

Abstract
The ability of sulfate aerosols to reflect solar radiation and simultaneously act as cloud condensation nuclei renders them central players in the global climate system. The oxidation of S(IV) compounds and their transport as stable S(VI) in the Earth’s system are intricately linked to planetary scale processes, and precise characterization of the overall process requires a detailed understanding of the linkage between climate dynamics and the chemistry leading to the product sulfate. This paper reports a high-resolution, 22-y (1980–2002) record of the oxygen-triple isotopic composition of sulfate (SO4) aerosols retrieved from a snow pit at the South Pole. Observed variation in the O-isotopic anomaly of SO4 aerosol is linked to the ozone variation in the tropical upper troposphere/lower stratosphere via the Ozone El-Niño Southern Oscillations (ENSO) Index (OEI). Higher ∆17O values (3.3‰, 4.5‰, and 4.2‰) were observed during the three largest ENSO events of the past 2 decades. Volcanic events inject significant quantities of SO4 aerosol into the stratosphere, which are known to affect ENSO strength by modulating stratospheric ozone levels (OEI = 6 and ∆17O = 3.3‰, OEI = 11 and ∆17O = 4.5‰) and normal oxidative pathways. Our high-resolution data indicated that ∆17O of sulfate aerosols can record extreme phases of naturally occurring climate cycles, such as ENSOs, which couple variations in the ozone levels in the atmosphere and the hydrosphere via temperature driven changes in relative humidity levels. A longer term, higher resolution oxygen-triple isotope analysis of sulfate aerosols from ice cores, encompassing more ENSO periods, is required to reconstruct paleo-ENSO events and paleotropical ozone variations.
Keywords: Pinatubo, El-Chichón, Cerro Hudson, Intertropical Convergence Zone
Sulfate aerosols affect climate systems by altering radiation balance, temperature, precipitation, and atmospheric dynamics (1, 2). The overall effect of sulfate aerosols on the climate is estimated to be net cooling (−2.0 ± 0.2 Wm−2) (3). According to the Intergovernmental Panel on Climate Change fourth assessment report, aerosols are one of the largest sources of uncertainties in climate prediction models due to their temporal and spatial variability (4). Globally, anthropogenic sulfur exceeds natural sources by a factor of 3 to 4 (35 Tg⋅y−1 vs. 10 Tg⋅y−1 from 1990 to 2000) (5–8). Occasionally, volcanoes emit large quantities of sulfur dioxide (SO2) directly into the atmosphere. Pinatubo, for example, released ∼30 Tg of SO2 (5). In the troposphere, SO2 has an atmospheric residence time of ∼2–3 d, and it is oxidized to SO42− by homogeneous and heterogeneous pathways and removed via wet and dry deposition (9). Gas phase oxidation of SO2 by OH and subsequent reactions with water vapor yield sulfuric acid vapor [H2SO4(g)] (10). The oxidation of aqueous SO2 by O3 and H2O2 far exceeds gas phase rates and is pH-dependent (11, 12). The oxygen-triple isotopic composition of sulfate aerosols has been demonstrated to be a useful diagnostic tool to distinguish and quantify reaction pathways and to determine the paleooxidant levels on centennial to millennium (glacial period) time scales to present-day environments (13–18). Tropospheric SO2 has a mass-dependent oxygen isotopic composition (δ17O ≈ 0.52 δ18O) due to rapid isotopic equilibration with water vapor (19), which erases the source-derived oxygen isotopic signature. [The delta (δ) values denote the relative deviation of the isotope ratios 17R= (17O/16O) and 18R = (18O/16O) in a sample (Rs) with respect to standard material (Rst) in permill (‰) (e.g., δ17O [‰] = [17Rs/17Rst −1] * 1,000). Isotope abundance or depletion is measured with reference to a standard material (e.g., for oxygen isotopes, reference material is Vienna Standard Mean Ocean Water [VSMOW]).] S(IV) species (SO2aq, HSO3−, and SO3−2) are oxidized to stable sulfate [S(VI)] via OH radicals, H2O2 and O3 (19, 20). The ozone molecule is a unique quantitative tracer of oxidation reactions because it possesses the highest enrichment in the heavier isotopes of oxygen (70–120‰) and oxygen isotope anomaly (∆17O = 25–30‰). The anomalous oxygen isotopic distribution of ozone has been shown to be transferred to oxygen-carrying molecules, such as SOx-NOx-ClOx-HOx (21–25) (∆17O ≈ δ17O − 0.52 δ18O, a mass-dependent process, has ∆17O = 0; mass-independent processes have ∆17O ≠ 0). The positive ∆17O of sulfate derives from aqueous phase oxidation of SO2 by H2O2 and O3 via Reaction 2–Reaction 3 and involves transfer of the isotopic anomaly from the oxidant to the product sulfate (12, 26). All other sulfate sources, including gas-phase oxidation by OH in the troposphere via Reaction 1a and metal-catalyzed oxidation by atmospheric O2, possess mass-dependent signatures, as verified by laboratory and field measurements (12, 14, 27, 28). However, OH in the stratosphere has been suggested to possess an O-isotopic anomaly (28, 29), which can be transferred to sulfate produced in the stratosphere via Reaction 1b. The magnitude of the transfer of the ∆17O depends on the relative contribution (Reaction 1–Reaction 3):
