
). For the atmospheric fluxes specified in case D (Table 1), the value of 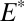 required to bracket the range of the late Archean
required to bracket the range of the late Archean 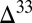 S record is ∼40‰. Propagation of S MIF in MSA and similar compounds to other marine sulfur reservoirs is expected to be minor because aqueous MSA is relatively unreactive, except with OH radicals (41), which likely were scarce in the Archean ocean. Therefore, dilution of the
S record is ∼40‰. Propagation of S MIF in MSA and similar compounds to other marine sulfur reservoirs is expected to be minor because aqueous MSA is relatively unreactive, except with OH radicals (41), which likely were scarce in the Archean ocean. Therefore, dilution of the  S of opposite sign in the sulfoxy anion pool is expected to be minor. Although no anaerobic bacteria are known to degrade MSA (42), this likely reflects an incomplete survey of existing microbial metabolisms and not the true absence of such organisms. Once in the sediments, MSA likely would undergo microbial reduction and end up in pyrite. A caveat to this statement, suggesting fertile ground for future research, is that anoxic aqueous chemistry of MSA and similar molecules is poorly characterized.
S of opposite sign in the sulfoxy anion pool is expected to be minor. Although no anaerobic bacteria are known to degrade MSA (42), this likely reflects an incomplete survey of existing microbial metabolisms and not the true absence of such organisms. Once in the sediments, MSA likely would undergo microbial reduction and end up in pyrite. A caveat to this statement, suggesting fertile ground for future research, is that anoxic aqueous chemistry of MSA and similar molecules is poorly characterized.
Conclusions
A model of the coupled sulfur and iron cycles constrains atmospheric S MIF production and delivery to the ocean. Either S8 or Sorg (or both) might have carried S MIF, generated by a moderately fractionating process (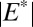 and
and 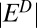 between 17‰ and 40‰). S MIF with an asymmetry similar to observations (strong positive and weak negative
between 17‰ and 40‰). S MIF with an asymmetry similar to observations (strong positive and weak negative 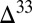 S) is preserved when (i)
S) is preserved when (i) 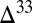 S
S  0 occurs in S8 and both the fractional fluxes of S8 and H2S from the atmosphere to the ocean are small, (ii)
0 occurs in S8 and both the fractional fluxes of S8 and H2S from the atmosphere to the ocean are small, (ii) 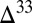 S
S  0 occurs in the combined sulfite–polythionate–thiosulfate pool (derived from atmospheric SO2) and
0 occurs in the combined sulfite–polythionate–thiosulfate pool (derived from atmospheric SO2) and 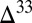 S
S  0 is in marine S8 (derived from atmospheric S8 with high negative
0 is in marine S8 (derived from atmospheric S8 with high negative 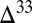 S and strongly diluted by inputs from H2S oxidation), and (iii)
S and strongly diluted by inputs from H2S oxidation), and (iii) 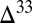 S
S  0 occurs in Sorg (derived from atmospheric MSA and similar molecules) and
0 occurs in Sorg (derived from atmospheric MSA and similar molecules) and 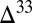 S
S  0 occurs in the combined sulfite–polythionate–thiosulfate pool. Options i and iii are consistent with existing observations of negative
0 occurs in the combined sulfite–polythionate–thiosulfate pool. Options i and iii are consistent with existing observations of negative 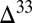 S in marine barite (17) and with a transition from negative
S in marine barite (17) and with a transition from negative 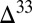 S to positive
S to positive  S accompanying a traverse from proximal to distal marine environments (15). As mentioned above, option i constrains a large fraction of atmospherically deposited sulfur to be in oxidized forms (SO2 and H2SO4).
S accompanying a traverse from proximal to distal marine environments (15). As mentioned above, option i constrains a large fraction of atmospherically deposited sulfur to be in oxidized forms (SO2 and H2SO4).
The model predicts a spatial dependence of S MIF variability due to differential incorporation of the various marine sulfur species, which is consistent with observations of the dependence of preserved S MIF on sedimentary facies (15, 36). To further constrain the characteristics of atmospheric S MIF and inform its production mechanism, future observations from nonmarine environments would be optimal. In marine rocks, the best-preserved primary atmospheric signal likely represents the combined sulfite–polythionate–thiosulfate pool or Sorg, both of which are undiluted by inputs from other marine sulfur species. Such a signal most likely will be found in pyrites from shallow-water environments hosting strong 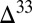 S gradients. Barite and carbonate-associated sulfate carry mixed S MIF, derived both from atmospheric H2SO4 and from oxidation and disproportionation of marine sulfite. These phases still may provide a good window into S MIF in atmospheric SO2, as most atmospheric H2SO4 and marine sulfate are derived from gas-phase and aqueous reaction of SO2 and should carry S MIF similar to SO2.
