

Significance
The generation of thrombin by prothrombinase, a complex composed of activated (a) factors X (FXa) and V (FVa), is a final step in blood coagulation. We demonstrate that tissue factor pathway inhibitor (TFPI) blocks thrombin generation by prothrombinase at physiologically relevant rates and concentrations, but only during the initiation of clot formation. TFPI mediates this inhibitory activity through two high-affinity interactions, one with FXa and one with FVa. This is the first description of an endogenous human protein that inhibits prothrombinase under physiological conditions and may prevent a full thrombotic response to subthreshold coagulant stimuli that otherwise could occlude blood vessels. It provides unique understanding of thrombotic disorders and has important implications for development of anti-TFPI agents to treat hemophilia.
Keywords: hemophilia, bleeding, thrombosis
Abstract
Tissue factor (TF) pathway inhibitor (TFPI) is a well-characterized activated factor X (FXa)-dependent inhibitor of TF-initiated coagulation produced in two alternatively spliced isoforms, TFPIα and TFPIβ. The TFPIα C terminus has a basic sequence nearly identical to a portion of the factor V (FV) B domain necessary for maintaining FV in an inactive conformation via interaction with an acidic region of the B domain. We demonstrate rapid inhibition of prothrombinase by TFPIα mediated through a high-affinity exosite interaction between the basic region of TFPIα and the FV acidic region, which is retained in FXa-activated FVa and platelet FVa. This inhibitory activity is not mediated by TFPIβ and is lost upon removal of the acidic region of FVa by thrombin. The data identify a previously undescribed, isoform-specific anticoagulant function for TFPIα and are a unique description of physiologically relevant inhibition of prothrombinase. These findings, combined with previous descriptions of differential expression patterns of TFPIα and TFPIβ in platelets and endothelial cells, suggest that the TFPI isoforms may act through distinct mechanisms to inhibit the initial stages of intravascular coagulation, with TFPIβ acting to dampen TF expressed on the surface of vascular cells, whereas TFPIα dampens the initial prothrombinase formed on the activated platelet surface.
The hemostatic response to injury is initiated when extravascular tissue factor (TF) binds to activated (a) factor VII (FVIIa) in plasma, which then activates factor X (FXa). In the presence of calcium ions, FXa binds to the cofactor protein factor Va (FVa) on a membrane surface, such as that provided by activated platelets, to form the powerful procoagulant complex, prothrombinase, which rapidly converts prothrombin to thrombin (1, 2). In plasma clotting assays initiated by activation of either the intrinsic or extrinsic pathway, the initial thrombin generation is slow; however, amplification mechanisms, including thrombin-mediated activation of platelets and factor V (FV), eventually produce a rapid thrombin burst (3).
The hemostatic response to injury is also mediated by platelets, which are activated upon exposure to collagen and other components of the extracellular matrix. Following activation, platelet α-granules release a heterogeneous mixture of FVa, differing only in the amount of the B domain that has been proteolytically removed. These forms of FVa readily assemble into prothrombinase and promote thrombin generation at the platelet surface (4, 5).
TF-initiated coagulation is regulated by two alternatively spliced isoforms of tissue factor pathway inhibitor (TFPI). TFPIα contains three Kunitz-type inhibitory domains (K1–K3) and a highly basic C terminus (6). K1 and K2 bind and inhibit FVIIa and FXa, respectively, forming the TF–FVIIa–FXa–TFPI quaternary complex (7, 8). Protein S binds K3 and enhances inhibition of FXa by K2 (9, 10); however, the physiological contributions of K3 and the basic C terminus to TFPI anticoagulant activity remain unclear. TFPIα is present within platelets and is released upon activation (11, 12). It is also present within cultured endothelial cells and can be released by treatment with heparin or thrombin (13–15). TFPIα efficiently inhibits FXa from cleaving amidolytic substrates (6, 7, 16); however, it is a poor inhibitor of thrombin generation by FXa assembled into prothrombinase with thrombin-activated FVa (FVaIIa) (17, 18). In fact, an endogenous, physiologically active inhibitor of prothrombinase has not been previously described. Incorporation of FXa into prothrombinase protects it from plasma protease inhibitors, including the protein Z/protein Z-dependent protease inhibitor complex (19) and antithrombin, even in the presence of heparin (20). Similarly, the presence of FXa and prothrombin protects FVa from degradation by the activated protein C/protein S complex (21). The resistance of prothrombinase to inhibition by plasma protease inhibitors is so profound that it remains active in TF-treated whole blood for at least 100 min, much longer than is necessary to convert all plasma prothrombin to thrombin (22).
TFPIβ is the predominant isoform expressed on endothelium and contains the same K1 and K2 domains as TFPIα but lacks the K3 domain and basic C terminus (6). Instead, the C terminus of TFPIβ encodes a glycosylphosphatidylinositol (GPI) anchor that attaches it to the surface of vascular endothelial cells. Its localization to the cell surface renders TFPIβ a highly efficient anticoagulant protein (16) and provides a logical explanation for its expression as the primary TFPI isoform on endothelium of both mice and humans (23, 24). However, TFPIα is the only isoform expressed within the platelets of either species (11, 25) and is evolutionarily conserved back to zebrafish, suggesting that it has an anticoagulant function distinct from TFPIβ (24). This notion is supported by recent studies revealing that hematopoietic cell TFPI, which is primarily TFPIα, modifies thrombus growth following vascular injury (25) and bleeding in mice with hemophilia (26), as well as demonstration that degradation of platelet TFPIα by neutrophil proteases produces a procoagulant state in vivo in mice and in vitro using human cells (27). Here, we define a TFPIα-specific anticoagulant function: the rapid inhibition of prothrombinase assembled with platelet-released FVa or FVa generated through limited proteolysis by FXa, a unique description of inhibition of this powerful procoagulant complex by any endogenous protein at physiologically relevant rates and concentrations.
Results
Platelet TFPI Inhibits Thrombin Generation in a TF-Independent Manner.