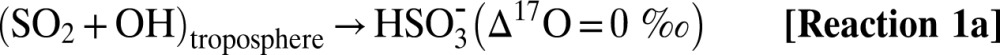 |
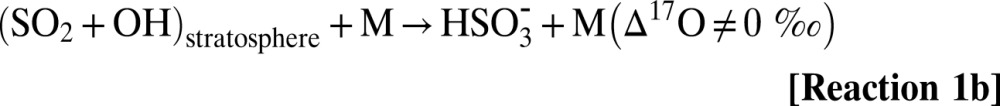 |
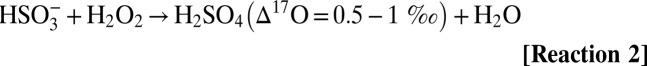 |
Tropospheric S(IV) oxidation by O3 (Reaction 3) is the only significant mechanism producing sulfate ∆17O values >1‰, therefore, ∆17O values greater than 1‰ quantitatively reflect the relative contribution of O3 during sulfate formation (12). S(IV) oxidized in the stratosphere acquires an anomalous signature via Reaction 1 to Reaction 3 (30). Once sulfate is formed, the isotopic signature is stable and permanently preserved in the aerosol. In addition to defining reaction pathways, both S and O isotopic anomalies of sulfate aerosols may determine paleovolcanic activities and reflect their upper atmospheric chemistry (28, 31, 32). It is argued that large volcanic eruptions in the Anthropocene era are depleting stratospheric ozone by providing surfaces for heterogeneous chemical reactions (33). These variations in O3 can modulate the dynamic of the tropical stratosphere (quasibiennial oscillations); thus, volcanoes are considered responsible for the strongest El-Niño Southern Oscillations (ENSOs) (34). In this work, we investigate whether oxygen-triple isotopic composition of sulfate aerosols may serve as a unique fingerprint of ozone chemistry to parse out the effect of volcanoes and ENSO events based on their differing oxidative pathways. The polar ice caps are nature’s best archive of Earth’s atmospheric history and preserve a record of paleovolcanic activities (30, 35). A high-resolution seasonal record of sulfate aerosols is used to reconstruct atmospheric history on annual and decadal time scales. The oxygen-triple isotope data of sulfate aerosols from Greenland (monthly samples from July 1999–June 2000) have demonstrated its potential to constrain the transport and oxidation history of sulfate aerosols to polar regions (14). Here, we present a 22-y seasonally resolved profile of major ions and oxygen-triple isotope data of sulfate aerosols obtained from surface snow sampled in a snow pit (1 m × 1 m) at the South Pole to help elucidate the processes determining the observed global variation in sulfate aerosol from 1980 to 2002, as well as the oxidation history of S(IV), its transport to the South Pole, and its linkage with the dynamics of the upper atmosphere. Our data on the oxygen-triple isotope measurements of sulfate aerosol encompass three major volcanic events of the century [El-Chichόn (17.3° N, 93.2° W, 1,205 m), Pinatubo (15.13° N, 120.35° E, 1,745 m), and Cerro Hudson (45° S, 72° W, 1,905 m)] and three major ENSO events (1982–1983, 1991–1992, and 1997–1998).
Results
A high-resolution temporal record (1980–2002) of sulfate aerosols extracted from the snow pit at the South Pole and the associated oxygen isotopic composition (δ17O, δ18O, and ∆17O) are given in Fig. 1 and Table S1. The sulfate concentration in composite samples (details provided in Materials and Methods) ranged from 36 to 165 parts per billion (ppb). The highest sulfate concentration was observed in ice layers deposited in 1991 and 1992 (Fig. 1A). The single oxygen isotope ratio of sulfate aerosol showed significant variation, ranging from 1980 to 2002 (δ18O = −2 to +12‰). Increases in sulfate concentrations due to volcanic activities show a corresponding decrease in δ18O (average δ18O = 3.1‰ and 2.3‰ in 1983 and 1992, respectively). The oxygen isotopic anomaly (∆17O) varied from 0.4 to 4.5‰ (Fig. 1B). A high-resolution concentration profile of sulfate aerosols indicated that volcanic sulfate layers from Pinatubo and Cerro Hudson eruptions were deposited from 1991–1992 (36) and El-Chichón from 1982–1983 with corresponding oxygen isotope anomaly of 4.5‰ and 3.3‰. The oxygen isotopic anomaly in sulfate aerosols observed during three major El-Nino events (ENSO-I = 1982–1983, ENSO-II = 1991–1992, ENSO-III 1997–1998) and a moderate event (ENSO-IV = 1986–1987) track the Ozone ENSO Index (OEI; Fig. 1B). An unusually high enrichment in oxygen-triple isotopic composition (δ18O =12‰, ∆17O = 4.1‰), along with a higher OEI of 6 is observed in 1990 and labeled as an unknown event in Fig. 1A.
Fig. 1.

(A) Oxygen isotopic composition (brown squares) and sulfate concentration profile (green diamonds) of composite aerosol sulfate samples extracted from the snow pit (6 m high) at the South Pole, Antarctica. The increase in sulfate concentration due to El-Chichón and Pinatubo + Cerro Hudson is also shown. UE, unknown event. (B) Comparison of oxygen isotope anomaly (red lines) and OEI (blue lines) obtained by Ziemke et al. (65) from the deseasonalized trend in total O3 column measured at the equatorial Eastern and Western Pacific, an El-Niño region. Violet bars indicate three major ENSO events: ENSO-I (1982–1983), ENSO-II (1991–1992), and ENSO-III (1997–1998). A moderate event, ENSO-IV (1986–1987), is also shown. (The scale of ENSO events is defined by the National Oceanic and Atmospheric Administration and is available at www.cpc.noaa.gov/products/analysis_monitoring/ensostuff/ensoyears.shtml).
