

Significance
Squamous cell carcinoma (SCC) is one of the most common human tumor types with high malignancy. Most SCC mouse models involve multiple genetic mutations. Here we present the ASPP2Δexon3/+ Balb/c mouse as a haploinsufficient SCC mouse model and identify ASPP2 as a suppressor of SCC and a potent repressor of p63 expression. The finding that ASPP2 represses p63 expression via NF-κB provides a molecular insight into how inflammatory signaling may affect SCC development. All these demonstrate that ASPP2 may serve as a molecular signature of SCC and the characterized ASPP2Δexon3/+ Balb/c mouse may provide a much-needed experimental model to extend our understanding of SCC as well as enable us to develop better strategies to treat SCC.
Keywords: inflammation, T53BP2, stratified epithelial cell tumor
Abstract
Squamous cell carcinoma (SCC) is highly malignant and refractory to therapy. The majority of existing mouse SCC models involve multiple gene mutations. Very few mouse models of spontaneous SCC have been generated by a single gene deletion. Here we report a haploinsufficient SCC mouse model in which exon 3 of the Tp53BP2 gene (a p53 binding protein) was deleted in one allele in a BALB/c genetic background. Tp53BP2 encodes ASPP2 (ankyrin repeats, SH3 domain and protein rich region containing protein 2). Keratinocyte differentiation induces ASPP2 and its expression is inversely correlated with p63 protein in vitro and in vivo. Up-regulation of p63 expression is required for ASPP2Δexon3/+ BALB/c mice to develop SCC, as heterozygosity of p63 but not p53 prevents them from developing it. Mechanistically, ASPP2 inhibits ΔNp63 expression through its ability to bind IκB and enhance nuclear Rel/A p65, a component of the NF-κB transcription complex, which mediates the repression of p63. Reduced ASPP2 expression associates with tumor metastasis and increased p63 expression in human head and neck SCCs. This study identifies ASPP2 as a tumor suppressor that suppresses SCC via inflammatory signaling through NF-κB–mediated repression of p63.
Approximately 80% of human cancers are epithelial in origin, with squamous cell carcinoma (SCC) one of the most common tumor types. SCCs predominantly derive from the squamous epithelia of the skin, oral cavity (including the esophagus), and cervix. Mouse genetics have played a key role in our understanding of the molecular pathways involved in SCC development. Most existing SCC mouse models require multistage genetic changes, the best studied being the chemically induced multistage carcinogenesis skin model that induces papillomas that contain Ras mutation, and increases inflammation (1). An inducible mutant Ras knock-in mouse model confirmed that Ras mutation is an early molecular event in SCC initiation (2). Additional mutations such as loss of p53 function are required to convert papillomas to SCCs and enhance malignant progression (3). Simultaneous deletion of p53 and Rb in mouse epidermis also results in spontaneous SCC (4), indicating that p53, Ras, or Rb mutations alone are insufficient to induce it. Only a few mouse models of spontaneous SCC have been generated by a single gene deletion. One such example is the deletion of Smad4 in skin epithelial cells, which results in spontaneous SCC with severe inflammation (5). Inflammation’s role in SCC development remains a subject of intense interest largely because NF-κB, one of the most important signaling molecules of the inflammatory response, acts as a tumor suppressor in the epidermis.
In hematopoietic tissue NF-κB is antiapoptotic and proproliferative and positively contributes to tumorigenesis. In epidermis, however, mice overexpressing the NF-κB inhibitory protein IκBα develop spontaneous SCCs (6, 7). NF-κB inhibition via IκB cooperates with RAS oncogene to promote SCC development (8). Reduced p65/NF-κB function has been observed in human SCC (9). Why p65/NF-κB acts as a tumor suppressor in the epidermis but an oncogene in other tissues (i.e., hematopoietic tissues) remains unknown. A possible explanation is its ability to regulate the expression of p63 (10, 11), a member of the p53 family and master transcription factor of epithelial stratification (12, 13) that is often overexpressed in SCC (14).
Six p63 isoforms have been identified to date, with three different C termini: TAp63α, TAp63β, and TAp63γ. The N-terminal transactivation domain-deleted isoforms are ΔNp63α, ΔNp63β, and ΔNp63γ. ΔN isoforms are functionally different from their full-length counterparts, mainly behaving as dominant-negative toward TAp63 isoforms (15). In the epidermis, TAp63 is expressed at low levels compared with ΔNp63 and is required for epidermal stem cell self-renewal (16, 17). ΔNp63 is restricted to transit-amplifying and stem cells residing in the basal layer. Its expression is down-regulated during skin differentiation and ΔNp63 regulates genes that result in basal cell proliferation (17, 18). Hence, ΔNp63 is considered an oncogene (19). It is well established that overexpression of p63 is a hallmark of SCCs, as nearly 90% express high p63 levels, particularly ΔNp63 (20). Nonetheless, we know very little about how its expression is regulated. An experimental model system is also urgently needed to determine the requirement for p63 expression in SCC development in vivo.
Here we identify ASPP2 as a repressor of SCC and p63. ASPP2 is a member of the ASPP family of proteins, which consists of three members: ASPP1, ASPP2, and iASPP. ASPP1 and ASPP2 stimulate, whereas iASPP inhibits, the activities of p53 and its family members p63 and p73 (21–23). Studies with genetically modified mice have shown that ASPP2 is a haploinsufficient tumor suppressor, and that ASPP2 and p53 cooperate to suppress the onset of lymphomas and sarcomas (24, 25). Recent studies have also shown that ASPP2 is a key mediator of RAS-induced senescence, a property independent of p53 (26, 27), suggesting that ASPP2 can suppress tumor growth through p53-dependent and -independent pathways. In contrast to ASPP2, iASPP (the inhibitory ASPP) is mainly expressed in basal epithelial cells. In vitro, iASPP is down-regulated together with p63 upon keratinocyte differentiation. Nuclear iASPP colocalizes with p63 to maintain stratified epithelial homeostasis (28, 29), and is a potent inhibitor of p53, apoptosis, and cellular senescence (23, 28, 30). These findings suggest that the ASPPs may play a key role in epithelial tumor development. We show here that ASPP2Δexon3/+ BALB/c mice represent a haploinsufficient spontaneous SCC mouse model. Mechanistically, ASPP2 suppresses SCC by inducing nuclear p65–mediated repression of p63.