Calibrated automated thrombography (CAT) experiments were performed to examine the inhibitory function of platelet TFPIα (Fig. 1A). Platelet-rich plasma (PRP) was prepared from blood anticoagulated with citrate and corn trypsin inhibitor, and coagulation was initiated by a mixture of collagen and FXa. Thrombin was not produced in reactions where FXa was omitted, demonstrating that TF–FVIIa did not initiate coagulation in these assays. In the presence of FXa, thrombin generation occurred. Blocking TFPI activity with monoclonal antibodies directed against the active site region of K2 (anti-K2), or against K3 and the two lysine residues immediately following it (anti-K3C), produced faster thrombin generation, decreasing the lag time by 9.6 ± 1.7 min or 5.8 ± 3.3 min, respectively, and the time to peak thrombin by 11.3 ± 1.4 min or 7.3 ± 2.9 min. Neither antibody altered peak thrombin nor total thrombin generated. A monoclonal antibody directed against the active site region of K1 (anti-K1) had no effect on thrombin generation, suggesting a TF–FVIIa-independent inhibitory function for TFPI in these assays. This was confirmed using an inhibitory polyclonal antibody against FVII (Fig. S1A), which did not influence thrombin generation in this system, either in the presence or absence of the TFPI antibodies (Fig. S1B).
Fig. 1.

TFPI inhibits thrombin generation in a TF-independent manner. (A) Platelet-rich plasma, anticoagulated with citrate and corn trypsin inhibitor, was incubated with collagen (0.1 mg/mL) and FXa (0.1 nM). Thrombin generation was initiated by addition of CaCl2 and a thrombin fluorogenic substrate and measured by calibrated automated thrombography. For some experiments, the incubation mixture contained anti-K1, -K2, or -K3C antibody (50 nM). As a control, experiments were also performed in the absence of FXa. Shown are the average chromatograms from three independent sets of experiments. (B and C) Washed platelets stimulated with collagen (0.1 mg/mL) (B) or collagen followed by thrombin (0.1 nM) (C) (1 × 108 per mL) were incubated with DAPA (3 µM) and, where indicated, anti-K1, -K2, or -K3C antibody (50 nM), TFPI-BR peptide (1 µM), and/or purified TFPIα (1 nM). Prothrombin (1.4 µM) and FXa (5 nM) were added, and the rate of prothrombin conversion to thrombin is reported as a percent of control (no additions) (mean ± SD, n = 3 platelet donors).
These data suggested that TFPI dampens initial thrombin generation on collagen-activated platelets through a previously undescribed mechanism that is dependent upon K2, K3, and/or the basic C terminus. Although TFPIα is a poor inhibitor of prothrombinase assembled with FVaIIa (17, 18), we considered the possibility that it inhibits prothrombinase assembled with FVa released from the α-granules of activated platelets. This hypothesis was evaluated by measuring prothrombinase activity in reactions using isolated, collagen-activated platelets and purified FXa and prothrombin. Consistent with the CAT data, the initial rate of thrombin generation by prothrombinase assembled with platelet FVa increased ∼2- to 2.5-fold in reactions performed in the presence of anti-K2 or anti-K3C antibody (Fig. 1B). Again anti-K1 did not alter thrombin generation, consistent with the presence of a TF–FVIIa-independent anticoagulant activity for TFPIα. The effects of anti-K2 and anti-K3C could be reversed by a peptide mimicking the TFPIα basic region (residues 240–265; TFPI-BR). Addition of TFPI-BR (1 µM) or TFPIα (1 nM) to collagen-activated platelets produced further inhibition of thrombin generation to ∼25% of the baseline prothrombinase activity. Finally, the inhibitory effect of platelet TFPIα was eliminated by pretreatment of the collagen-activated platelets with thrombin (Fig. 1C), consistent with the described inability of TFPIα to inhibit prothrombinase assembled with FVaIIa (17, 18).
The TFPIα Basic C Terminus Binds to an Acidic Region Within the FV B Domain.
Inhibition of platelet prothrombinase was mediated by both K2 and the TFPIα C terminus, which contains a 10-amino-acid stretch that is nearly identical to a basic region within the FV B domain. Both sequences are well conserved across mammalian species (Fig. 2). The sequence of Leu-Ile-Lys-Thr followed by at least five Lys and Arg residues is rare in the human proteome. A search using the National Center for Biotechnology Information (NCBI) BLASTp tool revealed that only FV and TFPIα contain this exact sequence. The FV basic region interacts with an acidic region, also located within the B domain, to maintain an inactive procofactor protein conformation. Removal of either region of the B domain activates FV to FVa (28, 29). Interestingly, platelets release multiple forms of FVa from their α-granules upon activation, some containing the acidic region of the B domain attached to the light chain (Fig. S2) (4, 5). Therefore, we hypothesized that inhibition of prothrombinase by TFPIα is mediated both through the K2 domain binding the FXa active site and the basic C terminus binding to the acidic-region–containing forms of FVa (FVaAR). In support of this hypothesis, TFPIα has been shown to bind FV through its C terminus, although the binding region on FV was not defined (30). Binding of the TFPIα C terminus to the FV acidic region was measured by fluorescence anisotropy, using Oregon Green 488 (OG488)-labeled TFPI-BR and unlabeled FV810, an altered form of FVa with a truncated B domain that contains the acidic region but lacks the basic region (28, 29). Consistent with the hypothesis, TFPI-BR bound to FV810 with high affinity (Kd = 0.43 ± 0.06 nM), but did not bind to FV810 treated with thrombin to completely remove the B domain (Fig. 3A). In addition, TFPI-BR displaced the corresponding OG488-labeled FV basic peptide (residues 951–1008; FV-BR) with comparable affinity to unlabeled FV-BR in competitive binding assays (Kd = 1.49 ± 0.13 nM vs. 1.9 ± 0.16 nM, respectively) (Fig. 3B). Finally, TFPIα bound FV810 with approximately fivefold higher affinity than did TFPI-BR (Kd = 88 ± 37 pM) (Fig. 3C). Binding of TFPIα was completely blocked by anti-K3C, suggesting a mechanism for the increased thrombin generation effect observed with this antibody (Fig. 1). By contrast, anti-K1 and anti-K2 had no effect on this interaction.
Fig. 2.