A plot of Δ17O vs. δ18O indicated a very weak (r2 = 0.2) inverse correlation (Fig. 2). In Fig. 2, the maximum Δ17O achievable via tropospheric ozone and peroxide aqueous phase oxidation is represented by red and green rectangles and gas phase oxidation via stratospheric OH/HO2 is shown in blue. A high-resolution (∼1-cm sampling interval) concentration measurement of major ions [sulfate, methane sulfonic acid (MSA), nitrate, and chloride] obtained from the snow pit indicated no significant relation to the sulfate concentration (Fig. S1). A higher resolution sulfate concentration profile revealed two distinct peaks 1 and 2 (Fig. 3) with a fivefold and 3.6-fold increase, respectively, in sulfate (SO4) concentration in ice layers deposited during 1992. Peak 3 appeared during a volcanically quiescent period (1990) and showed an approximately fourfold increase in sulfate concentration (190 ppb) compared with an average background value of 50 ppb observed in this study. The persistent background during the study period is defined as the time period when natural and anthropogenic emissions of S compounds are maintained at a relatively quasi-steady state (1999–2002). Sulfate from the El-Chichόn volcanic activity indicated as peak 4 (1982–1983) did not appear as a sharp peak; rather, it is more spread out in time. In the high-resolution profile, an increase in non-sea salt (nss) SO4 (peak 3 in 1990) is potentially associated with a significant increase in MSA, although they are not temporally identical. MSA concentrations varied from 2 to 50 ppb, with a maximum increase in1985–1986 and 1990 (Fig. 3).
Fig. 2.

Four-isotope plot shows Δ17O and δ18O of sulfate aerosols extracted from the snow pit at the South Pole. A weak observed correlation indicates mixing of various sulfates from different sources. Red and green rectangles display variation in δ18O and Δ17O sources of sulfate. The blue rectangle is an oxidation source with stratospheric (Strat.) OH/HO2 radicals (28, 39). The green rectangle shows the range of pure hydrogen peroxide oxidation. The red square denotes the value of atmospheric (Atm.) oxygen. Max., maximum. Primary sulfate produced during fossil fuel combustion at high temperature has been shown to possess δ18O values close to the atmospheric oxygen (51); however, sulfate produced during biomass burning showed a range of δ18O values (50) depending on biomass type. The observed dataset reflects the range of processes contributing to the observed oxygen isotopic composition of sulfate.
Fig. 3.

Concentration profile (1977–2003) of nss sulfate aerosol and MSA extracted from the snow pit samples at the South Pole. Peaks 1, 2, and 4 represent Pinatubo, Cerro Hudson, and El-Chichón volcanic sulfates, respectively. Peak 3 represents an unknown event.
Discussion
The dataset presented here is the longest (1980–2002) and maximum time-resolved record of chemical and isotopic composition of sulfate aerosols retrieved at the South Pole, and it encompasses three major volcanic events (El-Chichόn, Pinatubo, and Cerro Hudson eruptions in April 1982, June 1991, and October 1991, respectively). These volcanic activities introduced significant quantities of SO2 (Pinatubo = 30 Tg SO2, Cerro Hudson = 10 Tg SO2, and El-Chichόn = 7 Tg SO2) (5, 37, 38) into the stratosphere, which can be observed as distinct sulfate peaks as labeled in Figs. 1A and 3. The volcanic sulfate aerosol showed less enrichment in the heavy isotopes of oxygen (δ18Oaverage = 2.6 ± 1‰) but possesses a mass-independent anomaly (El-Chichόn: ∆17O =3.3 ‰ and Pinatubo + Cerro Hudson: ∆17O = 4.5‰). The oxygen isotope anomaly reported here for the composite Pinatubo and Cerro Hudson sulfate sample in 1992 is similar to the previously reported higher resolution signal for Pinatubo (δ18O = 5.1–9.5‰, Δ17O = 3.8–4.7‰) (28). Higher ∆17O values (3.3–4.5‰) of volcanic sulfate and nonvolcanic ENSO events (3.8–4.2 ‰) compared with the lower tropospheric ∆17O values (0.4–1.6‰) (13, 18) indicate the predominant role of stratospheric OH and HO2 radicals (30, 39). The concentration of these radicals in the stratosphere depends on ozone, water vapor, CH4, and NOx concentrations (40), and it is suggested to be 1.5 ± 0.3 × 106 molecules per cubic centimeter in the tropics using the global chemistry transport model (41). Numeric simulations have also indicated that the OH in the stratosphere acquires an oxygen isotope anomaly (∆17O = 2–40‰) by means of exchange with NOx (39, 42). The anomalous signal of OH and HO2 in the stratosphere is preserved (39, 42) due to the extremely low water content in the stratosphere (∼5–10 ppm by volume) (43, 44), which is normally erased in the troposphere due to rapid isotope exchange with water vapor (∼3% in the tropics to 0.1% in the cold polar regions) (20).