Results
Keratinocyte Differentiation Induces ASPP2 Expression, Which Inversely Associates with p63 Expression in Vitro and in Vivo.
To clarify ASPP2’s role in regulating the homeostasis of squamous epithelia, we first examined its expression in the epidermis. ASPP2 was mainly detected in the differentiated layers of human and mouse skin. Double immunofluorescence (IF) staining showed that ASPP2 expression was almost mutually exclusive from that of p63. This was more pronounced in human skin (Fig. 1A), which has more extensive stratification than murine skin (Fig. S1A). This expression pattern was also observed in human cervical squamous epithelia (Fig. S1B), in which ASPP2 and iASPP expression was inversely correlated (Fig. 1B). In murine esophageal squamous epithelium ASPP2-expressing cells coexpressed keratin-4, a marker of differentiation, and were almost absent from the proliferative keratin-14 (K14)-expressing basal layer (Fig. S1C). In human esophagus ASPP2 was also mainly detected in differentiated suprabasal epithelial cells (Fig. S1D).
Fig. 1.

Mutually exclusive expression of ASPP2 and p63 in squamous epithelia and PKs. (A) Double staining of human skin squamous epithelium using anti-ASPP2 and anti-p63 antibodies. BL, basal layer; GL, granular layer; SC, stratum corneum; SL, spinous layer. (B) ASPP2 and iASPP double IF staining of human cervical epithelium sections. Nuclei were counterstained with DAPI. (C) Immunoblot shows expression levels of ASPP2, p63, and envoplakin (EVPL) in Ca2+-induced mouse PK with β-tubulin as loading control. (Scale bars: 50 µm.)
The impact of keratinocyte differentiation on ASPP2 expression was examined in mouse primary keratinocytes (PKs) in vitro. High calcium-induced mouse PK differentiation induced ASPP2 expression, the timing of which coincided with p63 down-regulation, and up-regulation of the differentiation marker envoplakin (Fig. 1C and Fig. S1E). In contrast, iASPP was down-regulated upon PK differentiation (28). These data suggest that ASPP2 may promote epithelial differentiation.
ASPP2Δexon3/+ BALB/c Mice Develop Spontaneous SCC with High Frequency and Early Onset.
p53 deficiency caused more epithelial tumors in mice with a BALB/c background than a C57BL6 background (31). We thus examined the tumor suppressive properties of ASPP2 in ASPP2Δexon3/+ and ASPP2Δexon3/Δexon3 BALB/c mice (32). The genotype birth ratio followed the expected Mendelian segregation (Fig. S2A). The overall survival rate of ASPP2Δexon3/Δexon3 mice was significantly reduced compared with heterozygous and WT littermates, with only approximately 30% surviving to 20 wk of age (Fig. S2B). Two of 13 ASPP2Δexon3/Δexon3 mice developed spontaneous SCC at 19 to 20 wk. As a result of early mortality of the ASPP2Δexon3/Δexon3 mice, tumor studies were carried out between WT and ASPP2Δexon3/+ BALB/c mice. ASPP2Δexon3/+ BALB/c mice started to develop spontaneous SCC as early as 20 wk of age. Over 80 wk, almost 50% of ASPP2Δexon3/+ mice developed tumors, compared with fewer than 10% of WT mice. The difference in tumor-bearing frequency was statistically significant (P = 0.0002; Fig. 2A), whereas tumor frequency in male vs. female ASPP2Δexon3/+ mice was not, suggesting that ASPP2’s tumor-suppressive function has no sex bias (Fig. S2C). All tumors found in ASPP2Δexon3/+ BALB/c mice were classified as carcinomas, contrasting with those observed in ASPP2 WT mice: retinoblastoma (one of 37), lymphomas (two of 37) and carcinoma (one of 37; Fig. 2B). Most of the ASPP2Δexon3/+ BALB/c tumors were located in the neck, abdomen, flanks, and chest (Fig. S2D). Mice were killed when tumors reached approximately 1 cm diameter in size (Fig. S2E). Histological analysis of the ASPP2Δexon3/+ tumors revealed that they all belonged to poorly differentiated SCCs, the main histological characteristics being barrel-shaped masses of tumoral cells (Fig. S2 F, i) and desmoplastic stroma (Fig. S2G). Necrosis was frequently observed in groups of proliferating cells (Fig. S2 F, n). Poor differentiation was underscored by a high number of pleomorphic cells with vesicular nuclei (Fig. S2 F, i′) and no keratinous pearls. Invasion of the blood vessels was also found (Fig. S2H). Large numbers of mitotic nuclei were observed, indicating a high proliferative index (Fig. S2I). Immunohistological analysis showed that the majority of tumor cells expressed high K14 and keratin-1 (K1) levels, two well-known markers for SCC, but were negative for vimentin, a mesenchymal cell marker. High levels of nuclear p63 were detected in all SCCs derived from ASPP2Δexon3/+ BALB/c mice (Fig. 2C). Double IF analysis also confirmed that K14, K1, and p63 proteins were coexpressed in the tumor cell population (Fig. S2J), whereas vimentin and keratin-18 were mainly found in the surrounding stromal compartment and normal sweat ducts within the tumor mass, respectively (Fig. S2K). By using antibodies and primers able to distinguish between the two N-terminal p63 isoforms, it was found that tumors analyzed mainly over-expressed ΔNp63 rather than TAp63 (Fig. S2 L and N). These results demonstrate that ASPP2 is a haploinsufficient tumor suppressor, and reduced ASPP2 expression associates with increased ΔNp63 expression.
