

Significance
Cellulose is the most abundant biopolymer on Earth, primarily formed by vascular plants, but also by some bacteria. Bacterial extracellular polysaccharides, such as cellulose and alginate, are an important component of biofilms, which are multicellular, usually sessile, aggregates of bacteria. Biofilms exhibit a greater resistance to antimicrobial treatments compared with isolated bacteria and thus are a particular concern to human health. Cellulose synthases synthesize cellulose by polymerizing UDP-activated glucose and transport the growing polymer across the cell membrane during its synthesis. Despite numerous attempts, reconstituting cellulose synthesis in vitro from purified components has been unsuccessful. Here we present the complete reconstitution of bacterial cellulose synthesis from components from Rhodobacter sphaeroides, thereby establishing an experimental basis for cellulose and biofilm research.
Keywords: membrane transport, biopolymer, glycobiology, in vitro reconstitution
Abstract
Cellulose is a linear extracellular polysaccharide. It is synthesized by membrane-embedded glycosyltransferases that processively polymerize UDP-activated glucose. Polymer synthesis is coupled to membrane translocation through a channel formed by the cellulose synthase. Although eukaryotic cellulose synthases function in macromolecular complexes containing several different enzyme isoforms, prokaryotic synthases associate with additional subunits to bridge the periplasm and the outer membrane. In bacteria, cellulose synthesis and translocation is catalyzed by the inner membrane-associated bacterial cellulose synthase (Bcs)A and BcsB subunits. Similar to alginate and poly-β-1,6 N-acetylglucosamine, bacterial cellulose is implicated in the formation of sessile bacterial communities, termed biofilms, and its synthesis is likewise stimulated by cyclic-di-GMP. Biochemical studies of exopolysaccharide synthesis are hampered by difficulties in purifying and reconstituting functional enzymes. We demonstrate robust in vitro cellulose synthesis reconstituted from purified BcsA and BcsB proteins from Rhodobacter sphaeroides. Although BcsA is the catalytically active subunit, the membrane-anchored BcsB subunit is essential for catalysis. The purified BcsA-B complex produces cellulose chains of a degree of polymerization in the range 200–300. Catalytic activity critically depends on the presence of the allosteric activator cyclic-di-GMP, but is independent of lipid-linked reactants. Our data reveal feedback inhibition of cellulose synthase by UDP but not by the accumulating cellulose polymer and highlight the strict substrate specificity of cellulose synthase for UDP-glucose. A truncation analysis of BcsB localizes the region required for activity of BcsA within its C-terminal membrane-associated domain. The reconstituted reaction provides a foundation for the synthesis of biofilm exopolysaccharides, as well as its activation by cyclic-di-GMP.
Polysaccharides are essential biopolymers performing diverse biological functions, ranging from energy storage to osmoregulation and cell wall formation. Extracellular polysaccharides, including cellulose, chitin, and alginate, are synthesized inside the cell from nucleotide-activated sugars and are transported across the cell membrane during their synthesis. This remarkable task is performed by membrane-integrated glycosyltransferases (GTs) that couple polymer elongation with translocation (1, 2).
Cellulose is a linear polymer of glucose molecules linked via β-1,4 glycosidic linkages (3, 4) and is primarily formed by vascular plants, but also by some algae, protists, and bacteria (4–6). Cellulose is synthesized by cellulose synthase (CesA), a family 2 member of GTs (7) that processively polymerizes UDP-activated glucose via an evolutionarily conserved mechanism (2). CesAs contain eight predicted transmembrane (TM) segments and at least one extended intracellular domain adopting a GT-A fold (2, 8). The intracellular GT-A domain is responsible for donor and acceptor sugar binding, as well as for catalyzing the GT reaction, and the membrane-embedded part forms a TM pore in close juxtaposition with the catalytic site, thereby allowing translocation of the nascent polysaccharide (2).
Although most eukaryotic CesAs are believed to form supramolecular complexes that organize the secreted glucans into cable-like structures, i.e., the cellulose microfibrils (9), many Gram-negative bacteria synthesize cellulose as a biofilm component (10, 11). Biofilm formation is stimulated by the bacterial messenger cyclic-di-GMP (c-di-GMP) (12), which affects a diverse group of enzymes via interaction with either covalently or noncovalently attached c-di-GMP-binding domains, such as PilZ (13–15).
Bacterial cellulose synthase (Bcs) is a multicomponent protein complex encoded in an operon containing at least three genes, bcsA, B, and C (16, 17). BcsA is the catalytic subunit that synthesizes cellulose and forms the TM pore across the inner membrane and BcsB is a large periplasmic protein that is anchored to the inner membrane via a single C-terminal TM helix. BcsB may guide the polymer across the periplasm toward the outer membrane via two carbohydrate-binding domains (CBDs) (2). BcsA and BcsB are fused into a single polypeptide chain in some species (18). BcsC is predicted to form a β-barrel in the outer membrane, preceded by a large periplasmic domain containing tetratricopeptide repeats likely involved in complex assembly (16). Most cellulose synthase operons also code for a periplasmic cellulase, BcsZ, whose biological function is unknown, yet it appears to enhance cellulose production in vivo (19, 20). Although most biofilm-forming bacteria likely produce amorphous cellulose that is embedded in a 3D matrix of polysaccharides, proteinaceous fibers, and nucleic acids (21), some bacteria produce cellulose microfibrils resembling those synthesized by eukaryotic cells (22). In such bacteria, CesA complexes are linearly arranged along the cell axis, and the CesA operons encode at least one additional subunit, BcsD, that might facilitate the linear organization of the synthases (18).
Despite the numerous studies available on a large number of pro- and eukaryotic model systems, revealing the mechanism of cellulose synthesis and translocation has been hampered by difficulties in reconstituting functional CesAs in a purified system, either from eukaryotic or prokaryotic enzymes (23–26). To date, cellulose biosynthetic activities have only been recovered from detergent extracts of native membranes (24–26).
