

Abstract
Vinyl silyl ethers and disiloxanes can now be prepared from aryl-substituted alkenes and related substrates using a silyl-Heck reaction. The reaction employs a commercially available catalyst system and mild conditions. This work represents a highly practical means of accessing diverse classes of vinyl silyl ether substrates in an efficient and direct manner with complete regio- and geometric selectivity.
Vinyl silanes are ubiquitous reagents in organic synthesis,1 in part because their high utility is complemented by low toxicity and high stability.2 Among vinyl silanes, those bearing oxygen substitution at silicon are particularly useful, as these reagents consistently provide superior reactivity compared to their trialkylsilyl counterparts, especially when used in Hiyama-Denmark cross-coupling,3 or in Tamao-Fleming oxidation.4
Though alkoxyvinyl silane reagents are highly useful, the traditional methods to access them all carry significant drawbacks, which can limit their utility. These reagents are most often prepared by either silylation of a vinyl organomagnesium or –lithium reagent with an electrophilic silane,5 or through hydrosilylation of an alkyne.6 The first method requires the corresponding organometallic reagent, which has limited functional group compatibility, and as significantly, are often derived from relatively complex starting materials (i.e., vinyl bromides), which can take many steps to access. Similarly, while hydrosilylation is milder in nature, the reaction requires access to the corresponding alkyne, which also poses synthetic challenges and have limited commercial availability. Indeed, the limitations in accessing unsaturated silanes have been attributed to be one of the primary reasons for the slow adoption of Hiyama-type cross-coupling reactions compared to other cross-couplings.7
Alkenes are widely abundant, stable starting materials. However, to date, no direct methods exist to convert these highly attractive starting materials into vinyl silyl ethers and disiloxanes.8,9 A mild, functional group tolerant method to achieve this conversion would greatly increase the synthetic accessibility of oxygen-substituted vinyl silanes.
Herein we report the preparation of vinyl silyl ethers and disiloxanes using silylditriflates (Fig 1). We show that these reagents participate in the palladium-catalyzed silyl-Heck reaction with terminal aryl-substituted alkenes and, upon quenching with alcohol or water, provide trans-vinyl silyl ethers in high yield as single regio- and geometric isomers. This method uses a commercially available catalyst system and requires only catalytic amounts of iodide additives. When combined with Hiyama-Denmark cross-coupling, this chemistry provides superior yields and regioselectivities in the preparation of stilbene derivatives compared to direct Heck arylation. Furthermore, by varying the nature of the quenching reagent, this strategy allows the direct preparation of a wide variety of vinyl silanes with tunable reactivity.
Figure 1.
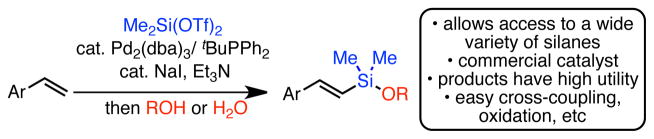
Preparation of Vinyl Silyl Ethers From Alkenes
We recently reported a palladium-catalyzed synthesis of vinyl and allyl trimethylsilanes from terminal alkenes and trimethylsilyl halides (X = Cl, I), which we believe proceeds through a Heck-type mechanism.10 Although this method displayed good functional group tolerance, it was limited to the preparation of trimethylsilanes. We recognized that an analogous reaction that allowed for the preparation of vinyl siloxyethers from simple alkenes would significantly increase the utility of this silyl-Heck reaction and allow facile access to this important class of reagents. A possible entry into silyl ethers would involve the use of dihalo or di(pseudo)halo silanes, however such substrates have not previously been examined in Heck-type reactions.11