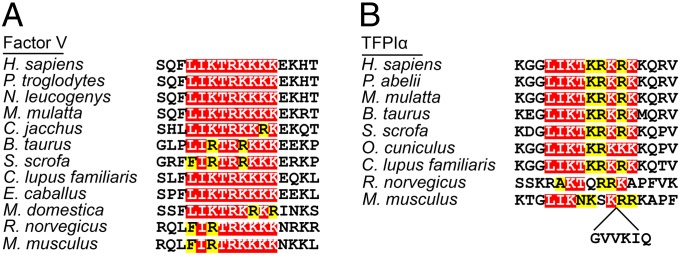
The basic regions of FV and TFPIα are conserved across mammalian species. Residues 995–1010 of human FV (A) and 249–264 of human TFPIα (B) are shown with the corresponding sequences from other species. Residues identical or homologous to the human FV sequence are highlighted red and yellow, respectively. Mouse TFPIα contains the indicated six-residue insertion.
Fig. 3.
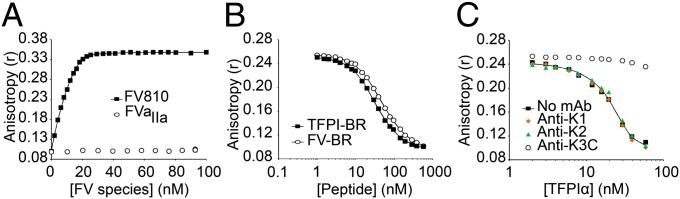
The basic region of TFPIα binds the acidic region in the B domain of FV with high affinity. (A) Oregon Green 488 (OG488)-labeled TFPI-BR (30 nM) was incubated with phospholipid vesicles (50 µM) and anisotropy measured following addition of FV810 (■) or FVaIIa (○). (B and C) OG488-labeled FV-BR (30 nM) and unlabeled FV810 (20 nM) were incubated with phospholipid vesicles (50 µM) and anisotropy measured following addition of unlabeled TFPI-BR (B, ■), FV-BR (B, ○), or TFPIα (C, ■). In C, experiments were also performed in the presence of anti-K1 (orange diamonds), -K2 (green triangles), or -K3C (○) (200 nM).
Inhibition of Prothrombinase by TFPIα Requires the FV Acidic Region.
Prothrombinase activity assays were performed in a purified system using FV variant molecules (Fig. 4A), FXa, and prothrombin in the presence of varying concentrations of TFPIα. It has previously been demonstrated that FV-BR inhibits thrombin generation by prothrombinase only if the acidic region of the B domain is attached to the light chain of FVa (28). Similar results were observed when prothrombinase activity was measured in the presence of TFPIα (Fig. 4B). TFPIα inhibited thrombin generation by prothrombinase assembled with FV810QQ (Fig. 4B, green; IC50 = 5.0 nM), in which the two thrombin cleavage sites (Arg709 and Arg1545) have been mutated to Gln, ensuring that the acidic region remains covalently associated with both the heavy and light chains. TFPIα similarly inhibited thrombin-treated FV810R1545Q (Fig. 4B, purple; IC50 = 8.2 nM), in which the heavy chain has been proteolytically detached, but the acidic region remains connected to the light chain. In contrast, TFPIα was a significantly weaker inhibitor of prothrombinase assembled with thrombin-treated FV810R709Q (Fig. 4B, blue; IC50 = 36.3 nM), in which the acidic region has been separated from the light chain but remains attached to the heavy chain. TFPIα also was a much weaker inhibitor of prothrombinase containing FV-B152, an altered form of FV whose B domain contains the basic region but not the acidic region (Fig. 4B, black; IC50 = 39.0 nM), or thrombin-treated FV810 (Fig. 4B, red; IC50 = 38.1 nM), which lacks the B domain entirely.
Fig. 4.

TFPIα inhibits prothrombinase assembled with FV variants containing the acidic region attached to the light chain. (A) The domain structures of the FV variants are diagrammed. The locations of the acidic (red) and basic (blue) regions of the FV B domain, the thrombin cleavage sites (black), mutations (green), and deletions (purple) are indicated. FV810 lacks residues 811–1491 of the B domain. FV810R1545Q, FV810R709Q, and FV810QQ have the indicated Arg→Gln mutations that alter thrombin cleavage. FV-B152 lacks residues 811–963 and 1008–1538 of the B domain. (B) FV-B152 (black), FV810QQ (green), thrombin-treated FV810 (red), FV810R709Q (blue), or FV810R1545Q (purple) (0.5 nM), was incubated with phospholipid vesicles (20 µM), DAPA (3 µM), and TFPIα. Reactions were initiated with prothrombin (1.4 µM) and FXa (5 nM). The initial rate of thrombin generation is reported as percent of control (mean ± SD; n ≥ 3).
Inhibition of FVaAR Prothrombinase by TFPIα Is Dependent upon K2 and the Basic C Terminus.
FVaAR can be generated in vitro from plasma FV by limited proteolysis with FXa, under conditions described by Monkovic and Tracy (31) (Fig. 5A), whereas similar treatment of plasma FV with thrombin results in complete removal of the B domain and generation of FVaIIa (31) (Fig. 5B). Treatment of purified plasma FV with these two proteases thus allowed for direct comparison of the ability of TFPIα to inhibit prothrombinase assembled with either FVaAR or FVaIIa (Fig. 5C). TFPIα was a 42-fold more potent inhibitor of thrombin generation by FVaAR prothrombinase than FVaIIa prothrombinase (IC50 = 0.9 nM vs. 37.5 nM, respectively). The IC50 for FVaAR prothrombinase inhibition is below the average plasma TFPI concentration of ∼2.3 nM (32), ∼10–40% of which is full-length TFPIα (33), suggesting that this inhibition may occur physiologically, particularly when there is also TFPIα released from platelets. In contrast to the differences observed in prothrombin activation assays, TFPIα equally inhibited cleavage of an amidolytic substrate by prothrombinase assembled with FVaAR or FVaIIa (Fig. S3), suggesting that the type of FVa present does not alter direct access of K2 to the FXa active site. As observed in experiments using PRP or isolated platelets, the anti-K2 and anti-K3C antibodies, but not anti-K1, blocked the inhibition of FVaAR prothrombinase by TFPIα (Fig. S4).
Fig. 5.