To overcome these challenges, we reconstituted an active cellulose synthetic system in vitro from a purified Rhodobacter sphaeroides BcsA-B complex (27). The purified complex efficiently synthesizes amorphous, high-molecular-weight (HMW) cellulose on incubation with UDP-glucose (UDP-Glc) and c-di-GMP, both in a detergent-solubilized state and after reconstitution into proteoliposomes (PLs). We show that cellulose elongation occurs directly from UDP-Glc without lipid-linked intermediates, reveal that c-di-GMP activates the synthase, and demonstrate the strict substrate specificity of BcsA for UDP-Glc. Furthermore, we demonstrate that BcsB is crucial for the catalytic activity of BcsA and localize the region required for cellulose synthesis within BcsB’s C-terminal, membrane-associated domain that packs against the TM region of BcsA.
Results
BcsB Is Required for Catalytic Activity of BcsA.
Gram-negative bacteria transport cellulose across the inner and outer bacterial membranes. Most likely this is achieved by associating the catalytic BcsA-B complex in the inner membrane with the pore-forming outer membrane BcsC subunit. To identify the complex components required for cellulose synthesis and translocation across the inner bacterial membrane, we expressed BcsA and BcsB from Escherichia coli K12 and Rhodobacter sphaeroides in E. coli C43 (28), prepared inverted membrane vesicles (IMVs), and analyzed them for cellulose synthesis activity. To this end, the membrane vesicles were incubated at 37 °C with the substrate UDP-Glc, the activator c-di-GMP, as well as 3H-labeled UDP-Glc as a radiotracer. The reaction was terminated by addition of 2% (wt/vol) SDS, and the water-insoluble, HMW polymer was sedimented by centrifugation. Subsequently, the obtained product was further purified by descending paper chromatography (1) and quantified by scintillation counting. Fig. 1 shows that only IMVs containing BcsA and BcsB produce a HMW polymer. IMVs containing only BcsA or BcsB are catalytically inactive. Combining IMVs containing BcsA or BcsB does not restore catalytic activity, suggesting that both subunits have to be in the same membrane to form a functional complex (Fig. 1 A and B). Polymer synthesis strictly depends on the presence of c-di-GMP as well as Mg2+, as expected for bacterial cellulose synthesis.
Fig. 1.

BcsA and BcsB are necessary for cellulose synthesis. (A) IMVs containing either BcsA, BcsB, or both subunits were incubated for 60 min at 37 °C in the presence and absence of c-di-GMP and 0.25 µCi UDP-[3H]-Glc. The synthesized polymer was purified by descending paper chromatography and quantified by scintillation counting. The amount of product obtained is shown relative to the product formed from IMVs containing BcsA and BcsB. Control reactions were performed with IMVs derived from cells containing an empty pET28a vector or in the absence of c-di-GMP or the presence of 20 mM EDTA. Results are shown in light and dark gray for components from E. coli and R. sphaeroides, respectively. (B) Nonreducing immuno-blotting of IMVs containing C-terminally poly-His and FLAG-tagged BcsA and BcsB, respectively. The full-length proteins are indicated by arrowheads. (C) Western analysis of oxidized and reduced BcsB. IMVs containing R. sphaeroides BcsB were solubilized in 2% (wt/vol) SDS and incubated with 20 mM DTT before SDS/PAGE.
BcsB carries an N-terminal secretion signal sequence and, depending on the species, has a predicted molecular weight of 75–83 kDa after signal peptide cleavage. Under nonreducing conditions, BcsB migrates at ∼100 kDa on an SDS/PAGE. Upon reduction, however, BcsB’s electrophoretic mobility significantly increases, suggesting that the protein forms an intramolecular disulfide bond (Fig. 1C). Indeed, most BcsBs contain only two invariant Cys residues (Fig. S1), one in each CBD, which are in close proximity to one another at the CBD interface and are thus likely to form a disulfide bond (2).
Purified BcsA-B Synthesizes HMW Cellulose.
The R. sphaeroides BcsA-B complex was purified to homogeneity in the detergent LysoFosCholine Ether 14 (LFCE14) via metal affinity and gel filtration chromatography and was reconstituted into PLs formed from the E. coli total lipid extract. In PLs, the cellulose synthase activity displays a similar dependence on activation by c-di-GMP as in IMVs, suggesting that the complex retained its native-like activity during purification (Fig. 2). To further confirm that the synthesized polymer represents a β-1,4-linked glucan, we tested whether a β-1,4 or β-1,3 specific endo glucanase degrades the water-insoluble polymer. As expected for cellulose, β-1,3 glucanase does not degrade the synthesized polymer, whereas β-1,4 glucanase does (Fig. 2). In addition, permethylation glycosyl linkage analysis of the glucan obtained from a 60-min synthesis reaction reveals that the in vitro synthesized polymer consists exclusively of 1,4-linked glucosyl residues (Fig. S2). The alditolacetates corresponding to the residues at the nonreducing ends of the chains represent no more than 0.3–0.5% of the total derivatives obtained from the in vitro product (Fig. S2). Altogether, these data confirm the cellulosic nature of the polysaccharide formed by the BcsA-B complex and indicate that the chains formed in vitro exhibit a degree of polymerization of at least 200, most likely within the range 200–300.
Fig. 2.

In vitro cellulose synthesis from purified and reconstituted BcsA-B. PLs containing 1 µM of purified R. sphaeroides BcsA-B complex were incubated at 37 °C for the indicated time, and the synthesized polymer was quantified as described in Fig. 1. The polymer is degraded by endo-β-1,4- but not by endo-β-1,3 glucanase (β-1,4, β-1,3). (Right) Coomassie-stained SDS/PAGE of reconstituted BcsA-B. DPM, disintegrations per minute.