Fig. 2.

ASPP2Δexon3/+ BALB/c mice develop spontaneous SCC with high frequency and early onset. (A) Tumor-free survival and (B) tumor incidence and spectrum spontaneously developed in ASPP2+/+ (WT) and ASPP2Δexon3/+ (HET) BALB/c mice over 88 wk [***P = 0.0002, log-rank (Mantel–Cox) test]. (C) Immunohistochemical staining of vimentin, K14, K1, and p63 in tumor sections. (Scale bars: 50 µm.)
Heterozygosity of p63, but Not p53, Prevents ASPP2Δexon3/+ BALB/c Mice from Developing SCC.
Mutational inactivation of p53 is frequently observed in human SCC (33). ASPP2 was originally identified as an activator of p53 (21) and cooperates with it to suppress tumor growth in vivo (24). Thus, reduced ASPP2 expression could dampen p53’s activity and predispose to SCC. The onset of SCC formation in ASPP2Δexon3/+ BALB/c mice should be enhanced in a p53+/− background. On the contrary, ASPP2 and p63 expression are mutually exclusive in normal epithelia, and all tumors derived from ASPP2Δexon3/+ BALB/c mice express high levels of nuclear p63. ASPP2 may suppress SCC by repressing p63’s expression in stratified epithelia. Reduced p63 expression might prevent SCC development in ASPP2Δexon3/+ BALB/c mice. To test these two hypotheses, ASPP2Δexon3/+ BALB/c mice were crossed with p53+/− or p63+/− BALB/c mice to generate a compound mouse. ASPP2Δexon3/+;p53+/− or ASPP2Δexon3/+;p63+/− mice were then intercrossed. As reported by Vives et al., ASPP2Δexon3/Δexon3;p53−/− mice were rarely obtained after birth (24). Thus, the ASPP2/p53 tumor study was performed with the eight available genotypes (Fig. S3 A and B). In the WT p53 background, ASPP2Δexon3/Δexon3;p53+/+ mice mainly developed carcinomas (66% carcinomas, 34% sarcomas), and tumor onset was earlier than in ASPP2Δexon3/+;p53+/+mice, but the ASPP2Δexon3/Δexon3;p53+/+ cohort was too small to reach statistical significance as a result of premature lethality. ASPP2Δexon3/+;p53+/+ mice had significantly higher carcinoma incidence (90% carcinomas, 10% lymphomas) than ASPP2+/+;p53+/+ mice and poorer tumor-free survival (P = 0.03). In a p53 heterozygous background, ASPP2Δexon3/Δexon3;p53+/− mice started to develop tumors at 9 wk (75% carcinomas, 25% lymphomas), and tumor-free survival rate was significantly worse than ASPP2Δexon3/+;p53+/− mice (P < 0.0001). ASPP2Δexon3/+;p53+/− mice developed tumors (45% carcinomas, 55% of lymphomas and sarcomas) faster than ASPP2+/+;p53+/− mice (P = 0.03). This confirms our earlier finding that ASPP2 and p53 cooperate in tumor suppression. In a p53-null background, ASPP2Δexon3/+;p53−/− and ASPP2+/+;p53−/− mice developed lymphomas and sarcomas exclusively with similar onset regardless of ASPP2 status, supporting the notion that p53 is the dominant tumor suppressor and is downstream of ASPP2 (Fig. S3 B–G). This study shows that p53 heterozygosity failed to enhance SCC in ASPP2Δexon3/+ BALB/c mice, as the percentage of carcinomas observed in ASPP2Δexon3/+;p53+/+, ASPP2Δexon3/+;p53+/−, and ASPP2Δexon3/+;p53−/− mice were 26%, 19%, and zero, respectively (Fig. S3C, bars marked by triangles).
The ability of ASPP2 to interact with p63 to suppress SCC was next tested in compound mice obtained from an intercross of ASPP2Δexon3/+;p63+/− BALB/c mice. Because of the early lethality of p63−/− and ASPP2Δexon3/Δexon3 mice (12, 13, 24), the study was performed in the remaining four genotypes. Remarkably, none of the ASPP2Δexon3/+;p63+/− mice developed any spontaneous tumor by 80 wk (Fig. 3A), demonstrating that p63 heterozygosity is sufficient to prevent ASPP2Δexon3/+ BALB/c mice from developing spontaneous SCC. Approximately 24% of the ASPP2Δexon3/+;p63+/+ mice developed spontaneous tumors, all of which were SCCs (Fig. 3B and Fig. S3H). Hence, ASPP2 is likely to suppress SCC development by repressing p63 expression.
Fig. 3.
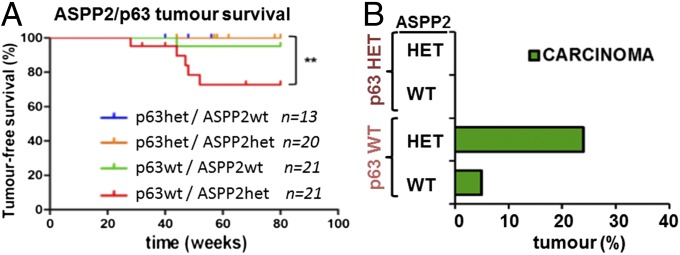
Heterozygosity of p63 prevents ASPP2Δexon3/+ BALB/c mice from developing SCC. (A) Tumor-free survival and (B) tumor incidence and spectrum in ASPP2/p63 compound mice with indicated genotypes over 80 wk [**P = 0.0086 by log-rank (Mantel–Cox) test].