S gradients. Barite and carbonate-associated sulfate carry mixed S MIF, derived both from atmospheric H2SO4 and from oxidation and disproportionation of marine sulfite. These phases still may provide a good window into S MIF in atmospheric SO2, as most atmospheric H2SO4 and marine sulfate are derived from gas-phase and aqueous reaction of SO2 and should carry S MIF similar to SO2.
Materials and Methods
The geochemical model is described fully in SI Materials and Methods.
Described here is the parameterization of S MIF in atmospheric fluxes. Internally consistent fluxes of S MIF-bearing material were calculated on the basis of photolysis experiments, SO2 cross-section measurements, and isotopic mass balance (Fig. 2). Photodissociation of SO2 at wavelengths  220 nm was taken to generate products that are fractionated relative to the residual SO2 by an S MIF factor,
220 nm was taken to generate products that are fractionated relative to the residual SO2 by an S MIF factor,  . Oxidation of SO2 to SO3 and subsequent hydration do not generate additional S MIF, and the H2SO4 carries S MIF similar to the residual SO2. Photoexcitation of SO2 at wavelengths between 260 nm and 340 nm and the subsequent collision of photoexcited SO2 with other gases results in quenching or reaction. Possible reactions include oxidation to ultimately form H2SO4, reduction to ultimately form S, or collision with hydrocarbon molecules to form CH3SO3H and other organosulfur molecules. These products were taken to be fractionated relative to the residual SO2 by a second S MIF factor,
. Oxidation of SO2 to SO3 and subsequent hydration do not generate additional S MIF, and the H2SO4 carries S MIF similar to the residual SO2. Photoexcitation of SO2 at wavelengths between 260 nm and 340 nm and the subsequent collision of photoexcited SO2 with other gases results in quenching or reaction. Possible reactions include oxidation to ultimately form H2SO4, reduction to ultimately form S, or collision with hydrocarbon molecules to form CH3SO3H and other organosulfur molecules. These products were taken to be fractionated relative to the residual SO2 by a second S MIF factor, 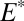 . The ultimate magnitude and sign of S MIF in the carriers depends on the relative proportions of the carriers generated by photodissociation and photoexcitation and by the factors
. The ultimate magnitude and sign of S MIF in the carriers depends on the relative proportions of the carriers generated by photodissociation and photoexcitation and by the factors 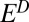 and
and  . Within this framework, isotopic mass balance dictates that the total flux of S MIF carriers to the surface has a weighted average S MIF of exactly zero:
. Within this framework, isotopic mass balance dictates that the total flux of S MIF carriers to the surface has a weighted average S MIF of exactly zero:
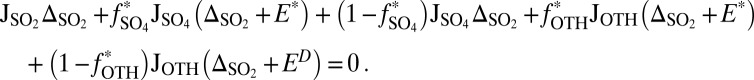 |
J denotes fluxes to the surface; 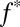 denotes the fraction of the flux from photoexcited SO2, with the rest produced by photodissociation in the case of S8 and by direct oxidation in the case of H2SO4;
denotes the fraction of the flux from photoexcited SO2, with the rest produced by photodissociation in the case of S8 and by direct oxidation in the case of H2SO4; 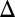 denotes mass-independent isotopic compositions; and the SO2, SO4, and OTH subscripts denote values associated with the fluxes of SO2, H2SO4, and the third carrier (S8 or S
denotes mass-independent isotopic compositions; and the SO2, SO4, and OTH subscripts denote values associated with the fluxes of SO2, H2SO4, and the third carrier (S8 or S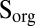 ), respectively. For S8,
), respectively. For S8, 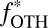 = 0.6% was calculated using SO2 absorption cross-section and the solar spectrum (SI Materials and Methods and Fig. S1). The remaining parameters (
= 0.6% was calculated using SO2 absorption cross-section and the solar spectrum (SI Materials and Methods and Fig. S1). The remaining parameters (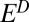 ,
, 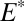 ,
,  ,
, 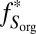 ) were varied over a range of values.
) were varied over a range of values.
Supplementary Material
Acknowledgments
Discussions with Woodward Fischer, David Johnston, and Alexey Kamyshny, as well as constructive reviews from Boswell Wing and an anonymous reviewer, improved this work. I.H. acknowledges support from Sir Charles Clore Prize for Outstanding Appointment in the Experimental Sciences at the Weizmann Institute of Science and from Israel Science Foundation Grant 1133/12.
Footnotes
The author declares no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1213148110/-/DCSupplemental.