The most striking feature of the present data (Fig. 1B) is that variables OEI and Δ17O of sulfate aerosol track each other during the strong El- Niño events of 1982–1983 (ENSO-I), 1991–1992 (ENSO-II), and 1997–1998 (ENSO-III) as well as during the moderate event of 1986–1987 (ENSO-IV). These ENSO time periods are defined according to the National Oceanic and Atmospheric Administration as sea surface temperature anomalies (El-Niño = warm and La-Niña = cool) in the tropical Pacific (5° N–5° S, 120°–170° W) (www.cpc.noaa.gov/products/analysis_monitoring/ensostuff/ensoyears.shtml). The OEI plotted in Fig. 1B, as discussed, is obtained from variation in ozone concentrations at tropical latitudes. The OEI and Δ17O data in Fig. 1B track each other but are slightly shifted, which may derive from two factors. First, the necessity to combine samples for the nitrate (45) and sulfate measurements introduces a modest time uncertainty. The average of the combined depth is used to specify sample time and assumes a uniform sulfate distribution throughout that time period. Consequently, there is an uncertainty in time of the sulfate peak, which, at maximum, is a few months. Second, there are different indices used to capture El-Niño events [e.g., OEI, Oceanic Niño Index (ONI)], which use a variety of differing geophysical observations to document El-Niño events, and they are not necessarily exactly temporally equivalent to one another. The comparison of the oxygen isotopic anomaly of sulfate aerosols with the OEI reveals that higher Δ17O values are associated with elevated ozone column densities measured by different satellites. The ENSO signal during two earlier events, ENSO-I [1982–1983 (El Chichόn: OEI = 6.5, Δ17O = 3.3‰)] and ENSO-II [1991–1992 (Pinatubo and Cerro Hudson: OEI = 6, Δ17O = 4.5‰)], may have been confounded due to the intense volcanic activities, which introduced, in addition to the SO2, significant amounts of sulfate oxidized in the troposphere with less Δ17O, thus diluting the higher Δ17O signal of S(IV) oxidation in the stratosphere via OH radicals. The Δ17O of sulfate and OEI track each other fairly well despite higher concentrations of volcanic sulfate. The ENSO events in volcanically quiescent periods manifested a higher O-isotopic anomaly and OEI [strongest ENSO-III (1997–1998): OEI = 10.8, Δ17O= 4.2‰; moderate ENSO-IV (1986–1987): OEI = 5, Δ17O= 3.8‰]. A significant increase of total ozone during ENSO-III (1997–1998) was accompanied by decreased precipitation, producing extensive forest fires in Indonesia, Australia, and South America (46, 47). The influence of ENSOs on total columnar ozone has been attributed to the variation in the tropopause height driven by changes of tropical deep convection and alteration of Brewer–Dobson circulation (14, 48). These observations suggest that the Δ17O of sulfate aerosols can be used to track moderate to strong ENSO events and the variation in ozone concentration. To develop this possibility, the following points must be addressed: Where does ozone-driven oxidation of S(IV) occur, and how is the observed variation in the tropical upper tropospheric O3 (OEI) and ENSOs chemically associated with Δ17O of sulfate aerosols? It is known that tropospheric air enters the stratosphere principally in the tropics within the Intertropical Convergence Zone (ITCZ, a thin and dynamic region along the equator separating trade winds between the Southern and Northern Hemispheres) and transports poleward in the stratosphere as shown in Fig. 4 (11). The ITCZ thus links the troposphere to the stratosphere and is an important corridor for the transport of aerosol and trace gases to the stratosphere (49). The maximum transport of water vapor also occurs in the ITCZ (44) in the vicinity of the ENSO region, which also corresponds to the region of the OEI measurements used in Fig. 1B. A comparison between stratospheric water vapor and tropical sea surface temperatures has demonstrated a strong correlation and an impact on the strength of El-Niño (49). The influence of ENSOs on the total column of ozone is also linked to the variation in the tropopause height. Tropical deep convection and changes in Brewer–Dobson circulation may account for the observed ozone enhancement during ENSO periods (14, 48). Cumulatively, all components needed to provide ENSO-related Δ17O enrichments are present. There is coexistence of increased ozone and water content and a dynamic mechanism of transport of aerosol into the stratosphere with poleward migration. The ozone-induced oxidation of S(IV) via OH radicals is likely stratospheric and occurs during transport (Fig. 4). In this model, large sulfate concentrations are not required, simply a preferential ozone oxidation channel that provides the heavy isotope-enriched sulfate. A detailed model and discussion are far beyond the scope of this paper, but the isotope measurements suggest that such experiments are warranted. Our data indicate that the O-isotope anomaly of sulfate is sensitive to changes in atmospheric dynamics and is faithfully preserved in this record. A quasibiennial oscillation signal in the Δ17O of nitrate extracted from the same set of aerosol samples from the South Pole has shown a similar linkage (45).
Fig. 4.

Schematic depicts transport and transformation of sulfur species into the stratosphere and deposition of aged sulfate aerosol in the ice at the South Pole. The red-shaded area indicates a significant contribution of SO2 and SO4 aerosols to the SSA in the lower stratosphere, whereas the gray-shaded region represents carbonyl sulfide (OCS) photolysis and contribution to the SSA. The blue area sandwiched between these layers represents the ozone layer. Although O3 production is maximum in the tropics, it is transported to the poles, as shown by the dynamics of the stratosphere with magenta lines. SSA, stratospheric sulfate aerosols; UT-LS exchange, air mass exchange at midlatitude between the upper troposphere and lower stratosphere.
In the coordinate system of Fig. 2, a source of sulfate with higher δ18O and low Δ17O is required. An ozone-rich source would have high δ18O values and also high Δ17O values. Various oxidants of different oxygen isotopic composition (OH, HO2, H2O2, and O3) can oxidize SO2 to SO4 (Fig. 2, red and green rectangles are guidelines to demonstrate variations in δ18O with the maximal Δ17O obtainable via aqueous phase O3 and H2O2 oxidation, and the blue rectangle indicates stratospheric OH and HO2 radical reactions, as well as the associated maximum isotopic anomaly). Fig. 2 requires the presence of a specific source, but it must be of higher δ18O and low Δ17O. Laboratory experiments, field observations, and numeric simulation of sulfur oxidation (14, 28, 50, 51) have shown that the δ18O of sulfate coupled with Δ17O can be used to distinguish between primary and secondary sulfates. Primary sulfate [i.e., S(VI) produced at the emission source] exhibits a higher enrichment in δ18O = 20–45‰ and Δ17O = 0 (50–52). Secondary sulfate is formed when SO2 is oxidized in the atmosphere via homogeneous and heterogeneous pathways (51). The δ18O of secondary sulfate thus represents a juxtaposition of the highly variable water isotopic signature derived during SO2 oxidation processes at varying latitudes and altitudes (10, 53, 54). A moderate correlation between δ18O of rainwater and δ18O(SO4) (55) also indicated the complexity of using δ18O to predict sources of sulfate in rainwater. Most primary sulfate is removed via wet and dry deposition in the free troposphere; however, a fraction of S(IV), carbonyl sulfide (OCS), and traces of S(VI) are transported to the stratosphere from the ITCZ and at midlatitudes via deep convection (7, 56), thus permitting gas phase oxidation via OH/HO2 radicals in the stratosphere (30).