Inhibition of prothrombinase by TFPIα requires K2 and the basic C terminus. (A) FXa generated FVaAR through cleavages following Arg709 and Arg1018, leaving the acidic region of the FV B domain (red) attached to the light chain, but removing the basic region (blue). (B) Thrombin generated FVaIIa through cleavages following Arg709, Arg1018, and Arg1545, completely removing the B domain and generating the heavy and light chains. (C–G) FVaAR or FVaIIa (0.5 nM), phospholipid vesicles (20 µM), and the thrombin inhibitor DAPA (3 µM) were incubated with varying concentrations of TFPIα (C), TFPI-BR (D), K1K2 (E), K1K2C (F), or TFPIβ-expressing CHO cells (G). For G, phospholipids were omitted. Reactions were initiated by addition of prothrombin (1.4 µM) and FXa (5 nM). The initial rate of thrombin generation is reported as percent of control. All data points are shown as mean ± SD (n ≥ 3 independent experiments for each). Lines represent best-fit curves. In G, data from C are shown for reference.
The inhibition of FVaAR prothrombinase by TFPIα was absolutely dependent upon the TFPIα basic region, as TFPI-BR inhibited FVaAR prothrombinase (IC50 = 93.9 nM) but had no effect on FVaIIa prothrombinase activity at concentrations up to 10 µM (Fig. 5D), and an altered form of TFPI containing K1 and K2 (K1K2) was a poor inhibitor of either form of prothrombinase (Fig. 5E). Although the C terminus and K1K2 individually were substantially weaker inhibitors of FVaAR prothrombinase than TFPIα, an altered form of TFPI containing K1, K2, and the C terminus (K1K2C) effectively inhibited FVaAR prothrombinase (IC50 = 2.9 nM) (Fig. 5F). Thus, it appears that both K2 and the TFPIα C terminus are required for high-affinity inhibition of prothrombinase. TFPIβ expressed on the surface of CHO cells, which lack the basic C-terminal region, was an equally poor inhibitor of both forms of prothrombinase (Fig. 5G).
Inhibition of Prothrombinase by TFPIα Is Blocked with Heparin, Polyphosphate, and Fucoidan.
A number of reports have shown that heparin, polyphosphate, and fucoidan block the inhibitory function of TFPIα in assays that rely on in situ generation of FVa (18, 34–37). However, these properties are lost when FVaIIa is present. In light of our findings, we hypothesized that these molecules, each of which is highly negatively charged, act by blocking the charge-dependent interaction between the TFPIα basic C terminus and the acidic region of FVaAR. To test this hypothesis, prothrombinase activity assays were repeated in the presence of heparin (Fig. 6A), polyphosphate (Fig. 6B), and fucoidan (Fig. 6C). Each of these molecules blocked the inhibition of FVaAR prothrombinase by TFPIα, shifting the inhibition curve toward that obtained when using FVaIIa prothrombinase, while having little effect on the inhibition of FVaIIa prothrombinase.
Fig. 6.
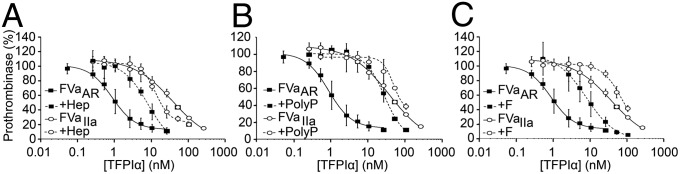
Heparin, polyphosphate, and fucoidan block the inhibition of FVaAR prothrombinase by TFPIα. Prothrombinase activity assays were performed as in Fig. 5C with heparin (0.5 units/mL) (A), polyphosphate (150 µM) (B), or fucoidan (10 nM) (C) included in the incubation mixture (dashed lines). In all panels, data from Fig. 5C (solid lines) are shown for reference.
Discussion
This research gives a unique description of a mechanism for inhibition of prothrombinase by an endogenous human protein, TFPIα, at physiologically relevant rates and concentrations, a finding that is directly relevant to understanding the molecular pathogenesis of bleeding disorders and thrombotic disease. This previously unrecognized anticoagulant activity of TFPIα is mediated through both active site inhibition of FXa by K2 and a high-affinity exosite interaction between the basic C-terminal region of TFPIα and the acidic region of the B domain of FV. Loss of either interaction results in substantially reduced inhibitory activity, and TFPIα is a very poor inhibitor of prothrombinase assembled with FVa in which the B domain has been proteolytically removed by thrombin (17, 18). Therefore, prothrombinase inhibition by TFPIα is only observed under conditions early in the procoagulant response when prothrombinase assembles with forms of FVa that retain the acidic region of the B domain, such as those released from activated platelets (4, 5) or generated through limited proteolysis with FXa (Fig. 7A). The essential nature of FXa activation of FV during the initiation of coagulation, before thrombin-mediated FV activation, was recently demonstrated by Schuijt and coworkers using a tick salivary protein that specifically blocks this reaction (38), supporting the physiological importance of TFPIα-mediated inhibition of prothrombinase assembled with FXa-activated FVa. Additionally, Vincent and coworkers (39) have described a FV mutation that results in circulating FV that contains the acidic region but lacks the basic region, binds tightly to TFPIα, and is associated with significant bleeding.
Fig. 7.

TFPIα inhibits thrombin generation during the initiation phase of coagulation. (A) Prothrombinase, consisting of FVa (green) and FXa (yellow) assembled on a membrane surface, is inhibited during the initiation phase of coagulation by TFPIα through binding of the K2 domain to the active site of FXa and an exosite interaction between the TFPIα basic region (BR, blue) and the acidic region of the FV B domain (AR, red). For clarity, FVa and FXa are shown dissociated, although TFPIα may be inhibiting them in complex. During the propagation phase of coagulation, thrombin removes the acidic region of FVa and prothrombinase is not inhibited by TFPIα. (B) TFPIα consists of three Kunitz-type inhibitor domains (K1–K3) and a highly basic C terminus. The “left side” of TFPIα inhibits the TF–FVIIa–FXa complex, with K1 and K2 binding the active sites of FVIIa and FXa, respectively. This function is mediated by TFPIα and TFPIβ. The TFPIα-specific “right side” inhibits thrombin generation by FVaAR prothrombinase, with K2 and the C terminus binding FXa and the acidic region of FVaAR, respectively. Red, blue, and yellow circles indicate acidic residues, basic residues, and cysteines, respectively.