ASPP2 Represses p63 Expression.
Double IF staining of p63 and ASPP2 in SCC tumors from ASPP2Δexon3/+ mice showed very few ASPP2-positive cells (5%) scattered within the tumor mass, whereas most tumor cells expressed p63 (78%; Fig. S4A). These tumors retained the WT ASPP2 allele (Fig. S4B), supporting the notion that ASPP2 is a haploinsufficient suppressor of SCC. Importantly, in ASPP2-enriched tumor regions, ASPP2 and p63 expression were mutually exclusive, and no cells coexpressed ASPP2 and p63 (Fig. 4A). Double IF staining of human cutaneous SCC confirmed that ASPP2 and p63 expression were again mutually exclusive. ASPP2 was expressed in only a few cells within the tumor mass or in residual normal stratified epithelium, whereas most tumor cells expressed p63 (Fig. S4C). The molecular mechanisms controlling ASPP2 and p63’s mutually exclusive expression were analyzed in ASPP2+/+ and ASPP2Δexon3/Δexon3 mouse PKs and mouse embryonic fibroblasts (MEFs). In both cell types, the amount of ΔNp63 transcript was much higher than that of TAp63, measured by real-time quantitative PCR (RT-qPCR) using isoform-specific primers (Fig. S4D). An approximately three- or sixfold increase in ΔNp63 mRNA was observed in ASPP2Δexon3/Δexon3 compared with ASPP2+/+ PKs and MEFs, respectively, using RT-qPCR. Very little change was detected in TAp63 expression levels under the same conditions (Fig. 4B and Fig. S4 E and F). A small increase in ΔNp63 protein was also observed (Fig. S4G). These findings suggest that ASPP2 may repress p63 expression. To investigate how, the expression levels of ASPP2, iASPP, and p63 were analyzed in a panel of 34 human SCC cell lines by immunoblotting. Interestingly, iASPP was expressed at similar levels in nearly all lines tested, most of which also expressed p63, although expression levels varied. Most of the lines expressed low levels of ASPP2, and the mean of p63 expression was significantly different and higher than that of ASPP2 (P = 0.0009; Fig. S4 H and I). An inverse correlation between p63 and ASPP2 expression was also observed in some cell lines (Fig. S4H, red and green frames). The signal intensity of ASPP2 and p63 (normalized by using iASPP levels) was quantified in each cell line and is shown in Fig. S4J. The values obtained from the ratios between p63 and ASPP2 expression levels for each line were used to produce a scatter-plot graph (Fig. S4K). Approximately 22% of the cell lines had similar p63:ASPP2 expression ratios (∼1; Fig. S4K, yellow dots), whereas the remaining 78% had imbalanced expression. Most of the SCC cell lines (59%) had a high p63:ASPP2 ratio (Fig. S4K, red dots). In approximately 19% of the lines, values of the p63:ASPP2 ratios were less than 1 (Fig. S4K, blue dots). The cell lines with profoundly imbalanced ASPP2 and p63 expression patterns provided us with an experimental system with which to investigate how ASPP2 may negatively regulate p63 expression in vitro. UPCI-SCC-040 cells (henceforth “040 cells”; SI Materials and Methods) express high levels of p63 in almost all cells, whereas ASPP2 expression is barely detectable (Fig. 4D and Fig. S4H). The 040 cells were used to test whether increased ASPP2 level could repress p63 expression. V5-tagged ASPP2 was transiently transfected into 040 cells, and anti-V5 and anti-p63 antibodies were used for double IF staining to detect the exogenously introduced V5-ASPP2 and endogenous p63. In empty vector-transfected V5− cells, fewer than 1% of cells lacked p63 expression. Remarkably, in approximately 70% of transfected cells expressing detectable ASPP2-V5, endogenous p63 expression was undetectable. Exogenously expressed iASPP-V5 failed to affect endogenous p63 expression, with only approximately 2% of iASPP-V5–expressing cells losing endogenous p63 expression (Fig. 4 C and D). Similar results were obtained in another human SCC cell line, HSC3 (Fig. S4L). A reduction of ΔNp63 mRNA of approximately 40% was detected in 040 cells upon transient expression of ASPP2-V5 (Fig. S4M). The ability of increased iASPP or reduced ASPP2 expression to influence p63 expression was further tested in the immortalized human keratinocyte cell line HaCat. As earlier, iASPP overexpression did not affect p63 expression, whereas ASPP2 RNAi induced endogenous p63 expression in HaCat cells (Fig. S4 N and O). These data illustrate that ASPP2 but not iASPP is able to specifically repress ΔNp63 expression.
Fig. 4.

ASPP2 represses p63 expression in vivo and in vitro. (A) Double IF staining of ASPP2 and p63 in a tumor section derived from an ASPP2Δexon3/+ BALB/c mouse. (Scale bar: 20 µm.) (B) RT-qPCR expression analysis of ΔNp63 and TAp63 mRNA in mouse PKs (whole epidermis), with GAPDH mRNA as internal control. (*P = 0.034; n indicates the number of littermate-paired PKs used). Bar graph values are the mean ± SD from three different experiments. (C and D) Double IF staining of 040 cells to detect transfected ASPP2-V5 (red) or iASPP-V5 (red) and endogenous p63 (green). TO-PRO was used to visualize nuclei. ASPP2-V5+ and iASPP-V5+ cells are labeled with white arrows (D). The percentage of V5+/p63− cells in indicated transfected samples is shown in C (***P < 0.0001). Values are mean ± SD from three different experiments. (Scale bars: 10 µm.)
ASPP2 Represses p63 Expression by Binding IκB and Inducing Nuclear p65/NF-κB.