An unusual increase in SO4 concentration (∼100 ppb) and oxygen isotope enrichment (δ18O ∼12‰ and Δ17O ∼3.7‰) in 1990 are immediately followed by an increase in MSA (Fig. 3). To explain this peak, we consider two possible scenarios. The first is stratospheric volcanic emissions due to the presence of a higher oxygen isotope anomaly in the sulfate. Total ozone mapping spectrometer (TOMS) and stratospheric aerosol and gas experiment (SAGE) satellite data indicated no significant increase in the global inventory of stratospheric aerosols loading during 1989–1990 (57), ruling out stratospheric volcanic emissions, and this leaves us with one option, a local sulfate source such as Mount Erebus (167° 25′ E, 77° 30′ S). The Smithsonian database reports a significant increase in SO2 emissions, up to 100 t/d during this period, which could be a potential source of SO4 peak (www.volcano.si.edu/reports/bulletin/contents.cfm?issue=3609). Volcanic emissions, despite their complexity, change mostly SO4 concentration (58). The second possible scenario is that biogenic sources, such as phytoplankton, emit DMS, which, on oxidation with O3, H2O2, NOx, or ClOx, can produce MSA, and ultimately an increase in nss-SO4 (59, 60). This observed spike in MSA temporally followed by an increase in nss-SO4 is consistent with a biological source. Higher resolution measurements of MSA on the high Antarctic Plateau (61) (both inland and coastal sites) indicated postdepositional losses of MSA and nss-SO4 production via MSA oxidation. Future sulfur isotope measurements of sulfate aerosols in this time period may help to elucidate and quantify this very specific source and oxidation process further.
Conclusion
The most significant observation of the sulfate multioxygen isotopic record reported here is that observed trends in the mass-independent O-isotopic anomaly are apparently linked to the ozone variation in the tropical upper troposphere and lower stratosphere via the OEI and sea surface temperature anomalies in the El-Niño 3.4 region. The combined enhanced ozone levels observed by satellites and elevated upward flow of air masses from the Intertropical Convergence Zone may provide a source of anomalous sulfate not present in non-ENSO years. The presence of stratospheric volcanic emissions in two ENSO time slots also shows that they exert a significant effect on the upper atmospheric odd oxygen cycle, which our study suggests is captured in the sulfate O-isotopic record. Future measurements of different ENSO periods in volcanically quiescent periods will help to quantify clearly the unique fingerprints of ENSOs on the sulfate O-isotopic anomaly. These results provide preliminary insight into ENSO-driven climate fluctuations on the concentration of ozone and S(IV) oxidation, and how this transported aerosol captures new facets of chemical and transport history during moderate and severe ENSO events. Understanding of the ENSO signal and its frequency is important to understand the perturbation in the tropical climate and its relevance to the global climate system. The ENSO-driven climate patterns had a significant impact on the proliferation and collapse of the Mayan civilization, as inferred from the rainfall patterns preserved in the δ18O of stalagmites (62). A higher resolution, multidecadal record of oxygen-triple isotopic composition of sulfate aerosol is needed to investigate ocean-atmosphere-biosphere interaction using a global comprehensive Earth system model. The information can be used to assess socioeconomic costs of climate vulnerabilities and to develop sustainable solutions.
Materials and Methods
The surface snow samples were acquired to analyze oxygen-triple isotope composition of both nitrate and sulfate [National Science Foundation polar program, project South Pole Atmospheric Nitrate Isotopic Analysis (SPANIA)] with the highest resolution (∼1-cm depth interval) from a snow pit (1 m × 1 m) at the South Pole (45). Organic impurities from each aliquot were removed from the composite samples (∼6-cm depth) by adding 2.0 mL of peroxide (30% by volume) and further passing through polyvinyl pyrrolidine C18 (Alltech) resins. Purified SO4 solution was converted to silver sulfate and pyrolysed at 1,050 °C (14, 63) using a quartz tube. These samples were combined in a prior study to obtain sufficient sample for the oxygen-triple isotope measurements of nitrates, and the remaining solution was used for oxygen-triple isotope analysis of sulfates. Oxygen-triple isotopic composition was measured using a Thermo Finnigan Mat-253 Isotope Ratio mass spectrometer and corrected for high-temperature oxygen-isotope exchange with quartz (64). The reported sample dates are calculated from an average annual snow accumulation rate; therefore, actual dates in composite samples may be shifted by ±4 mo, which defines the maximal uncertainty in time. The oxygen isotopic anomaly is based on the nss-SO4 concentration. The sea sulfate carries no O-isotopic anomaly (∆17O = 0), and this component was removed using sodium concentration as a tracer of sea salt (28). The sea salt contribution is ∼3–7% at maximum at the South Pole, and the correction factor is small.
The OEI is obtained by Ziemke et al. (65) from the variation in ozone concentrations at tropical latitudes (15° S–15° N). The following ozone data were acquired from four different satellites: TOMS, Earth probe TOMS, solar backscatter UV, and Aura ozone monitoring instrument. The measured zonal variability in ozone was verified with the O3 data obtained from the Goddard Earth Observing System chemistry climate model and from the microwave limb sounder vertical profile of O3, cloud ice, temperature, and pressure. This comparison also confirmed that zonal variability in total column ozone in the tropics is mostly caused by ENSO events and provides a direct measure of the changes in tropospheric ozone levels (65–67).
Supplementary Material
Acknowledgments
We thank A. Hill and S. Chakraborty for useful discussions about the OEI. We also thank the reviewers for valuable suggestions that helped to improve the manuscript. M.H.T. and R.S. thank the National Science Foundation Atmospheric Chemistry Division for the support through Award ATM0960594, which allowed the present measurements to be made. The National Science Foundation Office of Polar Programs is also acknowledged for its generous support, which permitted the snow pit work at the South Pole Atmospheric Nitrate Isotope Analysis through SPANIA Award OPP0125761. J.S. thanks the Agence Nationale de la Recherche [ANR-NT09-431976-volcanic and solar radiative forcing (VOLSOL)] and the Centre National de la Recherche Scientifique/Projet International de Coopération Scientifique (PICS) exchange program for their financial support for maintaining the collaboration with the University of California, San Diego.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1213149110/-/DCSupplemental.