Most inverting GT require an essential divalent cation for catalysis. The cation is coordinated by a conserved Asp-X-Asp motif at the active site to stabilize the nucleoside diphosphate leaving group during glycosyl transfer (2, 29). To determine cation preference, cellulose synthesis reactions were performed in the presence of Ba2+, Mn2+, Mg2+, and Ca2+. Only Mn2+ and Mg2+ enabled approximately equal activity levels (Fig. S3A), consistent with earlier observations on other processive GT (30, 31).
To facilitate glycosyl transfer, the acceptor 4′ hydroxyl group of the growing polysaccharide becomes deprotonated during the SN2 nucleophilic substitution reaction (32). Based on the crystal structure of R. sphaeroides BcsA-B, the general base catalyzing deprotonation (Asp343) is part of an invariant Thr-Glu-Asp motif within hydrogen bond distance to the nonreducing end of the polymer (2). Asp343 lies at the back of a deep substrate-binding groove between the donor and acceptor coordination sites. Cellulose synthesis assays performed in a pH range from 4.5 to 9.5 show that BcsA-B exhibits optimal activity at neutral pH (Fig. S3B), yet significant activity remains even at an alkaline pH of 9.5. This pH profile is consistent with a buried carboxylate as a catalytic base (33).
Kinetic Characterization and Activation of Cellulose Synthesis by c-di-GMP.
The GT reaction catalyzed by BcsA transfers the donor glucose from the donor substrate UDP-Glc to the nonreducing end of the acceptor glucan as also observed for plant and other bacterial CesAs (25, 26, 34). Thus, the second product of the cellulose synthesis reaction is UDP. To confirm that BcsA indeed forms UDP as a reaction product (and not, for example, UMP plus inorganic phosphate) and to obtain kinetic insights into the reaction, we coupled cellulose synthesis to the activities of pyruvate kinase (PK) and lactate dehydrogenase (LDH), thus monitoring polymer formation in real time by following the oxidation of reduced NADH spectrophotometrically (1, 35). Because PK recognizes UDP but not UMP as substrate (36), the successful coupling of cellulose synthesis with its activity implies the formation of UDP. To ensure that all BcsA-B complexes are accessible to the substrates and can contribute to the observed reaction, the complex was reconstituted into lipid nanodiscs (NDs) (Fig. S4), which are edge-stabilized, planar lipid bilayer discs ∼10 nm in diameter (37). At this size, each ND most likely contains a single BcsA-B complex (38).
As in PLs, the activity of BcsA-B in NDs strongly depends on activation by c-di-GMP, and the polymer is readily degraded by β-1,4 glucanase, suggesting that NDs provide a native-like environment (Fig. S5). As shown in Fig. 3A, the c-di-GMP–activated BcsA-B complex generates UDP at a constant rate for up to 45 min, indicating that the enzyme is not inhibited by the accumulating or aggregating cellulose. No UDP is formed in the absence of c-di-GMP, UDP-Glc, or BcsA-B.
Fig. 3.

Kinetic analysis of cellulose synthesis and stimulation by cyclic-di-GMP. (A) Cellulose synthesis reactions were performed in the presence of 1 U PK and LDH, 0.5 mM NADH, and 1 mM phosphoenolpyruvate (PEP). The LDH-catalyzed oxidation of NADH to NAD+ was monitored spectrophotometrically at 340 nm. Control reactions in the absence of UDP-Glc, c-di-GMP, PEP, or BcsA-B do not lead to oxidation of NADH. Addition of 1 mM UDP to the reaction rapidly depletes NADH by circumventing cellulose synthesis. (B and C) Titration of UDP-Glc and c-di-GMP. Apparent reaction rates at a constant c-di-GMP concentration of 30 µM and increasing UDP-Glc concentrations (B) or increasing concentrations of c-di-GMP at a constant UDP-Glc concentration of 5 mM (C). The data were fit to monophasic Michaelis-Menten kinetics. Km and KA are the apparent affinities for the substrate UDP-Glc and the activator c-di-GMP, respectively. (D) Titration of UDP-Glc from 0 to 7.5 mM at the indicated c-di-GMP concentrations. All reactions were performed at room temperature with 0.5 µM ND-reconstituted BcsA-B in a total volume of 20 µL.
The robust activity of BcsA-B in ND allows analyzing its activity at either varying UDP-Glc or varying c-di-GMP concentrations (Fig. 3 B and C). Under both conditions, the enzyme obeys monophasic Michaelis-Menten kinetics with apparent affinities of 0.5 mM and 1.8 µM for UDP-Glc and c-di-GMP, respectively, consistent with the estimated physiological concentrations of 1–2 mM for UDP-Glc and 1–10 µM for c-di-GMP (39, 40). Although the concentration of active BcsA-B in NDs is unknown, assuming that 100% of the enzyme is responsible for the observed activity suggests a minimal polymerization rate generating ∼90 UDP molecules/s per BcsA-B complex. This rate is about 10-fold higher than what has been observed in other model systems, perhaps due to the lack of higher-order organization of the synthesized glucan chains (25, 41, 42).
c-di-GMP strongly activates cellulose synthesis by an unknown mechanism (43). The tight association of BcsA’s PilZ and GT domains suggests that c-di-GMP controls the accessibility of the GT active site (2). Titrating UDP-Glc at different c-di-GMP concentrations shows that the maximum catalytic activity achieved depends on the overall c-di-GMP concentration, whereas the apparent affinity for UDP-Glc remains within 0.1–1.0 mM, comparable with the Km of 0.5 mM for UDP-Glc determined in the presence of 30 µM c-di-GMP (Fig. 3B and D).