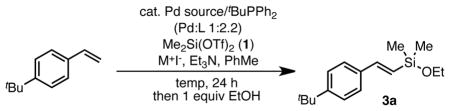
Our prior study demonstrated that Si-OTf bonds can participate in silyl-Heck reactions in the presence of an iodide additive.10 Thus, we began our studies by examining reaction of dimethylsilylditriflate (Me2Si(OTf)2, (1)),12,13 using model substrate 4-tert-butylstyrene and EtOH (Table 1). Our initial reaction conditions mirrored those developed in the reactions of trimethylsilyl halides (catalytic (COD)Pd(CH2TMS)2 (2) and tBuPPh2 with stoichiometric LiI and Et3N at 60 °C). When ethanol was added at the beginning of the reaction, modest yield of the desired ethoxyvinyl silane 3a resulted (26%, Table 1, entry 1). This reaction presumably proceeds via in situ formation of Me2Si(OEt)(OTf). Although promising, attempts to optimize these reaction conditions were not successful. In contrast, simply waiting to add the alcohol until the end of the reaction resulted in a dramatically improved yield of the desired product (81%, entry 2). In this latter case, we assume that the initially formed product is the corresponding dimethyl(vinyl)silyl(triflate), which is then converted to the silyl ether upon addition of the alcohol. Using this order of addition, commercially available Pd2(dba)3 can also be used as the precatalyst with equal success (entry 3). This result contrasts our earlier work wherein Pd2(dba)3 was ineffective. Modest decreases in the reaction temperature were tolerated without ill effect, but at room temperature the reaction was sluggish (entries 4 and 5). The reaction proved sensitive to the amount of 1 employed. A slight excess (1.1 equiv) of 1 proved optimal, but additional reagent rapidly eroded the yield (entry 6). As anticipated, iodide was a critical additive in the reaction (entry 7). However, only a catalytic amount was required and afforded superior yields (entry 8).14 Unlike our prior work, both LiI and NaI can be used with equal effect (entry 9). As NaI is both cheaper and more readily handled, we continued our studies with this additive. Finally, additional studies demonstrated that the reactions were best conducted at 35 °C and that only 2.5 mol % palladium was required to achieve quantitative yield (NMR).
Table 1.
Optimization of Silyl Ether Formation
| |||||
|---|---|---|---|---|---|
| entry | temp (°C) | Pd source (mol %) | 1 (equiv) | M+I− (mol %) | % yield (NMR) |
| 1 | 60 | 2 (5) | 1 | LiI (100) | 26a |
| 2 | 60 | 2 (5) | 1 | LiI (100) | 81 |
| 3 | 60 | Pd2(dba)3 (2.5) | 1 | LiI (100) | 81 |
| 4 | 40 | Pd2(dba)3 (2.5) | 1 | LiI (100) | 78 |
| 5 | rt | Pd2(dba)3 (2.5) | 1 | LiI (100) | 22 |
| 6 | 40 | Pd2(dba)3 (2.5) | 1.5 | LiI (100) | 24 |
| 7 | 40 | Pd2(dba)3 (2.5) | 1 | LiI (0) | 6 |
| 8 | 40 | Pd2(dba)3 (2.5) | 1.1 | LiI (5) | 98 |
| 9 | 40 | Pd2(dba)3 (2.5) | 1.1 | NaI (5) | 98 |
| 10 | 35 | Pd2(dba)3 (1.25) | 1.1 | NaI (5) | >99 |
EtOH added at start.
With these conditions in hand, we investigated the scope of the reaction (Table 2). On preparative scale (1–5 mmol), the model product 3a was isolated in 92% yield as the trans-vinyl silyl ether. A variety of other alcohols, including both secondary and tertiary alcohols, can also be used to quench the reaction, giving rise to a series of vinyl silyl ethers in similar isolated yields (3b and 3c). When water was used to quench the reaction, disiloxane 3d emerged in good isolated yield. In general, any of these quenching methods can be used to prepare vinyl silanes via this method, as demonstrated by the series of silanes 4–6 (exceptions noted below). For simplicity, we elected to study quenching with isopropanol and water in the remaining examples. In all cases, only trans-1,2-disubstituted vinyl silanes were observed. No trace of the cis-vinyl isomer, nor the 1,1-disubstituted regioisomeric silane has been detected.
Table 2.