References
- 1.Jones A, Haywood J, Boucher O. A comparison of the climate impacts of geoengineering by stratospheric SO2 injection and by brightening of marine stratocumulus cloud. Atmos Sci Lett. 2011;12(2):176–183. [Google Scholar]
- 2.Jones A, Haywood J, Boucher O, Kravitz B, Robock A. Geoengineering by stratospheric SO2 injection: Results from the Met Office HadGEM(2) climate model and comparison with the Goddard Institute for Space Studies ModelE. Atmos Chem Phys. 2010;10(13):5999–6006. [Google Scholar]
- 3.Leibensperger EM, et al. Climatic effects of 1950-2050 changes in US anthropogenic aerosols—Part 1: Aerosol trends and radiative forcing. Atmos Chem Phys. 2012;12(7):3333–3348. [Google Scholar]
- 4.Soloman SQD, Manning M. Intergovernmental Panel on Climate Change Fourth Assessment Report, Climate Change 2007: The Physical Science Basis. New York: Cambridge Univ Press; 2007. [Google Scholar]
- 5.Deshler T. A review of global stratospheric aerosol: Measurements, importance, life cycle, and local stratospheric aerosol. Atmos Res. 2008;90(2-4):223–232. [Google Scholar]
- 6.Halmer MM, Schmincke HU, Graf HF. The annual volcanic gas input into the atmosphere, in particular into the stratosphere: A global data set for the past 100 years. J Volcanol Geotherm Res. 2002;115(3-4):511–528. [Google Scholar]
- 7.Solomon S, et al. The persistently variable “background” stratospheric aerosol layer and global climate change. Science. 2011;333(6044):866–870. doi: 10.1126/science.1206027. [DOI] [PubMed] [Google Scholar]
- 8.Bauer E, Petoukhov V, Ganopolski A, Eliseev AV. Climatic response to anthropogenic sulphate aerosols versus well-mixed greenhouse gases from 1850 to 2000 AD in CLIMBER-2. Tellus B Chem Phys Meterol. 2008;60(1):82–97. [Google Scholar]
- 9.Miyakawa T, Takegawa N, Kondo Y. Removal of sulfur dioxide and formation of sulfate aerosol in Tokyo. Journal of Geophysical Research. Atmospheres. 2007 doi: 10.1029/2006JD007896. [DOI] [Google Scholar]
- 10.Alexander B, et al. Isotopic constraints on the formation pathways of sulfate aerosol in the marine boundary layer of the subtropical northeast Atlantic Ocean. Journal of Geophysical Research. Atmospheres. 2012 doi: 10.1002/rcm.6332. [DOI] [Google Scholar]
- 11.Seinfeld HJ, Pandis NS. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change. New York: Wiley Interscience; 2006. [Google Scholar]
- 12.Savarino J, Lee CCW, Thiemens MH. Laboratory oxygen isotopic study of sulfur (IV) oxidation: Origin of the mass-independent oxygen isotopic anomaly in atmospheric sulfates and sulfate mineral deposits on Earth. Journal of Geophysical Research. Atmospheres. 2000;105(D23):29079–29088. [Google Scholar]
- 13.Patris N, Cliff SS, Quinn PK, Kasem M, Thiemens MH. Isotopic analysis of aerosol sulfate and nitrate during ITCT-2k2: Determination of different formation pathways as a function of particle size. Journal of Geophysical Research. Atmospheres. 2007 doi: 10.1029/2005jd006214. [DOI] [Google Scholar]
- 14.McCabe JR, Savarino J, Alexander B, Gong S, Thiemens MH. Geophys Res Lett. 2006. Isotopic constraints on non-photochemical sulfate production in the Arctic winter. [DOI] [Google Scholar]
- 15.Alexander B, Savarino J, Barkov NI, Delmas RJ, Thiemens MH. Climate driven changes in the oxidation pathways of atmospheric sulfur. Geophys Res Lett. 2002;29(14):30–34. [Google Scholar]
- 16.Alexander B, Savarino J, Kreutz KJ, Thiemens MH. Impact of preindustrial biomass-burning emissions on the oxidation pathways of tropospheric sulfur and nitrogen. Journal of Geophysical Research. Atmospheres. 2004 doi: 10.1029/2003jd004218. [DOI] [Google Scholar]
- 17.Alexander B, et al. East Antarctic ice core sulfur isotope measurements over a complete glacial-interglacial cycle. Journal of Geophysical Research. Atmospheres. 2003 doi: 10.1029/2003jd003513. [DOI] [Google Scholar]
- 18.Jenkins KA, Bao H. Multiple oxygen and sulfur isotope compositions of atmospheric sulfate in Baton Rouge, LA, USA. Atmos Environ. 2006;40(24):4528–4537. [Google Scholar]
- 19.Holt BD, Kumar R, Cunningham PT. O-18 study of the aqueous-phase oxidation of sulfur-dioxide. Atmos Environ. 1981;15(4):557–566. [Google Scholar]
- 20.Holt BD, Cunningham PT, Engelkemeir AG, Graczyk DG, Kumar R. O-18 study of nonaqueous-phase oxidation of sulfur-dioxide. Atmos Environ. 1983;17(3):625–632. [Google Scholar]
- 21.Heidenreich JE, Thiemens MH. A non-mass-dependent isotope effect in the production of ozone from molecular-oxygen. J Chem Phys. 1983;78(2):892–895. [Google Scholar]
- 22.Heidenreich JE, Thiemens MH. A non-mass-dependent oxygen isotope effect in the production of ozone from molecular-oxygen—The role of molecular symmetry in isotope chemistry. J Chem Phys. 1986;84(4):2129–2136. [Google Scholar]