Feedback Inhibition of BcsA-B.
The reconstituted cellulose biosynthetic activity solely requires the presence of UDP-Glc and the activator c-di-GMP. The cellulose synthesis rate of microfibril-forming, oligomeric CesAs is influenced by the interaction of the individual glucans outside the cell, suggesting that cellulose microfibril formation is rate limiting (42, 44, 45). Thus, we further investigated whether the catalytic rate of BcsA-B is also influenced by the accumulating products, either HMW cellulose or UDP.
BcsZ is a periplasmic cellulase encoded in most bacterial cellulose synthase operons characterized to date (4). Although BcsZ exhibits low activity toward crystalline cellulose microfibrils (20), it efficiently degrades in vitro-synthesized cellulose in situ. Performing cellulose synthesis assays with ND-reconstituted BcsA-B in the presence of 0.1 mg/mL E. coli BcsZ prevents the accumulation of HMW cellulose (Fig. 4A), suggesting that BcsZ efficiently degrades the glucan as it emerges from the complex. Kinetically, however, BcsA-B's apparent reaction rate does not change in the presence of BcsZ, indicating that its activity is not affected by the accumulating polymer (Fig. 4A), perhaps because the emerging glucans lack higher-order organization. This observation is consistent with the higher apparent polymerization rate of BcsA-B compared with CesAs that form cellulose microfibrils (Fig. 3B) (25, 41, 42). The same results were obtained with PLs-reconstituted BcsA-B, arguing that the lack of product inhibition is not due to a different oligomeric state of the synthase in ND.
Fig. 4.
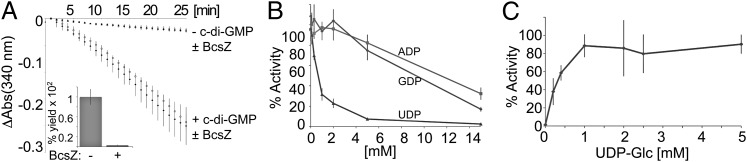
Product inhibition of cellulose synthase. (A) Enzyme-coupled cellulose synthesis assays were performed as described in Fig. 3 in the absence or presence of 0.1 mg/mL E. coli BcsZ. (Inset) Radioactive sedimentation assay performed with ND-reconstituted BcsA-B synthesizing cellulose in the absence or presence of BcsZ. (B) Inhibition of cellulose synthesis by UDP. Cellulose synthesis sedimentation assays were performed with 1 µM PL-reconstituted BcsA-B in the presence of increasing concentrations of the indicated nucleotides and 0.5 mM UDP-Glc for 60 min at 37 °C. The obtained product is quantified relative to the polymer formed in the absence of any nucleoside diphosphate. (C) UDP is a competitive inhibitor of cellulose synthase. Cellulose synthesis sedimentation assays performed (as in B) in the presence of 0.7 mM UDP and increasing concentrations of UDP-Glc. The products are quantified relative to the polymer formed in the absence of UDP and the presence of 5 mM UDP-Glc.
The glycosyl transfer reaction converts UDP-Glc to UDP, thereby releasing an important nucleotide whose physiological concentration is maintained in the low micromolar range (39). To analyze whether BcsA-B undergoes feedback inhibition by UDP, we first tested BcsA-B's activity in the presence of a constant 0.5 mM UDP-Glc concentration and increasing concentrations of UDP by the cellulose sedimentation assay. BcsA-B's activity is significantly inhibited by UDP, with only 50% of product formed in the presence of 0.7 mM UDP (Fig. 4B). In contrast, guanosine and adenosine diphosphates do not inhibit cellulose synthesis. However, inhibition by UDP is competitive as the enzyme reaches normal activity levels with increasing UDP-Glc to UDP ratios (Fig. 4C), suggesting a model by which UDP, at a high concentration, competitively competes with UDP-Glc for binding to BcsA’s active site. Under physiological conditions, however, it is unlikely that this inhibitory effect becomes rate limiting as the concentration of UDP-Glc exceeds that of UDP by about an order of magnitude (39).
Substrate Specificity of BcsA-B.
It is unknown how CesAs select their substrate UDP-Glc. Several UDP-activated sugars are common precursors for many physiological processes in pro- and eukaryotes, and substrate promiscuity is predicted to change the physico-chemical properties of the polymer formed or even terminate polymerization. To probe the substrate specificity of BcsA-B, we performed enzyme-coupled cellulose synthesis reactions in the presence of 5 mM UDP-galactose (Gal), -glucuronic acid (GA), -N-acetylglucosamine (NAG), -arabinose (Ara), or -xylose (Xyl) as the only carbohydrate source. As shown in Fig. 5A, none of the alternative substrates enables a reaction rate similar to UDP-Glc, giving rise to about 20% residual activity compared with the nonstimulated state in the absence of c-di-GMP, perhaps due to slow incorporation or hydrolysis of the alternative UDP-sugars.
Fig. 5.

Substrate specificity of BcsA-B. (A) Enzyme-coupled cellulose synthase assays performed with 0.5 µM PLs-reconstituted BcsA-B in the presence of 5 mM of the indicated substrates. The apparent activities are expressed relative to the activity in the presence of UDP-Glc. (B) Inhibition of cellulose synthesis by alternative substrates. Activity assays were performed for 90 min at 37 °C in the presence of 1 mM UDP-Glc and increasing concentrations of UDP-Xyl, -Gal, or -NAG. The apparent rates are shown relative to the activity in the presence of 1 mM UDP-Glc only. (C) As in B but shown are the linear decreases in absorbance at 340 nm for samples incubated with 1 mM UDP-Glc and the indicated concentrations of UDP-Xyl. (D) BcsA-B is catalytically active in a detergent-solubilized state. Cellulose synthesis is activated by c-di-GMP and the HMW product is degraded by endo β-1,4 glucanase (β-1,4).