References
- 1.Hulston JR, Thode HG. Variations in S33, S34, and S36 contents of meteorites and their relation to chemical and nuclear effects. J Geophys Res. 1965;70:3475–3484. [Google Scholar]
- 2.Romero AB, Thiemens MH. Mass-independent sulfur isotopic compositions in present-day sulfate aerosols. J Geophys Res. 2003;108:4524–4530. [Google Scholar]
- 3.Savarino J, Romero A, Cole-Dai J, Bekki S, Thiemens MH. UV induced mass-independent sulfur isotope fractionation in stratospheric volcanic sulfate. Geophys Res Lett. 2003;30:2131–2134. [Google Scholar]
- 4.Farquhar J, Bao H, Thiemens MH. Atmospheric influence of Earth’s earliest sulfur cycle. Science. 2000;289(5480):756–759. doi: 10.1126/science.289.5480.756. [DOI] [PubMed] [Google Scholar]
- 5.Farquhar J, Wing BA. Multiple sulfur isotopes and the evolution of the atmosphere. Earth Planet Sci Lett. 2003;213:1–13. [Google Scholar]
- 6.Pavlov AA, Kasting JF. Mass-independent fractionation of sulfur isotopes in Archean sediments: Strong evidence for an anoxic Archean atmosphere. Astrobiology. 2002;2(1):27–41. doi: 10.1089/153110702753621321. [DOI] [PubMed] [Google Scholar]
- 7.Ono S, et al. New insights into Archean sulfur cycle from mass-independent sulfur isotope records from the Hamersley Basin, Australia. Earth Planet Sci Lett. 2003;213:15–30. [Google Scholar]
- 8.Farquhar J, Savarino J, Airieau S, Thiemens MH. Observation of wavelength-sensitive mass-independent sulfur isotope effects during SO2 photolysis: Implications for the early atmosphere. J Geophys Res. 2001;106:32829–32839. [Google Scholar]
- 9.Masterson AL, Farquhar J, Wing BA. Sulfur mass-independent fractionation patterns in the broadband UV photolysis of sulfur dioxide: Pressure and third body effects. Earth Planet Sci Lett. 2011;306:253–260. [Google Scholar]
- 10.Whitehill AR, Ono S. Excitation band dependence of sulfur isotope mass-independent fractionation during photochemistry of sulfur dioxide using broadband light sources. Geochim Cosmochim Acta. 2012;94:238–253. [Google Scholar]
- 11.Danielache SO, Eskebjerg C, Johnson MS, Ueno Y, Yoshida N. High-precision spectroscopy of 32S, 33S, and 34S sulfur dioxide: Ultraviolet absorption cross sections and isotope effects. J Geophys Res. 2008;113:D17314. [Google Scholar]
- 12.Lyons J, Blackie D, Stark G, Pickering J. The origin of sulfur isotope mass-independent fractionation in Archean rocks. Mineral Mag. 2012;76:2044. (abstr) [Google Scholar]
- 13.Halevy I, Johnston DT, Schrag DP. Explaining the structure of the Archean mass-independent sulfur isotope record. Science. 2010;329(5988):204–207. doi: 10.1126/science.1190298. [DOI] [PubMed] [Google Scholar]
- 14.Johnston DT. Multiple sulfur isotopes and the evolution of Earth’s surface sulfur cycle. Earth Sci Rev. 2011;106:161–183. [Google Scholar]
- 15.Guy BM, et al. A multiple sulfur and organic carbon isotope record from non-conglomeratic sedimentary rocks of the Mesoarchean Witwatersrand Supergroup, South Africa. Precambrian Res. 2012;216–219:208–231. [Google Scholar]
- 16.Philippot P, van Zuilen M, Rollion-Bard C. Variations in atmospheric sulphur chemistry on early Earth linked to volcanic activity. Nat Geosci. 2012;5:668–674. [Google Scholar]
- 17.Roerdink DL, Mason PRD, Farquhar J, Reimer T. Multiple sulfur isotopes in Paleoarchean barites identify an important role for microbial sulfate reduction in the early marine environment. Earth Planet Sci Lett. 2012;331–332:177–186. [Google Scholar]
- 18.Ono S, Beukes NJ, Rumble D. Origin of two distinct multiple-sulfur isotope compositions of pyrite in the 2.5 Ga Klein Naute Formation, Griqualand West Basin, South Africa. Precambrian Res. 2009;169:48–57. [Google Scholar]
- 19.Jameison JW, Wing BA, Farquhar J, Hannington MD. Neoarchean seawater sulphate concentrations from sulphur isotopes in massive sulphide ore. Nat Geosci. 2013;6:61–64. [Google Scholar]
- 20.Guo Q, et al. Reconstructing Earth’s surface oxidation across the Archean-Proterozoic transition. Geology. 2009;37:399–402. [Google Scholar]
- 21.Paris G, et al. Profile of sulfate isotopic composition of Lake Matano, Indonesia. Mineral Mag. 2012;76:2204. (abstr) [Google Scholar]