- 23.Thiemens MH. History and applications of mass-independent isotope effects. Annu Rev Earth Planet Sci. 2006;34:217–262. [Google Scholar]
- 24.Thiemens MH, Chakraborty S, Dominguez G. The physical chemistry of mass-independent isotope effects and their observation in nature. Ann Rev Phys Chem. 2012;63:155–177. doi: 10.1146/annurev-physchem-032511-143657. [DOI] [PubMed] [Google Scholar]
- 25.Krankowsky D, et al. Stratospheric ozone isotope fractionations derived from collected samples. Journal of Geophysical Research. Atmospheres. 2007 doi: 10.1029/2006JD007855. [DOI] [Google Scholar]
- 26.Savarino J, Thiemens MH. Mass-independent oxygen isotope (O-16, O-17, O-18) fractionation found in H-x, O-x reactions. J Phys Chem A. 1999;103(46):9221–9229. [Google Scholar]
- 27.Alexander B, Park RJ, Jacob DJ, Sunling G. Transition metal-catalyzed oxidation of atmospheric sulfur: Global implications for the sulfur budget. Journal of Geophysical Research. Atmospheres. 2009 doi: 10.1029/2008JD010486. [DOI] [Google Scholar]
- 28.Savarino J, Slimane B, Cole-Dai J, Thiemens MH. Evidence from sulfate mass independent oxygen isotopic compositions of dramatic changes in atmospheric oxidation following massive volcanic eruptions. J Geophys Res. 2003;108(D21):ACH7-1–6. [Google Scholar]
- 29.Lyons JR. Mass-independent fractionation of oxygen isotopes in Earth’s atmosphere. Astrobiology. 2001;1(3):333–334. [Google Scholar]
- 30.Savarino J, Bekki S, Cole-Dai JH, Thiemens MH. Evidence from sulfate mass independent oxygen isotopic compositions of dramatic changes in atmospheric oxidation following massive volcanic eruptions. Journal of Geophysical Research. Atmospheres. 2003;108(D21):ACH7-1-6. [Google Scholar]
- 31.Baroni M, Savarino J, Cole-Dai J, Rai VK, Thiemens MH. Anomalous sulfur isotope compositions of volcanic sulfate over the last millennium in Antarctic ice cores. Journal of Geophysical Research. Atmospheres. 2008 doi: 10.1029/2008jd010185. [DOI] [Google Scholar]
- 32.Cole-Dai J, et al. Cold decade (AD 1810-1819) caused by Tambora (1815) and another (1809) stratospheric volcanic eruption. Geophys Res Lett. 2009 doi: 10.1029/2009gl040882. [DOI] [Google Scholar]
- 33.Halmer MM. Have volcanoes already passed their zenith influencing the ozone layer? Terra Nova. 2005;17(6):500–502. [Google Scholar]
- 34.Robock A, Adams T, Moore M, Oman L, Stenchikov G. Southern Hemisphere atmospheric circulation effects of the 1991 Mount Pinatubo eruption. Geophys Res Lett. 2007 doi: 10.1029/2007gl031403. [DOI] [Google Scholar]
- 35.Cole-Dai J. Volcanoes and climate. Wiley Interdiscip Rev Clim Change. 2010;1(6):824–839. [Google Scholar]
- 36.Cole-Dai J, Mosley-Thompson E, Qin DH. Evidence of the 1991 Pinatubo volcanic eruption in South Polar snow. Chin Sci Bull. 1999;44(8):756–760. [Google Scholar]
- 37.Sato M, Hansen JE, McCormick MP, Pollack JB. Stratospheric aerosol of optical depths, 1850-1990. Journal of Geophysical Research. Atmospheres. 1993;98(D12):22987–22994. [Google Scholar]
- 38.Chaochao G, Robock A, Ammann C. Volcanic forcing of climate over the past 1500 years: An improved ice core-based index for climate models. Journal of Geophysical Research. Atmospheres. 2008;113(D23):D23111. doi: 10.1029/2008jd010239. [DOI] [Google Scholar]
- 39.Zahn A, Franz P, Bechtel C, Grooss JU, Roeckmann T. Modelling the budget of middle atmospheric water vapour isotopes. Atmos Chem Phys. 2006;6:2073–2090. [Google Scholar]
- 40.Jones RL, et al. The water-vapor budget of the stratosphere studied using LIMS and SAMS satellite data. Q J R Meteorol Soc. 1986;112(474):1127–1143. [Google Scholar]
- 41.Crutzen PJ, Lawrence MG, Poschl U. On the background photochemistry of tropospheric ozone. Tellus Series A: Dynamic Meteorology and Oceanography. 1999;51(1):123–146. [Google Scholar]
- 42.Lyons JR. Transfer of mass-independent fractionation in ozone to other oxygen-containing radicals in the atmosphere. Geophys Res Lett. 2001;28(17):3231–3234. [Google Scholar]
- 43.Ravishankara AR. Atmospheric science. Water vapor in the lower stratosphere. Science. 2012;337(6096):809–810. doi: 10.1126/science.1227004. [DOI] [PubMed] [Google Scholar]
- 44.Anderson JG, Wilmouth DM, Smith JB, Sayres DS. UV dosage levels in summer: Increased risk of ozone loss from convectively injected water vapor. Science. 2012;337(6096):835–839. doi: 10.1126/science.1222978. [DOI] [PubMed] [Google Scholar]
- 45.McCabe JR, Thiemens MH, Savarino J. A record of ozone variability in South Pole Antarctic snow: Role of nitrate oxygen isotopes. Journal of Geophysical Research-Atmospheres. 2007 doi: 10.1029/2006JD007822. [DOI] [Google Scholar]
- 46.Takegawa N, et al. Photochemical production of O-3 in biomass burning plumes in the boundary layer over northern Australia. Geophys Res Lett. 2003 doi: 10.1029/2003gl017017. [DOI] [Google Scholar]