These findings and conclusions are based on the following: CAT experiments with PRP, which revealed TFPIα-mediated inhibition of thrombin generation that is dependent on its K2 and K3C regions but independent of its ability to inhibit TF-FVIIa; fluorescence anisotropy experiments, demonstrating high-affinity binding between the C-terminal region of TFPIα and the acidic region of the FV B domain; and prothrombinase activity experiments, using a purified system with various altered forms of FV/FVa, which demonstrate unequivocally that the inhibitory effect is dependent upon K2 and the exosite interaction between the C terminus of TFPIα and the acidic region of the B domain of FV. The inhibition of prothrombinase by TFPIα is blocked by antibodies directed against its K2 and K3C domains, but not its K1 domain, ascribing physiological anticoagulant activity to regions of TFPIα that are not present in TFPIβ. Thus, the results provide logical explanations for the presence of TFPIα, but not TFPIβ, within platelets (11, 25); the evolutionary conservation of TFPIα back to zebrafish (24); and the retention of amino acid sequence identity of the basic regions of TFPIα and the FV B domain among mammalian species. Interestingly, murine TFPIα contains a six-amino-acid insertion in the basic region (Fig. 2). As mouse models are commonly used in hemostasis and thrombosis studies, it will be important to determine if the basic region of murine TFPIα interacts with the acidic region of murine factor Va in a manner similar to the human proteins. Interestingly, the exosite interaction between TFPIα and the FVa acidic region is only required to inhibit cleavage of prothrombin, an observation that may be explained by the recent structure of prothrombinase from the venom of Pseudonaja textilis (40). This structure revealed an extended hydrophobic channel on FVa, which was proposed to be a binding site for prothrombin. Therefore, disruption of prothrombin binding and cleavage by prothrombinase may require that TFPIα interact with both FXa and FVa.
FV maintains its procofactor conformation through an intramolecular interaction between the basic and acidic regions of its B domain (28). TFPIα mimics this interaction to inhibit prothrombinase assembled with forms of FVa that retain the acidic region in two important ways. First, the binding affinity of TFPI-BR to the acidic region of FV810 is as tight or tighter than that of FV-BR; second, the inhibition of prothrombinase by TFPIα requires that the FVa acidic region be covalently linked to the light chain, as has been described for FV-BR (28). Neither TFPIα nor FV-BR inhibits prothrombinase if the acidic region is disconnected from the light chain or absent altogether. Studies using FV-BR and FV810 have demonstrated that the acidic and basic regions of FV combine to prevent prothrombinase assembly by disrupting the binding of FXa (28). The C-terminal region of TFPIα may mediate anticoagulant activity by disrupting prothrombinase assembly in a similar manner; however, further studies are needed to define and characterize this effect.
The inhibition of prothrombinase by TFPIα is blocked by the negatively charged molecules heparin, polyphosphate, and fucoidan. Each has previously been shown to inhibit TFPIα in purified or plasma-based assays where FV is converted to FVa during the course of the reaction, but not in assays where FVaIIa is added to the system (18, 34–36). It has been proposed that these molecules act by promoting FV activation (36, 37). However, based on the data presented here, we propose an alternative: these molecules block TFPIα-mediated inhibition of prothrombinase assembled with forms of FVa containing the acidic region of the B domain but not prothrombinase assembled with FVa totally lacking the B domain, resulting in faster thrombin generation in the experimental systems studied. Thus, the mechanism for inhibition of prothrombinase by TFPIα provides a plausible biochemical explanation for (i) the procoagulant effect of heparin that is observed in the absence of antithrombin (18, 36); (ii) the therapeutic efficacy of fucoidan in a dog model of hemophilia (41); and (iii) the increased thrombin generation observed in PRP stimulated with histones, which is driven by polyphosphate released from platelet-dense granules (42, 43).
Polyphosphates, FV/Va, and TFPIα are released from different storage granules within platelets [dense granules (43), α-granules (44), and an unidentified granule (11), respectively]. Therefore, different platelet agonists may result in differential release of these three molecules, allowing for dynamic regulation of prothrombinase assembly and function that depends on the circumstances of activation.
Based on the studies reported here, it is proposed that TFPIα inhibits early events in blood coagulation through two separate mechanisms, thereby preventing small procoagulant or inflammatory stimuli from inducing intravascular thrombosis (Fig. 7B). First, TFPIα and TFPIβ rapidly inhibit small amounts of intravascular tissue factor on activated endothelial cells or monocytes, thereby preventing “subthreshold” intravascular inflammatory stimuli from developing into occlusive thrombi, a function mediated by K1 and K2 (7, 16). Second, platelet and plasma TFPIα rapidly inhibit early forms of prothrombinase that assemble on the surface of platelets activated by subthreshold stimuli within the vasculature, a function mediated by K2 and the basic C terminus. In both cases, the inhibitory capacity of TFPI is overcome by strong prothrombotic stimuli, producing feedback activation of factors VIIIa and Va, removal of the entire B domain from FVa, and a resultant thrombin burst.
These findings lend unique insight into therapeutic agents to treat hemophilia that are directed against different structural regions of TFPI, some of which are currently in phase 1 clinical trials (26, 45, 46). As hemostasis in hemophilia is modulated by the TFPIα present in plasma (47) or in platelets (26), agents designed to specifically block TFPIα-mediated inhibition of prothrombinase may be therapeutically effective as they would promote clot formation at the site of the platelet plug, while leaving functional TFPIβ-mediated anticoagulant activity throughout the vasculature.
Materials and Methods
Materials.
Detailed information regarding the source or production of all materials used in these studies, including the FV/Va species, peptides, TFPI species, platelets and platelet-rich plasma, and phospholipid vesicles is available in SI Materials and Methods. All experiments using human subjects were approved by the institutional review board at the Blood Center of Wisconsin (Milwaukee), and informed consent was obtained from all subjects.
Fluorescence Binding Assays.