Scope of Substrates for Silyl Ether and Disiloxane Formation

|
Isolated yields (average of two runs), 1 mmol scale unless otherwise noted;
5 mmol scale;
2.5 mol % Pd2(dba)3, 11 mol %
48 h;
50 °C;
60 °C
A wide variety of vinyl arenes also participate in the reaction. Both electron-donating and electron-withdrawing ethereal substrates generate products in high yields (5 and 6, Table 2). Dioxolane and silyl ether functionalities were well tolerated (7–9), as were fluorides, chlorides, esters, and tertiary amines (10–13). In general, substrates bearing electron-withdrawing substituents were slower to react than their electron-rich counter-parts and required slightly elevated reaction temperature and time. In the extreme, substrates bearing highly electron-withdrawing groups, such as p-nitro styrene (not shown) were unreactive. Steric hindrance on the arene did not adversely affect the reaction (14); however increased substitution on the alkene was not tolerated using the current catalyst (not shown).15
Alkenes bearing heterocycles are also substrates. Silyl ether 15 was formed from 2-vinylbenzofuran in good yield, and the reaction of N-tosyl 3-vinylindole led to vinyl silane 16 without incident. Finally, the reaction N-vinyl carbazole provided silyl ether 17 in high yield. In the case of these heterocyclic substrates, alcohol quench proved to be superior to those using water. In the case of both 15 and 17, water quench led to complex mixtures of products.
This silyl-Heck protocol is also compatible with a range of more complex vinylic substrates. For example, vinylferrocene can be silylated in near quantitative yield to give redox-active vinyl silanes 18a and 18b. We anticipate that these products will be highly useful in the preparation of topologically defined redox probes.16 Similarly, 4-vinylbenzocyclobutene is also an excellent substrate. Isopropyl silyl ether 19a was isolated in 91% yield. Water quench led to the formation of divinyldisiloxane bis-benzocyclobutene (19b) in similar yield. This latter product is the key monomer of Cyclotene® photoresist resins.17
Bis-functionalized silyl boronic esters, 20a and 20b were readily prepared from 4-vinyl phenyl pinacolboronic ester. In this reaction, no C-B bond activation was observed. These products provide a lynch-pin substrate for orthogonal Suzuki and Hiyama-Denmark coupling reactions.18
As a final example of a complex substrate, we examined the preparation of estradiol-derived vinyl silanes 21a and 21b. As in the previous examples, these complex materials can be prepared without incident in high yield with either isopropanol or water quench. Not only do these results demonstrate that highly complex substrates are compatible with the silyl-Heck reaction conditions, but the products for this and similar transformations will be highly useful in preparing bimolecular conjugates.
 |
(1) |
We also have begun to explore the scope of the groups on the silylditriflate. We were pleased to find that diethylsilylditriflate is also an excellent silylating agent. Under the optimized reaction conditions, similar yields were observed for diethylvinylsilanes with this reagent. For example, 4-tert-butyl styrene can be silylated in 88% yield, after isopropanol quench (eq 1).19 This strategy will allow for tuning of the reactivity of the vinyl silane products.
The formation of vinyl silyl ethers presented herein has numerous advantages. First, the fact that only a single regio- and geometric isomer is obtained provides a significant advantage over competing technologies. For example, it is well known that direct Heck arylation of electron-rich styrenes leads to a mixture of regioisomeric products.20 Selectivity is particularly poor for triflate electrophiles. For example, arylation of p-methoxystyrene with phenyltriflate gives a 57:43 mixture of α and β products, which in our hands were obtained in 81% yield (Scheme 1, top).21 In contrast, Hiyama-Denmark coupling of vinyl silyl ethers 5a and 5b proceeded in 95% and 84% yield, respectively, to give exclusively stilbene product 23 (Scheme 1, bottom).22 As 5a and 5b are obtained in high yields as single isomers from p-methoxystyrene in the silyl-Heck reaction, this two-step protocol is vastly superior in overall yield and avoids the difficult separation of regioisomeric products.
Scheme 1.

Silyl-Heck/Hiyama-Denmark vs. Direct Heck
In addition, because of the intermediacy of the vinylsilyltriflate, this strategy provides a divergent approach to differentially substituted vinyl silanes. For example, quenching with allyl or benzyl magnesium chloride leads to the corresponding dimethylvinyl silanes 25 and 26 in high yields (Scheme 2). Allyl- and benzylsilanes have proven highly useful in a variety of applications,3c, 4, 23 and this method provides a convenient method for accessing these materials via the silyl-Heck reaction.