- 47.Kita K, et al. Photochemical production of ozone in the upper troposphere in association with cumulus convection over Indonesia. Journal of Geophysical Research. Atmospheres. 2002 doi: 10.1029/2001jd000844. [DOI] [Google Scholar]
- 48.Hood LL, Soukharev BE. Interannual variations of total ozone at northern midlatitudes correlated with stratospheric EP flux and potential vorticity. J Atmos Sci. 2005;62(10):3724–3740. [Google Scholar]
- 49.Solomon S, et al. Contributions of stratospheric water vapor to decadal changes in the rate of global warming. Science. 2010;327(5970):1219–1223. doi: 10.1126/science.1182488. [DOI] [PubMed] [Google Scholar]
- 50.Lee CCW, Savarino J, Cachier H, Thiemens MH. Sulfur (S-32, S-33, S-34, S-36) and oxygen (O-16, O-17, O-18) isotopic ratios of primary sulfate produced from combustion processes. Tellus B Chem Phys Meterol. 2002;54(3):193–200. [Google Scholar]
- 51.Dominguez G, et al. Discovery and measurement of an isotopically distinct source of sulfate in Earth’s atmosphere. Proc Natl Acad Sci USA. 2008;105(35):12769–12773. doi: 10.1073/pnas.0805255105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Holt BD, Cunningham PT, Kumar R. Oxygen isotopy of atmospheric sulfates. Environ Sci Technol. 1981;15(7):804–808. [Google Scholar]
- 53.Kunasek SA, et al. Sulfate sources and oxidation chemistry over the past 230 years from sulfur and oxygen isotopes of sulfate in a West Antarctic ice core. Journal of Geophysical Research. Atmospheres. 2010 doi: 10.1029/2010JD013846. [DOI] [Google Scholar]
- 54.Sofen ED, Alexander B, Kunasek SA. The impact of anthropogenic emissions on atmospheric sulfate production pathways, oxidants, and ice core Delta O-17(SO42−) Atmos Chem Phys. 2011;11(7):3565–3578. [Google Scholar]
- 55.Tichomirowa M, Heidel C. Regional and temporal variability of the isotope composition (O, S) of atmospheric sulphate in the region of Freiberg, Germany, and consequences for dissolved sulphate in groundwater and river water. Isotopes Environ Health Stud. 2012;48(1):118–143. doi: 10.1080/10256016.2011.624273. [DOI] [PubMed] [Google Scholar]
- 56.Bruehl C, Lelieveld J, Crutzen PJ, Tost H. The role of carbonyl sulphide as a source of stratospheric sulphate aerosol and its impact on climate. Atmos Chem Phys. 2012;12(3):1239–1253. [Google Scholar]
- 57.Sunilkumar SV, Parameswaran K, Thampi BV, Ramkumar G. Variability in background stratospheric aerosols over the tropics and its association with atmospheric dynamics. Journal of Geophysical Research. Atmospheres. 2011 doi: 10.1029/2010jd015213. [DOI] [Google Scholar]
- 58.Legrand M, Wagenbach D. Impact of the Cerro Hudson and Pinatubo volcanic eruptions on the Antarctic air and snow chemistry. Journal of Geophysical Research. Atmospheres. 1999;104(D1):1581–1596. [Google Scholar]
- 59.Legrand M, Wolff E, Wagenbach D. Antarctic aerosol and snowfall chemistry: Implications for deep Antarctic ice-core chemistry. Annals of Glaciology. 1999;29(1):66–72. [Google Scholar]
- 60.Jourdain B, Legrand M. Seasonal variations of atmospheric dimethylsulfide, dimethylsulfoxide, sulfur dioxide, methanesulfonate, and non-sea-salt sulfate aerosols at Dumont d’Urville (coastal Antarctica) (December 1998 to July 1999) Journal of Geophysical Research. Atmospheres. 2001;106(D13):14391–14408. [Google Scholar]
- 61.Preunkert S, et al. Seasonality of sulfur species (dimethyl sulfide, sulfate, and methanesulfonate) in Antarctica: Inland versus coastal regions. Journal of Geophysical Research. Atmospheres. 2008;113(D15) [Google Scholar]
- 62.Kennett DJ, et al. Development and disintegration of Maya political systems in response to climate change. Science. 2012;338(6108):788–791. doi: 10.1126/science.1226299. [DOI] [PubMed] [Google Scholar]
- 63.Savarino J, Alexander B, Darmohusodo V, Thiemens MH. Sulfur and oxygen isotope analysis of sulfate at micromole levels using a pyrolysis technique in a continuous flow system. Anal Chem. 2001;73(18):4457–4462. doi: 10.1021/ac010017f. [DOI] [PubMed] [Google Scholar]
- 64.Schauer AJ, et al. Oxygen isotope exchange with quartz during pyrolysis of silver sulfate and silver nitrate. Rapid Commun Mass Spectrom. 2012;26(18):2151–2157. doi: 10.1002/rcm.6332. [DOI] [PubMed] [Google Scholar]
- 65.Ziemke JR, Chandra S, Oman LD, Bhartia PK. A new ENSO index derived from satellite measurements of column ozone. Atmos Chem Phys. 2010;10(8):3711–3721. [Google Scholar]
- 66.Chandra S, Ziemke JR, Bhartia PK, Martin RV. Tropical tropospheric ozone: implications for dynamics and biomass burning. J Geophys Res. 2002;107(D14):ACH3-1–17. [Google Scholar]
- 67.Oman LD, et al. The response of tropical tropospheric ozone to ENSO. Geophys Res Lett. 2011 doi: 10.1029/2011gl047865. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.