Cellulose syntheses in the presence of 1 mM UDP-Glc and increasing concentrations of UDP-Gal, -NAG, or -Xyl reveal a concentration-dependent inhibition of polymer synthesis, with UDP-Xyl as strongest inhibitor. At 5 mM concentration, UDP-NAG reduces the apparent reaction rate by only 20%, whereas UDP-Xyl reduces the activity by about 60%, suggesting a UDP- and sugar-specific effect on inhibition (Figs. 4 B and C and 5B). Interestingly, the reactions proceed for at least 90 min at a constant rate, indicating that the alternative sugars are either not at all incorporated or limited incorporation does not have a cumulative adverse effect on the overall reaction rate (Fig. 5C).
Several capsular exopolysaccharides are assembled from lipid-bound intermediates (46). Based on radiotracer labeling, a similar mechanism has been proposed for cellulose synthesis in Agrobacterium tumefaciens, by which cellulose would be assembled from short, lipid-linked oligosaccharides (47). To investigate whether BcsA-B’s activity requires any components provided by the E. coli total lipid extract used for reconstitution, we performed cellulose synthesis reactions in a detergent-solubilized state, in the absence of any additional phospholipids. BcsA-B purified in the detergent lauryl N,N-dimethylamine oxide (LDAO) or LFCE14 robustly synthesizes cellulose in a c-di-GMP–dependent manner, which is degraded by cellulase, consistent with the synthesis of an authentic β-1,4 glucan (Fig. 5D). We note that, in a detergent-solubilized state, the enzyme displays ∼10–20% residual activity in the absence of c-di-GMP, perhaps due to an increased conformational flexibility of the “gating loop” controlling access to BcsA’s active site (2). Although the presence of lipids arising from the E. coli expression system cannot be fully ruled out, these results strongly suggest that the involvement of lipid-linked oligosaccharides in the elongation reaction is unlikely. This conclusion is further supported by the large distance between the active site and the putative lipid–water interface (∼25Å) (2).
The Membrane-Associated Domain of BcsB Is Essential for Cellulose Synthesis.
Based on the architecture of the BcsA-B complex, the strict dependence of BcsA’s catalytic activity on BcsB is surprising. Although BcsB shares a large interface with BcsA, none of its domains are in close proximity to the active site (2). Likewise, BcsB only interacts with the translocating glucan on the periplasmic side of the membrane, thus it is unlikely that it participates in the translocation reaction. BcsB is a multidomain protein containing a repeat of a CBD linked to a flavodoxin-like domain (FD) (2). The N-terminal CBD-1, which forms the membrane distal part, is located at the tip of the dome-shaped molecule, followed by FD-1. This organization is repeated with CBD-2 and FD-2 before BcsB forms a short amphipathic helix followed by its C-terminal TM anchor. The TM anchor packs into a deep groove formed by BcsA’s TM helices 1, 2, and 3.
To identify the core region of BcsB required for catalytic activity of BcsA, we systematically truncated BcsB N-terminally starting either at Gly190 (after CBD-1), at Thr309 (after FD-1), at Met456 (after CBD-2), or at Ser684 (after FD-2) (Fig. 6A). The constructs were coexpressed with BcsA, and the cellulose biosynthetic activity of the truncated complexes was analyzed in IMVs. While IMVs containing only BcsA fail to produce any HMW polymer, essentially all BcsB truncations supported a comparable catalytic activity of BcsA, demonstrating that only the C-terminal membrane-associated region of BcsB is required for function (Fig. 6B).
Fig. 6.

The membrane-associated domain of BcsB is required for cellulose synthesis. (A) N-terminal deletion mutants progressively truncating BcsB. The TM and cytosolic domains of BcsA are shown as a surface in dark and light gray, respectively, and the translocating glucan is shown as red spheres. (B) Cellulose synthase activity of the truncated BcsA-B complexes. IMVs containing WT BcsA and the indicated BcsB constructs were tested for cellulose synthase activity by quantifying the accumulation of radioactively labeled cellulose. IMVs were incubated for 60 min at 37 °C. The purified BcsA-B-S684 complex retains activity after reconstitution into PLs (S684-r). (C) Western analysis of IMVs and PLs. BcsA and BcsB carry a C-terminal poly-His and FLAG-tag, respectively. Red arrowheads highlight the positions of the BcsB constructs.
To further confirm that the catalytic activity of the BcsA-B-S684 complex is indeed due to the interaction of BcsA with the BcsB fragment, we purified the truncated complex by Ni-affinity and gel filtration chromatography via the C-terminal poly-histidine tag on BcsA. As shown in Fig. 6 B and C, the FLAG-tagged BcsB-S684 fragment copurifies with BcsA, and the purified complex is catalytically active after reconstitution into PLs, demonstrating that residues 684–725 of BcsB suffice to mediate the interaction with and maintain the catalytic activity of BcsA.
Discussion
The purified R. sphaeroides BcsA-B complex allows, for the first time, characterizing cellulose synthesis in a purified state. In vitro, BcsA-B synthesizes HMW cellulose in the presence of UDP-Glc and the allosteric activator c-di-GMP, thus providing a model system for not only cellulose synthesis but also for c-di-GMP–induced exopolysaccharide secretion, implicated in biofilm formation.
On activation by c-di-GMP, BcsA-B processively elongates the cellulose polymer, achieving a degree of polymerization in vitro in the range 200–300. This reaction proceeds at a similar rate in detergent-solubilized and membrane-reconstituted states, highly favoring a model by which BcsA catalyzes the stepwise transfer of UDP-activated glucose to the growing acceptor without the involvement of any lipid-linked reaction intermediates. Because glucan elongation is tightly coupled to its translocation through BcsA’s TM pore (2) and robust cellulose synthesis occurs in vitro in the absence of electrochemical gradients, the GT reaction must suffice to energize cellulose translocation.