Scheme 2. Preparation of Diverse Vinyl Silanes.
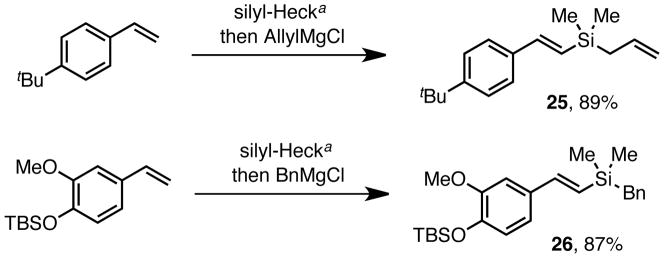
a1.25 mol % Pd2(dba)3, 5.5 mol % tBuPPh2, 1.1 equiv Me2Si(OTf)2 5 mol % NaI, Et3N, PhMe, 35 °C
In summary, we have developed a novel method to generate reactive and versatile vinyl silyl ethers and disiloxanes from aryl-substituted alkenes and related starting materials. This method exhibits broad functional group tolerance, and products are generated in good to excellent isolated yields and as single regio- and geometric isomers. The reaction proceeds with readily accessible reagents and an inexpensive, commercially available catalyst. Not only does this reaction dramatically expand the utility of the silyl-Heck reaction by allowing access to a wide variety of highly useful vinyl silyl ethers and related reagents, for the first time, it demonstrates that silane(bis)electrophiles can be used in a Heck-type process. Future studies will be directed towards expanding this transformation to other classes of alkenes and related molecules.
Supplementary Material
Acknowledgments
Mr. Scott Shuler is thanked for assistance. The University of Delaware (UD), the NSF (CAREER CHE1254360), and the Research Corporation (Cottrell Scholars Program) are gratefully acknowledged. SESM acknowledges graduate fellowship support from NIH 5T32 GM 08550-16. Data was acquired at UD on instruments obtained with the assistance of NSF and NIH funding (NSF CHE0421224, CHE1229234, & CHE0840401; NIH P20 GM103541 & S10 RR02692).
Footnotes
The authors declare no competing financial interest.
Supporting Information. Experimental procedures and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Fleming I, Barbero A, Walter D. Chem Rev. 1997;97:2063–2192. doi: 10.1021/cr941074u. [DOI] [PubMed] [Google Scholar]; (b) Brook MA. Silicon in Organic, Organometallic, and Polymer Chemistry. Wiley; Chichester: 2000. [Google Scholar]; (c) Fleming I, Dunoguès J, Smithers R. Organic Reactions. John Wiley & Sons, Inc; 2004. The Electrophilic Substitution of Allylsilanes and Vinylsilanes; pp. 57–575. [Google Scholar]
- 2.Nakao Y, Hiyama T. Chem Soc Rev. 2011;40:4893–4901. doi: 10.1039/c1cs15122c. [DOI] [PubMed] [Google Scholar]
- 3.(a) Denmark SE, Regens CS. Acc Chem Res. 2008;41:1486–1499. doi: 10.1021/ar800037p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Denmark SE, Liu JHC. Angew Chem Int Ed. 2010;49:2978–2986. doi: 10.1002/anie.200905657. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sore HF, Galloway WRJD, Spring DR. Chem Soc Rev. 2012;41:1845–1866. doi: 10.1039/c1cs15181a. [DOI] [PubMed] [Google Scholar]
- 4.Jones GR, Landais Y. Tetrahedron. 1996;52:7599–7662. [Google Scholar]
- 5.(a) Oshima K. Vinylsilanes. In: Fleming I, editor. Science of Synthesis. Vol. 4. Thieme; Stuttgart: 2001. pp. 713–754. [Google Scholar]; (b) Denmark SE, Neuville L, Christy MEL, Tymonko SA. J Org Chem. 2006;71:8500–8509. doi: 10.1021/jo061481t. [DOI] [PubMed] [Google Scholar]
- 6.(a) Denmark SE, Wang Z. Org Lett. 2001;3:1073–1076. doi: 10.1021/ol0156751. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Ball ZT. J Am Chem Soc. 2005;127:17644–17655. doi: 10.1021/ja0528580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corbet JP, Mignani G. Chem Rev. 2006;106:2651–2710. doi: 10.1021/cr0505268. [DOI] [PubMed] [Google Scholar]
- 8.Intramolecular alkene metathesis has been used to effectively prepare cyclic vinylsiloxy ethers, however, the intermolecular version of the reaction remains much less studied and is highly dependent on the nature of the starting alkenes. See: Pietraszuk C, Fischer H, Rogalski S, Marciniec B. J Organomet Chem. 2005;690:5912–5921.Denmark SE, Yang SM. Org Lett. 2001;3:1749–1752. doi: 10.1021/ol015950j.For a related reaction see: Wakatsuki Y, Yamazaki H, Nakano M, Yamamoto Y. J Chem Soc, Chem Commun. 1991:703–704. Also see reference 3b.