- 22.Zahnle K, Claire M, Catling D. The loss of mass-independent fractionation is sulfur due to a Palaeoproterozoic collapse of atmospheric methane. Geobiology. 2006;4:271–283. [Google Scholar]
- 23.Domagal-Goldman SD, Kasting JF, Johnston DT, Farquhar J. Organic haze, glaciations and multiple sulfur isotopes in the Mid-Archean Era. Earth Planet Sci Lett. 2008;269:29–40. [Google Scholar]
- 24.Watanabe Y, Farquhar J, Ohmoto H. Anomalous fractionations of sulfur isotopes during thermochemical sulfate reduction. Science. 2009;324(5925):370–373. doi: 10.1126/science.1169289. [DOI] [PubMed] [Google Scholar]
- 25.Oduro H, et al. Evidence of magnetic isotope effects during thermochemical sulfate reduction. Proc Natl Acad Sci USA. 2011;108(43):17635–17638. doi: 10.1073/pnas.1108112108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thiemens MH, Chakraborty S, Dominguez G. The physical chemistry of mass-independent isotope effects and their observation in nature. Annu Rev Phys Chem. 2012;63:155–177. doi: 10.1146/annurev-physchem-032511-143657. [DOI] [PubMed] [Google Scholar]
- 27.Lyons JR. Mass-independent fractionation of sulfur isotopes by isotope-selective photodissociation of SO2. Geophys Res Lett. 2007;34:L22811. [Google Scholar]
- 28.Lyons JR. Atmospherically-derived mass-independent sulfur isotope signatures, and incorporation into sediments. Chem Geol. 2009;267:164–174. [Google Scholar]
- 29.Ueno Y, et al. Geological sulfur isotopes indicate elevated OCS in the Archean atmosphere, solving faint young sun paradox. Proc Natl Acad Sci USA. 2009;106(35):14784–14789. doi: 10.1073/pnas.0903518106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeWitt HL, et al. The formation of sulfate and elemental sulfur aerosols under varying laboratory conditions: implications for early earth. Astrobiology. 2010;10(8):773–781. doi: 10.1089/ast.2009.9455. [DOI] [PubMed] [Google Scholar]
- 31.Oduro H, Whitehill A, Farquhar J, Summons RE, Ono S. Origin of anomalous isotope effects in photo- and thermo-chemical reactions of organosulfur compounds. Mineral Mag. 2012;76:2180. (abstr) [Google Scholar]
- 32.Ueno Y, Danielache S, Endo Y, Johnson M, Yoshida N. Photodissociation origin of Archean S-MIF and dynamical sulfur cycling under highly reducing atmosphere. Mineral Mag. 2012;76:2475. (abstr) [Google Scholar]
- 33.Canfield DE. The evolution of the Earth surf ate sulfur reservoir. Am J Sci. 2004;304:839–861. [Google Scholar]
- 34.Habicht KS, Gade M, Thamdrup B, Berg P, Canfield DE. Calibration of sulfate levels in the archean ocean. Science. 2002;298(5602):2372–2374. doi: 10.1126/science.1078265. [DOI] [PubMed] [Google Scholar]
- 35.Bontognali TRR, et al. Sulfur isotopes of organic matter preserved in 3.45-billion-year-old stromatolites reveal microbial metabolism. Proc Natl Acad Sci USA. 2012;109(38):15146–15151. doi: 10.1073/pnas.1207491109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ono S, Kaufman AJ, Fraquhar J, Sumner DY, Beukes NJ. Lithofacies control on multiple-sulfur isotope records and Neoarchean sulfur cycles. Precambrian Res. 2009;169:58–67. [Google Scholar]
- 37.Farquhar J, et al. Isotopic evidence for Mesoarchaean anoxia and changing atmospheric sulphur chemistry. Nature. 2007;449(7163):706–709. doi: 10.1038/nature06202. [DOI] [PubMed] [Google Scholar]
- 38.Ryabinina AF, Oshman VA. Thermal decomposition of aqueous sulfur dioxide solutions. Tr Ural Lesotekh Inst. 1972;28:182–189. [Google Scholar]
- 39.Guekezian M, Coichev N, Suarez-Iha MEV, de Almeida-Neves E. Stability of sulfur(IV) solutions in the presence of amines and the tendency of sulfite solution to disproportionate in stock solutions. Anal Lett. 1997;30:1423–1436. [Google Scholar]
- 40.Johnston F, McAmish L. A study of the rated of sulfate production in acid thiosulfate solutions using S-35. J Colloid Interf Res. 1973;42:112–119. [Google Scholar]
- 41.Saltzman ES, Savoie DL, Zika RG, Prospero JM. Methane sulfonic acid in the marine atmosphere. J Geophys Res. 1983;108:10897–10902. [Google Scholar]
- 42.Kelly DP, Murrell JC. Microbial metabolism of methanesulfonic acid. Arch Microbiol. 1999;172(6):341–348. doi: 10.1007/s002030050770. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.