In accordance with other biofilm polysaccharides (11), BcsA-B most likely produces amorphous cellulose, consisting of randomly oriented glucan chains. No cellulose microfibrils were observed by electron microscopy analyses, and the sensitivity of the synthesized cellulose toward cellulase digestion further indicates the loose organization of the individual glucan chains.
Cyclic-di-GMP activates cellulose synthesis allosterically and binds BcsA-B with high affinity. It is a potent inducer of biofilm formation, thus the mechanism by which it activates exopolysaccharide synthases is of particular importance. In contrast to other biofilm polysaccharide synthases, such as alginate- and poly-β-1,6 N-acetylglucosamine (Pga) synthase, the c-di-GMP-binding PilZ domain of cellulose synthase is a part of the catalytic BcsA subunit (Fig. S6). In alginate synthase, Alg44, resembling BcsB in its TM topology, contains an intracellular c-di-GMP–binding PilZ domain and associates with the catalytic Alg8 subunit (17). Pga synthases do not contain PilZ domains, but bind c-di-GMP at the interface between the catalytic PgaC and the associated PgaD subunits (Fig. S6) (48).
Titration of UDP-Glc at increasing c-di-GMP concentrations shows that c-di-GMP does not alter BcsA’s apparent affinity for UDP-Glc, yet it increases BcsA’s apparent catalytic rate in vitro at least 10-fold. These observations are consistent with a model by which c-di-GMP binding exposes BcsA’s active site, perhaps by removing a “lid” covering the opening of the GT domain (2), thereby directly allowing substrate binding to and product release from the active site. In the absence of c-di-GMP or under conditions where the concentration of c-di-GMP is rate limiting, only a fraction of the catalytic sites might be accessible, thus reducing the overall reaction rate.
With the exception of this study, no cellulose synthase activity has been recovered from purified components, neither from pro- nor eukaryotic sources (24, 25). Although BcsA requires BcsB for catalytic activity, only its C-terminal TM anchor together with a preceding amphipathic helix is necessary for activity. Its interaction with BcsA likely stabilizes the TM region of BcsA, such that the synthase is catalytically active. A destabilized TM region of BcsA would not only affect the glucan channel, but would also alter the localization of the signature pentapeptide (Gln-X-X-Arg-Trp) (49) that stabilizes the acceptor glucan at the active site (2). Thus, it is conceivable that eukaryotic cellulose synthases also require additional components for activity, which might dissociate during purification, leading to preparations with drastically reduced cellulose synthase rates. The described biochemical analysis of bacterial cellulose synthesis offers an alternative route to identify potential CesA interaction partners similar to BcsB.
Materials and Methods
The R. sphaeroides BcsA and BcsB cellulose synthase subunits were expressed in E. coli C43 and purified as described (2). The purified complex was reconstituted into proteoliposomes after incubation with detergent-solubilized E. coli total lipid extract and detergent removal by stepwise addition of SM-2 BioBeads. Cellulose synthesis was initiated by addition of UDP-Glc and c-di-GMP in the presence of MgCl2 and incubation at 37 °C. The synthesized cellulose was quantified after incorporation of 3H-labeled glucose as radiotracer by scintillation counting. Enzyme-coupled cellulose synthesis assays were performed spectrophotometrically by coupling cellulose synthesis to the reactions of pyruvate kinase and lactate dehydrogenase. Full experimental details are provided in SI Materials and Methods.
Supplementary Material
Acknowledgments
We are grateful to Joshua McNamara and Jacob Morgan for critical comments on the manuscript and thank Geoffrey Fincher for providing UDP-Xyl. A.N. was supported in part by the Robert Preston Pillow Scholarship, Kenneth C. Bass Research Scholarship, and the Ingrassia Family Echols Scholars Research Grant. This work was primarily supported by The Center for LignoCellulose Structure and Formation, Energy Frontier Research Center, US Department of Energy, Office of Science, Grant DE-SC0001090 and in part (BcsB truncation studies) by NIH Grant 1R01GM101001 (awarded to J.Z.). V.B. and H.M. were supported by the Royal Institute of Technology (KTH) Advanced Carbohydrate Materials Consortium (CarboMat) funded by the Swedish Research Council Formas.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1314063110/-/DCSupplemental.