Steady-state fluorescence anisotropy was measured at 25 °C in a QuantaMaster fluorescence spectrophotometer (Photon Technology International) using excitation and emission wavelengths of 480 nm and 520 nm, respectively, with long-pass filters (KV500; CVI Melles Griot) in the emission beam. For direct binding measurements, reaction mixtures (2.5 mL) containing OG488-TFPI-BR (30 nM) and phospholipid vesicles containing phosphatidylcholine (PC) and phosphatidylserine (PS) (PCPS) (50 µM) in HBS/Ca2+/PEG were prepared in 1-cm2 quartz cuvettes to which increasing concentrations of FV810 or thrombin-treated FV810 were added. For displacement experiments, TFPIα, FV-BR, or TFPI-BR was titrated into cuvettes containing OG488-FV-BR (30 nM), FV810 (20 nM), and PCPS (50 µM) in HBS/Ca2+/PEG. Where indicated, anti-K1, -K2, or -K3C (200 nM each) was added before titrating TFPIα. Fluorescence anisotropy measurements and data analyses were performed as described (48, 49).
Prothrombinase Activity Assays.
FVaAR, FVaIIa, FV810, FV810R709Q, FV810R1545Q, FV810QQ, or FV-B152 (0.5 nM) was diluted into HBS/Ca2+/PEG containing phospholipid vesicles containing PC, PS, and phosphatidylethanolamine (PE) (PCPSPE) (20 µM) and the thrombin inhibitor dansylarginine N-(3-ethyl-1,5-pentanediyl)amine (DAPA) (3 µM). TFPIα, K1K2, K1K2C, or TFPI-BR was added at varying concentrations and incubated for 20 min at 23 °C. Heparin (0.5 units/mL), polyphosphate (150 µM), or fucoidan (10 nM) was included in some reactions. For experiments using CHO-TFPIβ cells, PCPSPE was replaced with mixtures of CHO and CHO-TFPIβ cells holding the total cellular protein constant (900 µg/mL). In experiments using activated platelets (1 × 108 per mL), PCPSPE and FVa were omitted. For some platelet experiments, anti-K1, -K2, or -K3C (50 nM), TFPIα (1 nM), or TFPI-BR (1 µM) was included. Following incubation, prothrombin (1.4 µM) and FXa were added to initiate the reaction; aliquots were removed every 20 s for 2 min and quenched with HBS containing 0.1% PEG-8000 and EDTA (33 mM final concentration); thrombin was measured using a chromogenic thrombin substrate (0.32 mM; Spectrozyme TH; Sekisui Diagnostics) at 405 nm using a SpectraMax Plus microplate reader (Molecular Devices); and the rate of substrate cleavage was compared to a thrombin standard curve.
Factor Xa Chromogenic Assay.
TFPIα (varying concentrations), FVaAR, or FVaIIa (0.1 nM), PCPSPE (20 µM), and FXa chromogenic substrate (0.5 mM; Spectrozyme Xa) were diluted in HBS/Ca2+/PEG and incubated for 30 min at 23 °C. Reactions were initiated by addition of FXa (0.5 nM) and substrate cleavage monitored at 405 nm.
TF-FVIIa Activity Assay.
Relipidated TF (1/2,000 dilution), FVIIa (20 pM), PCPSPE (20 µM), and rabbit anti-FVII (0–100 nM) were incubated in HBS/Ca2+/PEG, and reactions were initiated by addition of factor X (20 nM) and quenched with EDTA (30 mM) following a 15-min incubation. The amount of FXa generated was determined using a FXa chromogenic substrate, as described above.
Western Blotting.
Washed platelets (1 × 109 per mL) were solubilized in Hepes-buffered Tyrode’s solution (5 mM Hepes, 137 mM NaCl, 2.68 mM KCl, 11.9 mM NaHCO3, 0.42 mM NaH2PO4, 1 mM MgCl2, 5 mM CaCl2, 0.2% dextrose), containing 0.35% BSA, 1% Triton X-100, 0.2 mM E-64, and 0.1 mM 3,4-dichloroisocoumarin, and immunoblotting was performed, as described (50), using anti-FVLC and anti-FVB monoclonal antibodies (5 µg/mL each). Antibody binding was detected using HRP-conjugated goat anti-rabbit IgG (1:4,000) (Jackson Immunoresearch) and SuperSignal West Pico chemiluminescent reagents (ThermoScientific).
Calibrated Automated Thrombography.
Platelet-rich plasma (80 µL) was incubated (10 min, 37 °C) with a mixture (20 µL total) of collagen (0.1 mg/mL) and FXa (0.1 nM). For some samples, anti-K1, -K2, or -K3C (50 nM each) or rabbit anti-FVII (100 nM) was included. Thrombin generation was initiated by addition of mixtures of calcium and fluorogenic thrombin substrate (FluCa; Diagnostica Stago) (20 µL). Fluorescence was read using a Fluoroskan Ascent microplate fluorometer (ThermoScientific). Thrombin concentrations were calculated using Thrombinoscope analysis software (Thrombinoscope).
Kinetic Analysis.
IC50 values were determined by fitting thrombin generation rates to a variable slope dose–response curve using GraphPad Prism version 5 (GraphPad software).
Sequence Alignments.
Protein sequences of mammalian FV and TFPIα were obtained from the NCBI database (www.ncbi.nlm.nih.gov). Sequence alignments were performed using ClustalW version 2 (51) (www.ebi.ac.uk/Tools/msa/clustalw2/).
Supplementary Material
Acknowledgments
This work was supported by National Heart, Lung, and Blood Institute (NHLBI) Grants HL068835 (to A.E.M.); HL880010 and HL74124, Project 2 (to R.M.C.); HL091469 (to S.A.M.); and HL046703, Project 3 (to P.B.T.). J.P.W. and M.W.B. were supported by NHLBI Training Grants HL007209 and HL07439, respectively. A.E.M. receives grant support from Novo Nordisk. R.M.C. receives research support from Pfizer and Alnylam Pharmaceuticals.
Footnotes
Conflict of interest statement: A.E.M. receives grant support from Novo Nordisk. R.M.C. receives research support from Pfizer and Alnylam Pharmaceuticals and royalties from Pfizer for technology related to factor Xa.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1310444110/-/DCSupplemental.