- 9.As this manuscript was in preparation, a report appeared demonstrating the preparation of predominately cis-vinyl silyl ethers from terminal alkenes using iridium-catalyzed hydrosilylation and a sacrificial oxidant. See: Cheng C, Simmons EM, Hartwig JF. Angew Chem Int Ed. 2013;52:8984–8989. doi: 10.1002/anie.201304084.
- 10.McAtee JR, Martin SES, Ahneman DT, Johnson KA, Watson DA. Angew Chem Int Ed. 2012;51:3663–3667. doi: 10.1002/anie.201200060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tanaka has demonstrated the oxidative addition of Pt(0) complexes to dichlorosilanes in stoichiometric reactions, see: Yamashita H, Tanaka M, Goto M. Organometallics. 1997;16:4696–4704.
- 12.Matyjaszewski K, Chen YL. J Organomet Chem. 1988;340:7–12. [Google Scholar]
- 13.This reagent is easily prepared from the reaction of neat Ph2SiMe2 and TfOH. It can be stored under nitrogen indefinitely after removal of the resulting PhH under vacuum, or used directly without diminished yield in the silyl-Heck reaction. See Supporting Information.
- 14.We believe iodide additives result in in situ generation of Si-I species, which enable oxidtive addition. See: Olah GA, Narang SC, Gupta BGB, Malhotra R. J Org Chem. 1979;44:1247–1251.
- 15.Terminal alkenes bearing allylic hydrogen atoms are also reactive but give poor allyl vs. vinyl selectivity. See Supporting Information for an example.
- 16.Gietter AAS, Pupillo RC, Yap GPA, Beebe TP, Rosenthal J, Watson DA. Chem Sci. 2013;4:437–443. doi: 10.1039/C2SC21413J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Gros WA. US4759874. Benzocyclobutene-based Die Attach Adhesive Compositions. 1988; (b) Kirchhoff RA, Bruza KJ. Prog Polym Sci. 1993;18:85–185. [Google Scholar]
- 18.Lim DSW, Anderson EA. Org Lett. 2011;13:4806–4809. doi: 10.1021/ol201833u. [DOI] [PubMed] [Google Scholar]
- 19.Some reactivity was also observed with diisopropylsilylditriflate. However, the yields remain low with the present catalyst system.
- 20.(a) Fristrup P, Le Quement S, Tanner D, Norrby PO. Organometallics. 2004;23:6160–6165. [Google Scholar]; (b) Oestreich M. The Mizoroki-Heck Reaction. John Wiley & Sons; Chichester, U.K: 2008. [Google Scholar]
- 21.With the use of phenyliodide, selectivity is only marginally better in this reaction (90:10 β:α).
- 22.(a) Denmark SE, Sweis RF. Org Lett. 2002;4:3771–3774. doi: 10.1021/ol026900x. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Regens CS. Tetrahedron Lett. 2011;52:2165–2168. doi: 10.1016/j.tetlet.2010.11.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bennetau B. Benzylsilanes. In: Fleming I, editor. Science of Synthesis. Vol. 4. Thieme; Stuttgart: 2002. pp. 825–836. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.