References
- 1.Hubbard C, McNamara JT, Azumaya C, Patel MS, Zimmer J. The hyaluronan synthase catalyzes the synthesis and membrane translocation of hyaluronan. J Mol Biol. 2012;418(1-2):21–31. doi: 10.1016/j.jmb.2012.01.053. [DOI] [PubMed] [Google Scholar]
- 2.Morgan JL, Strumillo J, Zimmer J. Crystallographic snapshot of cellulose synthesis and membrane translocation. Nature. 2013;493(7431):181–186. doi: 10.1038/nature11744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nishiyama Y, Sugiyama J, Chanzy H, Langan P. Crystal structure and hydrogen bonding system in cellulose I(alpha) from synchrotron X-ray and neutron fiber diffraction. J Am Chem Soc. 2003;125(47):14300–14306. doi: 10.1021/ja037055w. [DOI] [PubMed] [Google Scholar]
- 4.Römling U. Molecular biology of cellulose production in bacteria. Res Microbiol. 2002;153(4):205–212. doi: 10.1016/s0923-2508(02)01316-5. [DOI] [PubMed] [Google Scholar]
- 5.Somerville C. Cellulose synthesis in higher plants. Annu Rev Cell Dev Biol. 2006;22:53–78. doi: 10.1146/annurev.cellbio.22.022206.160206. [DOI] [PubMed] [Google Scholar]
- 6.Grimson MJ, Haigler CH, Blanton RL. Cellulose microfibrils, cell motility, and plasma membrane protein organization change in parallel during culmination in Dictyostelium discoideum. J Cell Sci. 1996;109:3079–3087. doi: 10.1242/jcs.109.13.3079. [DOI] [PubMed] [Google Scholar]
- 7.Cantarel BL, et al. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009;37(Database issue):D233–D238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sethaphong L, et al. Tertiary model of a plant cellulose synthase. Proc Natl Acad Sci USA. 2013;110(18):7512–7517. doi: 10.1073/pnas.1301027110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kimura S, et al. Immunogold labeling of rosette terminal cellulose-synthesizing complexes in the vascular plant Vigna angularis. Plant Cell. 1999;11(11):2075–2086. doi: 10.1105/tpc.11.11.2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jahn CE, Selimi DA, Barak JD, Charkowski AO. The Dickeya dadantii biofilm matrix consists of cellulose nanofibres, and is an emergent property dependent upon the type III secretion system and the cellulose synthesis operon. Microbiology. 2011;157:2733–2744. doi: 10.1099/mic.0.051003-0. [DOI] [PubMed] [Google Scholar]
- 11.McCrate OA, Zhou X, Reichhardt C, Cegelski L. Sum of the parts: Composition and architecture of the bacterial extracellular matrix. J Mol Biol. 2013 doi: 10.1016/j.jmb.2013.06.022. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cotter PA, Stibitz S. c-di-GMP-mediated regulation of virulence and biofilm formation. Curr Opin Microbiol. 2007;10(1):17–23. doi: 10.1016/j.mib.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 13.Ryjenkov DA, Simm R, Römling U, Gomelsky M. The PilZ domain is a receptor for the second messenger c-di-GMP: The PilZ domain protein YcgR controls motility in enterobacteria. J Biol Chem. 2006;281(41):30310–30314. doi: 10.1074/jbc.C600179200. [DOI] [PubMed] [Google Scholar]
- 14.Amikam D, Galperin MY. PilZ domain is part of the bacterial c-di-GMP binding protein. Bioinformatics. 2006;22(1):3–6. doi: 10.1093/bioinformatics/bti739. [DOI] [PubMed] [Google Scholar]
- 15.Römling U, Galperin MY, Gomelsky M. Cyclic di-GMP: The first 25 years of a universal bacterial second messenger. Microbiol Mol Biol Rev. 2013;77(1):1–52. doi: 10.1128/MMBR.00043-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keiski CL, et al. AlgK is a TPR-containing protein and the periplasmic component of a novel exopolysaccharide secretin. Structure. 2010;18(2):265–273. doi: 10.1016/j.str.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rehm BHA. Alginate production: Precursor biosynthesis, polymerization and secretion. Microbiol Monogr. 2009;13:55–71. [Google Scholar]
- 18.Saxena IM, Kudlicka K, Okuda K, Brown RM., Jr Characterization of genes in the cellulose-synthesizing operon (acs operon) of Acetobacter xylinum: Implications for cellulose crystallization. J Bacteriol. 1994;176(18):5735–5752. doi: 10.1128/jb.176.18.5735-5752.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Standal R, et al. A new gene required for cellulose production and a gene encoding cellulolytic activity in Acetobacter xylinum are colocalized with the bcs operon. J Bacteriol. 1994;176(3):665–672. doi: 10.1128/jb.176.3.665-672.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mazur O, Zimmer J. Apo- and cellopentaose-bound structures of the bacterial cellulose synthase subunit BcsZ. J Biol Chem. 2011;286(20):17601–17606. doi: 10.1074/jbc.M111.227660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mann EE, Wozniak DJ. Pseudomonas biofilm matrix composition and niche biology. FEMS Microbiol Rev. 2012;36(4):893–916. doi: 10.1111/j.1574-6976.2011.00322.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iguchi M, Yamanaka S, Budhiono A. Bacterial cellulose; a masterpiece of nature's arts. J Mater Sci. 2000;35:261–270. [Google Scholar]
- 23.Guerriero G, Fugelstad J, Bulone V. What do we really know about cellulose biosynthesis in higher plants? J Integr Plant Biol. 2010;52(2):161–175. doi: 10.1111/j.1744-7909.2010.00935.x. [DOI] [PubMed] [Google Scholar]
- 24.Aloni Y, Delmer DP, Benziman M. Achievement of high rates of in vitro synthesis of 1,4-beta-D-glucan: Activation by cooperative interaction of the Acetobacter xylinum enzyme system with GTP, polyethylene glycol, and a protein factor. Proc Natl Acad Sci USA. 1982;79(21):6448–6452. doi: 10.1073/pnas.79.21.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cifuentes C, Bulone V, Emons AMC. Biosynthesis of callose and cellulose by detergent extracts of tobacco cell membranes and quantification of the polymers synthesized in vitro. J Integr Plant Biol. 2010;52(2):221–233. doi: 10.1111/j.1744-7909.2010.00919.x. [DOI] [PubMed] [Google Scholar]