We showed previously that ASPP2 partially overlaps with β-catenin at cell–cell junctions and can be detected in the same protein complex (32). A more recent study showed that nuclear β-catenin can directly transactivate ΔNp63 expression (34). We tested whether ASPP2 could repress p63’s expression by inhibiting β-catenin–mediated induction of ΔNp63. The expression of endogenous p63 was assessed by IF staining of 040 cells transfected with plasmids expressing ASPP2 or β-catenin constitutively active mutant (ΔN89 β-catenin), separately or together. As expected, exogenous ASPP2 expression repressed endogenous p63 expression. Expression of ΔN89 β-catenin alone failed to affect p63 expression and when coexpressed with ASPP2, it failed to prevent ASPP2 from repressing p63’s expression, even though it was detected as a nuclear protein in the same cells in which ASPP2-V5 was expressed. Endogenous nuclear β-catenin expression was almost undetectable in 040 cells (Fig. S5A). These data suggest that the ability of ASPP2 to repress p63 expression is likely to be independent of nuclear β-catenin.
The Notch signaling pathway is one of the main pathways that negatively regulate p63’s expression in stratified epithelial cells (11). Notch1 is under positive transcriptional regulation by p53 (35) and is mainly expressed in the differentiated layers of the epithelium. Whether Notch1 could mediate ASPP2-induced p63 repression was tested by using Notch1 RNAi with an ASPP2-V5 expression plasmid. The presence of Notch1 RNAi almost abolished Notch1 expression, but failed to influence ASPP2’s ability to repress p63 expression in 040 cells (Fig. S5 B and C), suggesting that ASPP2 inhibits p63 expression independently of Notch1.
RelA/p65 has been shown to be nuclear in differentiated, but cytoplasmic in basal p63+, epidermal layers (6). RelA/p65 can also repress ΔNp63 expression in a transcription-dependent and -independent manner in epithelial cells (10, 11). As ASPP2 is mainly expressed in the differentiated layers of the epithelium, and a known binding protein of RelA/p65 (36), we tested its ability to repress p63 expression by binding and activating RelA/p65. Endogenous RelA/p65 was mainly cytoplasmic in 040 cells. Double IF staining showed that exogenously expressed cytoplasmic ASPP2-V5 induced nuclear accumulation of RelA/p65 in approximately 66% of ASPP2-V5–expressing cells, compared with fewer than 5% of empty vector-transfected V5− cells (Fig. 5A) Triple IF staining showed that ASPP2-V5–induced nuclear RelA/p65 expression associated with a significant reduction in p63 expression in approximately 64% of cells (Fig. 5B). Knockdown of RelA/p65 by RNAi largely prevented ASPP2-V5 from repressing p63 expression. The percentage of ASPP2-V5+/p63− cells was more than 60% in control RNAi-transfected cells, but reduced to approximately 20% in RelA/p65 RNAi-transfected cells (Fig. 5B). ASPP2-V5 was also transfected into 040 cells alone, or in the presence or absence of IκBβ, an inhibitor that binds and retains RelA/p65 in the cytoplasm to inhibit its transcriptional activity. When expressed alone, only 2% of transfected cells had reduced p63 expression. As before, ASPP2 reduced p63 expression in approximately 70% of transfected cells. When IκBβ was expressed with ASPP2-V5, down-regulation of p63 was observed in only 25% of cotransfected cells (Fig. 5C and Fig. S5D), an effect only observed in cells expressing high IκBβ levels (Fig. S5D, yellow arrows). In low IκBβ-expressing cells, ASPP2 still repressed p63 expression (Fig. S5D, white arrows). These results suggest that ASPP2 may counteract IκBβ to induce nuclear RelA/p65 to repress p63’s expression. It was confirmed that ASPP2 status has minimal impact on RelA/p65 and IκB expression in ASPP2+/+ and ASPP2Δexon3/Δexon3 MEFs (Fig. S5E). Interestingly, an anti-ASPP2 antibody coimmunoprecipitated IκBβ and p65/NF-κB in ASPP2+/+ MEFs. ASPP2 failed to coimmunoprecipitate p105/p50 under the same conditions (Fig. 5D). Only IκBβ and not RelA/p65 was able to complex with ASPP2 in a reciprocal immunoprecipitation assay (Fig. S5 F and G). IκBβ coimmunoprecipitated with RelA/p65 and p105/p50, but not with β-catenin (Fig. S5F) or iASPP (Fig. S5H) under the same conditions. The amount of the IκBβ–RelA/p65 complex detected in ASPP2Δexon3/Δexon3 MEFs was visibly higher than that in ASPP2+/+ MEFs (Fig. 5E and Fig. S5I). These results suggest that ASPP2 may repress p63’s expression by competing with RelA/p65 to bind IκB, thus inducing RelA/p65’s nuclear accumulation.
Fig. 5.

ASPP2 inhibits p63 by inducing nuclear p65/NFκB. (A) Double IF staining of 040 cells using anti-V5 (red) and anti-RelA/p65 (green) antibodies. Arrows label cells expressing ASPP2-V5 and nuclear RelA/p65. The graph shows the percentage of nuclear RelA/p65 expressing cells in indicated transfected samples (***P < 0.001). (B) Triple IF staining of 040 cells to detect transfected ASPP2-V5 (red), endogenous RelA/p65 (green), and endogenous p63 (magenta) in cells treated with control (CTR) or RelA/p65 (NFκB) RNAi. Arrows label ASPP2-V5–expressing cells. The bar graph shows the percentage of V5+/p63− cells in transfected samples as indicated. (**P = 0.0083). (C) Graph shows the percentage of cells with low or undetectable p63 in transfected samples as indicated. (**P = 0.001). (D and E) Lysates from ASPP2 WT and Δexon3 MEFs were immunoprecipitated with a control IgG (IP CTR) or indicated antibodies (IP ASPP2, IP IκB) and immunoblotted with antibodies as labeled. (Scale bars: 10 µm.) Bar graph values are the mean ± SD from three different experiments.