References
- 1.Miletich JP, Kane WH, Hofmann SL, Stanford N, Majerus PW. Deficiency of factor Xa-factor Va binding sites on the platelets of a patient with a bleeding disorder. Blood. 1979;54(5):1015–1022. [PubMed] [Google Scholar]
- 2.Mann KG, Nesheim ME, Church WR, Haley P, Krishnaswamy S. Surface-dependent reactions of the vitamin K-dependent enzyme complexes. Blood. 1990;76(1):1–16. [PubMed] [Google Scholar]
- 3.Lawson JH, Kalafatis M, Stram S, Mann KG. A model for the tissue factor pathway to thrombin. I. An empirical study. J Biol Chem. 1994;269(37):23357–23366. [PubMed] [Google Scholar]
- 4.Monković DD, Tracy PB. Functional characterization of human platelet-released factor V and its activation by factor Xa and thrombin. J Biol Chem. 1990;265(28):17132–17140. [PubMed] [Google Scholar]
- 5.Viskup RW, Tracy PB, Mann KG. The isolation of human platelet factor V. Blood. 1987;69(4):1188–1195. [PubMed] [Google Scholar]
- 6.Maroney SA, Ellery PE, Mast AE. Alternatively spliced isoforms of tissue factor pathway inhibitor. Thromb Res. 2010;125(Suppl 1):S52–S56. doi: 10.1016/j.thromres.2010.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Girard TJ, et al. Functional significance of the Kunitz-type inhibitory domains of lipoprotein-associated coagulation inhibitor. Nature. 1989;338(6215):518–520. doi: 10.1038/338518a0. [DOI] [PubMed] [Google Scholar]
- 8.Baugh RJ, Broze GJ, Jr, Krishnaswamy S. Regulation of extrinsic pathway factor Xa formation by tissue factor pathway inhibitor. J Biol Chem. 1998;273(8):4378–4386. doi: 10.1074/jbc.273.8.4378. [DOI] [PubMed] [Google Scholar]
- 9.Hackeng TM, Seré KM, Tans G, Rosing J. Protein S stimulates inhibition of the tissue factor pathway by tissue factor pathway inhibitor. Proc Natl Acad Sci USA. 2006;103(9):3106–3111. doi: 10.1073/pnas.0504240103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ndonwi M, Tuley EA, Broze GJ., Jr The Kunitz-3 domain of TFPI-alpha is required for protein S-dependent enhancement of factor Xa inhibition. Blood. 2010;116(8):1344–1351. doi: 10.1182/blood-2009-10-246686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maroney SA, et al. Active tissue factor pathway inhibitor is expressed on the surface of coated platelets. Blood. 2007;109(5):1931–1937. doi: 10.1182/blood-2006-07-037283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Novotny WF, Girard TJ, Miletich JP, Broze GJ., Jr Platelets secrete a coagulation inhibitor functionally and antigenically similar to the lipoprotein associated coagulation inhibitor. Blood. 1988;72(6):2020–2025. [PubMed] [Google Scholar]
- 13.Hansen JB, et al. Heparin induces synthesis and secretion of tissue factor pathway inhibitor from endothelial cells in vitro. Thromb Haemost. 2000;83(6):937–943. [PubMed] [Google Scholar]
- 14.Lupu C, Lupu F, Dennehy U, Kakkar VV, Scully MF. Thrombin induces the redistribution and acute release of tissue factor pathway inhibitor from specific granules within human endothelial cells in culture. Arterioscler Thromb Vasc Biol. 1995;15(11):2055–2062. doi: 10.1161/01.atv.15.11.2055. [DOI] [PubMed] [Google Scholar]
- 15.Lupu C, et al. Cellular effects of heparin on the production and release of tissue factor pathway inhibitor in human endothelial cells in culture. Arterioscler Thromb Vasc Biol. 1999;19(9):2251–2262. doi: 10.1161/01.atv.19.9.2251. [DOI] [PubMed] [Google Scholar]
- 16.Maroney SA, et al. Comparison of the inhibitory activities of human tissue factor pathway inhibitor (TFPI)α and TFPIβ. J Thromb Haemost. 2013;11(5):911–918. doi: 10.1111/jth.12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Franssen J, et al. Prothrombinase is protected from inactivation by tissue factor pathway inhibitor: Competition between prothrombin and inhibitor. Biochem J. 1997;323(Pt 1):33–37. doi: 10.1042/bj3230033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mast AE, Broze GJ., Jr Physiological concentrations of tissue factor pathway inhibitor do not inhibit prothrombinase. Blood. 1996;87(5):1845–1850. [PubMed] [Google Scholar]
- 19.Han X, Fiehler R, Broze GJ., Jr Characterization of the protein Z-dependent protease inhibitor. Blood. 2000;96(9):3049–3055. [PubMed] [Google Scholar]
- 20.Brufatto N, Ward A, Nesheim ME. Factor Xa is highly protected from antithrombin-fondaparinux and antithrombin-enoxaparin when incorporated into the prothrombinase complex. J Thromb Haemost. 2003;1(6):1258–1263. doi: 10.1046/j.1538-7836.2003.00254.x. [DOI] [PubMed] [Google Scholar]
- 21.Tran S, Norstrøm E, Dahlbäck B. Effects of prothrombin on the individual activated protein C-mediated cleavages of coagulation factor Va. J Biol Chem. 2008;283(11):6648–6655. doi: 10.1074/jbc.M708036200. [DOI] [PubMed] [Google Scholar]
- 22.Orfeo T, Brummel-Ziedins KE, Gissel M, Butenas S, Mann KG. The nature of the stable blood clot procoagulant activities. J Biol Chem. 2008;283(15):9776–9786. doi: 10.1074/jbc.M707435200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Girard TJ, Tuley E, Broze GJ., Jr TFPIβ is the GPI-anchored TFPI isoform on human endothelial cells and placental microsomes. Blood. 2012;119(5):1256–1262. doi: 10.1182/blood-2011-10-388512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maroney SA, et al. Temporal expression of alternatively spliced forms of tissue factor pathway inhibitor in mice. J Thromb Haemost. 2009;7(7):1106–1113. doi: 10.1111/j.1538-7836.2009.03454.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maroney SA, Cooley BC, Ferrel JP, Bonesho CE, Mast AE. Murine hematopoietic cell tissue factor pathway inhibitor limits thrombus growth. Arterioscler Thromb Vasc Biol. 2011;31(4):821–826. doi: 10.1161/ATVBAHA.110.220293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maroney SA, et al. Absence of hematopoietic tissue factor pathway inhibitor mitigates bleeding in mice with hemophilia. Proc Natl Acad Sci USA. 2012;109(10):3927–3931. doi: 10.1073/pnas.1119858109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Massberg S, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med. 2010;16(8):887–896. doi: 10.1038/nm.2184. [DOI] [PubMed] [Google Scholar]