- 26.Lai-Kee-Him J, et al. In vitro versus in vivo cellulose microfibrils from plant primary wall synthases: Structural differences. J Biol Chem. 2002;277(40):36931–36939. doi: 10.1074/jbc.M203530200. [DOI] [PubMed] [Google Scholar]
- 27.Pappas CT, et al. Construction and validation of the Rhodobacter sphaeroides 2.4.1 DNA microarray: Transcriptome flexibility at diverse growth modes. J Bacteriol. 2004;186(14):4748–4758. doi: 10.1128/JB.186.14.4748-4758.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wagner S, et al. Tuning Escherichia coli for membrane protein overexpression. Proc Natl Acad Sci USA. 2008;105(38):14371–14376. doi: 10.1073/pnas.0804090105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Charnock SJ, Henrissat B, Davies GJ. Three-dimensional structures of UDP-sugar glycosyltransferases illuminate the biosynthesis of plant polysaccharides. Plant Physiol. 2001;125(2):527–531. doi: 10.1104/pp.125.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ninomiya T, et al. Molecular cloning and characterization of chondroitin polymerase from Escherichia coli strain K4. J Biol Chem. 2002;277(24):21567–21575. doi: 10.1074/jbc.M201719200. [DOI] [PubMed] [Google Scholar]
- 31.DeAngelis PL, Jing W, Graves MV, Burbank DE, Van Etten JL. Hyaluronan synthase of Chlorella virus PBCV-1. Science. 1997;278(5344):1800–1803. doi: 10.1126/science.278.5344.1800. [DOI] [PubMed] [Google Scholar]
- 32.Lairson LL, Henrissat B, Davies GJ, Withers SG. Glycosyltransferases: Structures, functions, and mechanisms. Annu Rev Biochem. 2008;77:521–555. doi: 10.1146/annurev.biochem.76.061005.092322. [DOI] [PubMed] [Google Scholar]
- 33.Wallace JA, Shen JK. Predicting pKa values with continuous constant pH molecular dynamics. Methods Enzymol. 2009;466:455–475. doi: 10.1016/S0076-6879(09)66019-5. [DOI] [PubMed] [Google Scholar]
- 34.Koyama M, Helbert W, Imai T, Sugiyama J, Henrissat B. Parallel-up structure evidences the molecular directionality during biosynthesis of bacterial cellulose. Proc Natl Acad Sci USA. 1997;94(17):9091–9095. doi: 10.1073/pnas.94.17.9091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown C, Leijon F, Bulone V. Radiometric and spectrophotometric in vitro assays of glycosyltransferases involved in plant cell wall carbohydrate biosynthesis. Nat Protoc. 2012;7(9):1634–1650. doi: 10.1038/nprot.2012.089. [DOI] [PubMed] [Google Scholar]
- 36.Boehme C, et al. Chemical and enzymatic characterization of recombinant rabbit muscle pyruvate kinase. Biol Chem. 2013;394(5):695–701. doi: 10.1515/hsz-2012-0334. [DOI] [PubMed] [Google Scholar]
- 37.Denisov IG, Grinkova YV, Lazarides AA, Sligar SG. Directed self-assembly of monodisperse phospholipid bilayer nanodiscs with controlled size. J Am Chem Soc. 2004;126(11):3477–3487. doi: 10.1021/ja0393574. [DOI] [PubMed] [Google Scholar]
- 38.Park SH, et al. Nanodiscs versus macrodiscs for NMR of membrane proteins. Biochemistry. 2011;50(42):8983–8985. doi: 10.1021/bi201289c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buckstein MH, He J, Rubin H. Characterization of nucleotide pools as a function of physiological state in Escherichia coli. J Bacteriol. 2008;190(2):718–726. doi: 10.1128/JB.01020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simm R, Morr M, Kader A, Nimtz M, Römling U. GGDEF and EAL domains inversely regulate cyclic di-GMP levels and transition from sessility to motility. Mol Microbiol. 2004;53(4):1123–1134. doi: 10.1111/j.1365-2958.2004.04206.x. [DOI] [PubMed] [Google Scholar]
- 41.Benziman M, Haigler CH, Brown RM, White AR, Cooper KM. Cellulose biogenesis: Polymerization and crystallization are coupled processes in Acetobacter xylinum. Proc Natl Acad Sci USA. 1980;77(11):6678–6682. doi: 10.1073/pnas.77.11.6678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paredez AR, Somerville CR, Ehrhardt DW. Visualization of cellulose synthase demonstrates functional association with microtubules. Science. 2006;312(5779):1491–1495. doi: 10.1126/science.1126551. [DOI] [PubMed] [Google Scholar]
- 43.Ross P, et al. Regulation of cellulose synthesis in Acetobacter xylinum by cyclic diguanylic acid. Nature. 1987;325(6101):279–281. doi: 10.1038/325279a0. [DOI] [PubMed] [Google Scholar]
- 44.Harris DM, et al. Cellulose microfibril crystallinity is reduced by mutating C-terminal transmembrane region residues CESA1A903V and CESA3T942I of cellulose synthase. Proc Natl Acad Sci USA. 2012;109(11):4098–4103. doi: 10.1073/pnas.1200352109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen S, Ehrhardt DW, Somerville CR. Mutations of cellulose synthase (CESA1) phosphorylation sites modulate anisotropic cell expansion and bidirectional mobility of cellulose synthase. Proc Natl Acad Sci USA. 2010;107(40):17188–17193. doi: 10.1073/pnas.1012348107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Whitfield C. Biosynthesis and assembly of capsular polysaccharides in Escherichia coli. Annu Rev Biochem. 2006;75:39–68. doi: 10.1146/annurev.biochem.75.103004.142545. [DOI] [PubMed] [Google Scholar]
- 47.Matthysse AG, Thomas DL, White AR. Mechanism of cellulose synthesis in Agrobacterium tumefaciens. J Bacteriol. 1995;177(4):1076–1081. doi: 10.1128/jb.177.4.1076-1081.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Steiner S, Lori C, Boehm A, Jenal U. Allosteric activation of exopolysaccharide synthesis through cyclic di-GMP-stimulated protein-protein interaction. EMBO J. 2013;32(3):354–368. doi: 10.1038/emboj.2012.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pear JR, Kawagoe Y, Schreckengost WE, Delmer DP, Stalker DM. Higher plants contain homologs of the bacterial celA genes encoding the catalytic subunit of cellulose synthase. Proc Natl Acad Sci USA. 1996;93(22):12637–12642. doi: 10.1073/pnas.93.22.12637. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.