Down-Regulation of ASPP2 Associates with Increased p63 Expression and Tumor Metastasis in Human Head and Neck SCC.
In human samples, ASPP2’s expression was mainly confined to adjacent normal squamous epithelium, and was strongly decreased in the neighboring tumor mass (Fig. 6A). ASPP2 expression was subsequently examined in a cohort of 318 human head and neck SCCs (HNSCCs) from a tissue microarray, including nonmetastatic and metastatic tumors and lymph node metastases. Biopsies of nontransformed epithelia were used as a reference for ASPP2 expression in normal stratified epithelia, in which cytoplasmic ASPP2 was seen in all cases. In 26% of primary tumors, ASPP2 expression was undetectable. In the remaining ASPP2-expressing tumors, further ASPP2 down-regulation was observed in metastatic tumor samples, and the lowest expression levels were detected in lymph node metastases (P < 0.01; Fig. S6 A and B). Among the 74 primary HNSCC samples, we observed a significant inverse association between p63 and ASPP2 expression. High levels of ASPP2 tended to be detected in p63-negative samples (Fig. S6C). As most metastatic HNSCCs do not express p63 and many also have reduced ASPP2, we failed to see any association between p63 and ASPP2 expression in these samples. These results showed that down-regulation of ASPP2 frequently occurs in human HNSCC and associates with increased p63 expression and tumor metastasis. The findings presented in this study identify ASPP2 as a key suppressor of SCC.
Fig. 6.

ASPP2 expression is decreased in human SCC samples. (A) ASPP2 immunostain of human HNSCC. Areas of normal epithelium and tumor mass are shown at higher magnification. (Scale bars: 50 µm and 10 µm.) (B) Diagram to summarize the interplay between ASPP2, IκB, and RelA/p65 in regulating ΔNp63 expression.
Discussion
Mutation of p53, and overexpression of ΔNp63 and EGFR, are frequent events in the development and progression of human SCC. Existing mouse SCC models often involve multistage carcinogenesis or multigene mutations. The finding that ASPP2Δexon3/+ BALB/c mice spontaneously developed SCC, and p63 heterozygosity prevented ASPP2 haploinsufficiency-caused SCC, identifies ASPP2 as a key SCC suppressor, and its tumor suppressive function is partly a result of its ability to repress p63 expression. The exact role of p53 in mediating ASPP2’s ability to suppress SCC is less clear. Under the same conditions, p53 heterozygosity reduced the frequency of SCC, although this was not statistically significant (Fig. S3C). The interaction between ASPP2 and p53 in suppressing SCC could be masked by the early onset of lymphomas and sarcomas caused by p53 deficiency. Specific deletion of ASPP2 and p53 in keratinocytes is needed to confirm whether ASPP2 can indeed suppress SCC development through a p53-independent mechanism.
In a mixed genetic background of 129SvJ/C57BL6, ASPP2 heterozygosity mainly conferred susceptibility to lymphomas and sarcomas, with initial tumor onset at 60 or 80 wk of age (24, 25). Tumor onset of ASPP2Δexon3/+ BALB/c mice was more than 40 wk earlier, and the tumor spectrum altered from mainly lymphomas and sarcomas to SCCs. The dramatic difference in tumor latency and spectrum between the two backgrounds demonstrates that modifiers must cooperate with ASPP2 to suppress tumor development. ASPP2 mediates oncogenic RAS-induced senescence by preventing oncogenic RAS from inducing autophagy (27), and SUMO-modification and nuclear induction of Cyclin D1 (26), both required for RAS’ full oncogenic function. ASPP2 can also enhance oncogenic RAS-induced apoptosis in cancer cells (37). Hence, loss of ASPP2 potentiates the tumorigenic properties of oncogenic RAS. Interestingly, deregulated EGFR/RAS activity caused by overexpression or mutation is a common event in human SCC, and RAS mutations are frequently used to induce SCC in mice (2). Therefore, a significant difference in EGFR/RAS activity in the BALB/c, 129SvJ, and C57BL6 mouse strains may explain the observed differences in tumor onset and spectra between ASPP2Δexon3/+ BALB/c and 129SvJ/C57BL6 mice. Future studies are needed to test this hypothesis, by comparing tumor onset and spectra of ASPP2Δexon3/+ mice in BALB/c, C57BL6, and 129SvJ genetic backgrounds in the presence or absence of oncogenic RAS.