- 28.Bos MH, Camire RM. A bipartite autoinhibitory region within the B-domain suppresses function in factor V. J Biol Chem. 2012;287(31):26342–26351. doi: 10.1074/jbc.M112.377168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu H, Toso R, Camire RM. Inhibitory sequences within the B-domain stabilize circulating factor V in an inactive state. J Biol Chem. 2007;282(20):15033–15039. doi: 10.1074/jbc.M701315200. [DOI] [PubMed] [Google Scholar]
- 30.Ndonwi M, Girard TJ, Broze GJ., Jr The carboxy-terminus of TFPIalpha is required for its interaction with factor V and Va. J Thromb Haemost. 2012;10(9):1944–1946. doi: 10.1111/j.1538-7836.2012.04834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Monkovic DD, Tracy PB. Activation of human factor V by factor Xa and thrombin. Biochemistry. 1990;29(5):1118–1128. doi: 10.1021/bi00457a004. [DOI] [PubMed] [Google Scholar]
- 32.Novotny WF, Brown SG, Miletich JP, Rader DJ, Broze GJ., Jr Plasma antigen levels of the lipoprotein-associated coagulation inhibitor in patient samples. Blood. 1991;78(2):387–393. [PubMed] [Google Scholar]
- 33.Broze GJ, Jr, Lange GW, Duffin KL, MacPhail L. Heterogeneity of plasma tissue factor pathway inhibitor. Blood Coagul Fibrinolysis. 1994;5(4):551–559. [PubMed] [Google Scholar]
- 34.Liu T, et al. Improved coagulation in bleeding disorders by non-anticoagulant sulfated polysaccharides (NASP) Thromb Haemost. 2006;95(1):68–76. [PubMed] [Google Scholar]
- 35.Smith SA, et al. Polyphosphate exerts differential effects on blood clotting, depending on polymer size. Blood. 2010;116(20):4353–4359. doi: 10.1182/blood-2010-01-266791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith SA, Morrissey JH. Heparin is procoagulant in the absence of antithrombin. Thromb Haemost. 2008;100(1):160–162. doi: 10.1160/TH08-05-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith SA, et al. Polyphosphate modulates blood coagulation and fibrinolysis. Proc Natl Acad Sci USA. 2006;103(4):903–908. doi: 10.1073/pnas.0507195103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schuijt TJ, et al. Factor xa activation of factor v is of paramount importance in initiating the coagulation system: Lessons from a tick salivary protein. Circulation. 2013;128(3):254–266. doi: 10.1161/CIRCULATIONAHA.113.003191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vincent LM, et al. Coagulation factor VA2440G causes east Texas bleeding disorder via TFPIα. J Clin Invest. 2013;123(9):3777–3787. doi: 10.1172/JCI69091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lechtenberg BC, et al. Crystal structure of the prothrombinase complex from the venom of Pseudonaja textilis. Blood. 2013 doi: 10.1182/blood-2013-06-511733. 10.1182/blood-2013-06-511733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prasad S, et al. Efficacy and safety of a new-class hemostatic drug candidate, AV513, in dogs with hemophilia A. Blood. 2008;111(2):672–679. doi: 10.1182/blood-2007-07-098913. [DOI] [PubMed] [Google Scholar]
- 42.Semeraro F, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood. 2011;118(7):1952–1961. doi: 10.1182/blood-2011-03-343061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ruiz FA, Lea CR, Oldfield E, Docampo R. Human platelet dense granules contain polyphosphate and are similar to acidocalcisomes of bacteria and unicellular eukaryotes. J Biol Chem. 2004;279(43):44250–44257. doi: 10.1074/jbc.M406261200. [DOI] [PubMed] [Google Scholar]
- 44.Wencel-Drake JD, Dahlback B, White JG, Ginsberg MH. Ultrastructural localization of coagulation factor V in human platelets. Blood. 1986;68(1):244–249. [PubMed] [Google Scholar]
- 45.Gorczyca ME, et al. Inhibition of tissue factor pathway inhibitor by the aptamer BAX499 improves clotting of hemophilic blood and plasma. J Thromb Haemost. 2012;10(8):1581–1590. doi: 10.1111/j.1538-7836.2012.04790.x. [DOI] [PubMed] [Google Scholar]
- 46.Hilden I, et al. Hemostatic effect of a monoclonal antibody mAb 2021 blocking the interaction between FXa and TFPI in a rabbit hemophilia model. Blood. 2012;119(24):5871–5878. doi: 10.1182/blood-2012-01-401620. [DOI] [PubMed] [Google Scholar]
- 47.Knappe S, et al. Plasmatic tissue factor pathway inhibitor is a major determinant of clotting in factor VIII inhibited plasma or blood. Thromb Haemost. 2013;109(3):450–457. doi: 10.1160/TH12-07-0529. [DOI] [PubMed] [Google Scholar]
- 48.Betz A, Krishnaswamy S. Regions remote from the site of cleavage determine macromolecular substrate recognition by the prothrombinase complex. J Biol Chem. 1998;273(17):10709–10718. doi: 10.1074/jbc.273.17.10709. [DOI] [PubMed] [Google Scholar]
- 49.Buddai SK, Toulokhonova L, Bergum PW, Vlasuk GP, Krishnaswamy S. Nematode anticoagulant protein c2 reveals a site on factor Xa that is important for macromolecular substrate binding to human prothrombinase. J Biol Chem. 2002;277(29):26689–26698. doi: 10.1074/jbc.M202507200. [DOI] [PubMed] [Google Scholar]
- 50.Wood JP, Silveira JR, Maille NM, Haynes LM, Tracy PB. Prothrombin activation on the activated platelet surface optimizes expression of procoagulant activity. Blood. 2011;117(5):1710–1718. doi: 10.1182/blood-2010-09-311035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Larkin MA, et al. Clustal W and Clustal X Version 2.0. Bioinformatics. 2007;23(21):2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.