In addition to the p53/p63 and EGFR/RAS pathways, the association between SCC and inflammation is also emerging. It is currently unknown why increased NF-κB induces keratinocyte differentiation and represses SCC, or how NF-κB activity is regulated in squamous epithelia. The finding that ASPP2 could suppress SCC development through its ability to bind IκB, and induce nuclear RelA/p65-mediated repression of p63 expression, provides a molecular link between the NF-κB pathway and p63. ASPP2 was previously identified as an interacting protein of RelA/p65 in a yeast two-hybrid assay (36), and can also bind p63 and activate its apoptotic function in a cell-culture system (22). RelA/p65 has been shown to bind p63 and target it for degradation (10). In these studies, protein complex formation was needed to either influence the activity of p63 or RelA/p65. However, as ASPP2 is predominantly cytoplasmic in keratinocytes and SCC cells, whereas p63 and RelA/p65 are predominantly nuclear, the previously identified protein/protein interactions are unlikely to mediate ASPP2-induced repression of p63 expression and nuclear RelA/p65 induction. Cytoplasmic ASPP2 is more likely to function through its ability to bind IκB in the cytoplasm, and regulate the nuclear localization of RelA/p65 and p63’s expression (Fig. 6B). Overexpressing IκB can bypass ASPP2’s ability to repress p63 expression in the SCC cell line 040, consistent with the notion that constitutive IκB expression can block NF-κB signaling and induce SCC in vivo (6). Indeed, in suprabasal epithelial cells, ASPP2 cytoplasmic expression corresponds with nuclear expression of RelA/p65 NF-κB and low p63 expression. The finding that ASPP2 uses the IκB-RelA/p65 pathway to repress p63’s expression is of particular interest in light of the observed difference in ASPP2’s tumor suppressive function according to mouse strain used. Emerging evidence indicates differences between BALB/c, 129SvJ, and C57BL6 mice in their inflammatory response, such as their ability to heal corneal epithelia wounds (38). Thus, future investigations may elucidate whether differences in NF-κB signaling in the aforementioned mouse strains may influence ASPP2’s tumor-suppressive function. Finally, the observation that reduced ASPP2 expression is strongly associated with metastatic human HNSCCs agrees with a previous report of reduced ASPP2 mRNA expression in human breast cancer metastases (39). This finding also agrees with the ASPP2Δexon3/+ BALB/c mouse model, in which reduced levels of ASPP2 are sufficient to cause undifferentiated and often invasive spontaneous SCC. Together, these studies show that ASPP2 is a key haploinsufficient tumor suppressor of SCC.
Materials and Methods
ASPP2 Δexon3 C57BL/6Jx129SvJ mice (26) were backcrossed in a BALB/c background for nine generations. p53+/− and p63+/− BALB/c mice were generated by G.L. and F.D.M., respectively. All animal procedures were approved by the University of Oxford's ethical review committee and licensed by the UK Home Office (license number PPL 30/2862). Human tumor samples were obtained with full consent. Cell-culture experiments were performed by using MEFs, PKs, or SCC cell lines. More details are provided in SI Materials and Methods.
Supplementary Material
Acknowledgments
This work was mainly funded by the Ludwig Institute for Cancer Research and the Medical Research Council, UK.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1309362110/-/DCSupplemental.
References
- 1.Quintanilla M, Brown K, Ramsden M, Balmain A. Carcinogen-specific mutation and amplification of Ha-ras during mouse skin carcinogenesis. Nature. 1986;322(6074):78–80. doi: 10.1038/322078a0. [DOI] [PubMed] [Google Scholar]
- 2.Vitale-Cross L, Amornphimoltham P, Fisher G, Molinolo AA, Gutkind JS. Conditional expression of K-ras in an epithelial compartment that includes the stem cells is sufficient to promote squamous cell carcinogenesis. Cancer Res. 2004;64(24):8804–8807. doi: 10.1158/0008-5472.CAN-04-2623. [DOI] [PubMed] [Google Scholar]
- 3.Kemp CJ, Donehower LA, Bradley A, Balmain A. Reduction of p53 gene dosage does not increase initiation or promotion but enhances malignant progression of chemically induced skin tumors. Cell. 1993;74(5):813–822. doi: 10.1016/0092-8674(93)90461-x. [DOI] [PubMed] [Google Scholar]
- 4.Martínez-Cruz AB, et al. Spontaneous squamous cell carcinoma induced by the somatic inactivation of retinoblastoma and Trp53 tumor suppressors. Cancer Res. 2008;68(3):683–692. doi: 10.1158/0008-5472.CAN-07-3049. [DOI] [PubMed] [Google Scholar]
- 5.Qiao W, et al. Hair follicle defects and squamous cell carcinoma formation in Smad4 conditional knockout mouse skin. Oncogene. 2006;25(2):207–217. doi: 10.1038/sj.onc.1209029. [DOI] [PubMed] [Google Scholar]
- 6.Seitz CS, Lin Q, Deng H, Khavari PA. Alterations in NF-kappaB function in transgenic epithelial tissue demonstrate a growth inhibitory role for NF-kappaB. Proc Natl Acad Sci USA. 1998;95(5):2307–2312. doi: 10.1073/pnas.95.5.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Hogerlinden M, Rozell BL, Ahrlund-Richter L, Toftgård R. Squamous cell carcinomas and increased apoptosis in skin with inhibited Rel/nuclear factor-kappaB signaling. Cancer Res. 1999;59(14):3299–3303. [PubMed] [Google Scholar]
- 8.Dajee M, et al. NF-kappaB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature. 2003;421(6923):639–643. doi: 10.1038/nature01283. [DOI] [PubMed] [Google Scholar]
- 9.Liu B, et al. A critical role for I kappaB kinase alpha in the development of human and mouse squamous cell carcinomas. Proc Natl Acad Sci USA. 2006;103(46):17202–17207. doi: 10.1073/pnas.0604481103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sen T, Chang X, Sidransky D, Chatterjee A. Regulation of ΔNp63α by NFκΒ. Cell Cycle. 2010;9(24):4841–4847. doi: 10.4161/cc.9.24.14093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nguyen BC, et al. Cross-regulation between Notch and p63 in keratinocyte commitment to differentiation. Genes Dev. 2006;20(8):1028–1042. doi: 10.1101/gad.1406006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mills AA, et al. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature. 1999;398(6729):708–713. doi: 10.1038/19531. [DOI] [PubMed] [Google Scholar]
- 13.Yang A, et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature. 1999;398(6729):714–718. doi: 10.1038/19539. [DOI] [PubMed] [Google Scholar]
- 14.Di Como CJ, et al. p63 expression profiles in human normal and tumor tissues. Clin Cancer Res. 2002;8(2):494–501. [PubMed] [Google Scholar]
- 15.Stiewe T. The p53 family in differentiation and tumorigenesis. Nat Rev Cancer. 2007;7(3):165–168. doi: 10.1038/nrc2072. [DOI] [PubMed] [Google Scholar]
- 16.Su X, et al. TAp63 prevents premature aging by promoting adult stem cell maintenance. Cell Stem Cell. 2009;5(1):64–75. doi: 10.1016/j.stem.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Candi E, et al. TAp63 and DeltaNp63 in cancer and epidermal development. Cell Cycle. 2007;6(3):274–285. doi: 10.4161/cc.6.3.3797. [DOI] [PubMed] [Google Scholar]
- 18.Koster MI, Roop DR. Mechanisms regulating epithelial stratification. Annu Rev Cell Dev Biol. 2007;23:93–113. doi: 10.1146/annurev.cellbio.23.090506.123357. [DOI] [PubMed] [Google Scholar]
- 19.Keyes WM, et al. ΔNp63α is an oncogene that targets chromatin remodeler Lsh to drive skin stem cell proliferation and tumorigenesis. Cell Stem Cell. 2011;8(2):164–176. doi: 10.1016/j.stem.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sniezek JC, Matheny KE, Westfall MD, Pietenpol JA. Dominant negative p63 isoform expression in head and neck squamous cell carcinoma. Laryngoscope. 2004;114(12):2063–2072. doi: 10.1097/01.mlg.0000149437.35855.4b. [DOI] [PubMed] [Google Scholar]
- 21.Samuels-Lev Y, et al. ASPP proteins specifically stimulate the apoptotic function of p53. Mol Cell. 2001;8(4):781–794. doi: 10.1016/s1097-2765(01)00367-7. [DOI] [PubMed] [Google Scholar]
- 22.Bergamaschi D, et al. ASPP1 and ASPP2: Common activators of p53 family members. Mol Cell Biol. 2004;24(3):1341–1350. doi: 10.1128/MCB.24.3.1341-1350.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bergamaschi D, et al. iASPP oncoprotein is a key inhibitor of p53 conserved from worm to human. Nat Genet. 2003;33(2):162–167. doi: 10.1038/ng1070. [DOI] [PubMed] [Google Scholar]
- 24.Vives V, et al. ASPP2 is a haploinsufficient tumor suppressor that cooperates with p53 to suppress tumor growth. Genes Dev. 2006;20(10):1262–1267. doi: 10.1101/gad.374006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kampa KM, et al. Apoptosis-stimulating protein of p53 (ASPP2) heterozygous mice are tumor-prone and have attenuated cellular damage-response thresholds. Proc Natl Acad Sci USA. 2009;106(11):4390–4395. doi: 10.1073/pnas.0809080106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang XD, et al. SUMO-modified nuclear cyclin D1 bypasses Ras-induced senescence. Cell Death Differ. 2011;18(2):304–314. doi: 10.1038/cdd.2010.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, et al. Autophagic activity dictates the cellular response to oncogenic RAS. Proc Natl Acad Sci USA. 2012;109(33):13325–13330. doi: 10.1073/pnas.1120193109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Notari M, et al. Inhibitor of apoptosis-stimulating protein of p53 (iASPP) prevents senescence and is required for epithelial stratification. Proc Natl Acad Sci USA. 2011;108(40):16645–16650. doi: 10.1073/pnas.1102292108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chikh A, et al. iASPP/p63 autoregulatory feedback loop is required for the homeostasis of stratified epithelia. EMBO J. 2011;30(20):4261–4273. doi: 10.1038/emboj.2011.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu M, et al. Restoring p53 function in human melanoma cells by inhibiting MDM2 and cyclin B1/CDK1-phosphorylated nuclear iASPP. Cancer Cell. 2013;23(5):618–633. doi: 10.1016/j.ccr.2013.03.013. [DOI] [PubMed] [Google Scholar]
- 31.Kuperwasser C, et al. Development of spontaneous mammary tumors in BALB/c p53 heterozygous mice. A model for Li-Fraumeni syndrome. Am J Pathol. 2000;157(6):2151–2159. doi: 10.1016/S0002-9440(10)64853-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sottocornola R, et al. ASPP2 binds Par-3 and controls the polarity and proliferation of neural progenitors during CNS development. Dev Cell. 2010;19(1):126–137. doi: 10.1016/j.devcel.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 33.Ziegler A, et al. Sunburn and p53 in the onset of skin cancer. Nature. 1994;372(6508):773–776. doi: 10.1038/372773a0. [DOI] [PubMed] [Google Scholar]
- 34.Ruptier C, et al. TP63 P2 promoter functional analysis identifies β-catenin as a key regulator of ΔNp63 expression. Oncogene. 2011;30(46):4656–4665. doi: 10.1038/onc.2011.171. [DOI] [PubMed] [Google Scholar]
- 35.Lefort K, et al. Notch1 is a p53 target gene involved in human keratinocyte tumor suppression through negative regulation of ROCK1/2 and MRCKalpha kinases. Genes Dev. 2007;21(5):562–577. doi: 10.1101/gad.1484707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang JP, et al. NF-kappaB subunit p65 binds to 53BP2 and inhibits cell death induced by 53BP2. Oncogene. 1999;18(37):5177–5186. doi: 10.1038/sj.onc.1202904. [DOI] [PubMed] [Google Scholar]
- 37.Wang Y, et al. ASPP1 and ASPP2 bind active RAS, potentiate RAS signalling and enhance p53 activity in cancer cells. Cell Death Differ. 2013;20(4):525–534. doi: 10.1038/cdd.2013.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pal-Ghosh S, Tadvalkar G, Jurjus RA, Zieske JD, Stepp MA. BALB/c and C57BL6 mouse strains vary in their ability to heal corneal epithelial debridement wounds. Exp Eye Res. 2008;87(5):478–486. doi: 10.1016/j.exer.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sgroi DC, et al. In vivo gene expression profile analysis of human breast cancer progression. Cancer Res. 1999;59(22):5656–5661. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.