

Abstract
Lack of apoptotic cell death has been implicated in malignant transformation and resistance to anticancer therapies. The promotion of apoptosis in cancer cells could potentially lead to the regression and improved prognosis of refractory colorectal cancer. Synthetic triterpenoids have shown strong antitumorigenic activity towards diverse cancer cell types, but have not been investigated for colorectal cancer. In the present study, we tested the apoptosis-inducing activity of oleanane triterpenoid 2-cyano-3, 12-dioxooleana-1, 9(11)-dien-28-oic acid (CDDO) and its C-28 methyl ester (CDDO-Me) and C-28 imidazole (CDDO-Im) derivatives in colorectal cancer cells lines. Cell growth/viability assay (MTS) demonstrated that colorectal cancer cells are highly sensitive to CDDO-Me at concentrations of 1.25 to 10 μM. The primary mode of tumor cell destruction was apoptosis as demonstrated by the cleavage of PARP-1, activation of procaspases-3, -8, and -9 and mitochondrial depolarization. Induction of apoptosis by CDDO-Me was associated with the inhibition of pro-survival Akt, NF-κB and mTOR signaling proteins and NF-κB-regulated anti-apoptotic Bcl-2, Bcl-xL, Bad and survivin. These studies provide rationale for clinical evaluation of CDDO-Me for the treatment of advanced chemotherapy refractory colorectal cancer.
Keywords: Synthetic triterpenoids, colorectal cancer, apoptosis, signaling proteins
Colorectal cancer is the fourth most common cancer in men and women in the United States. It is more common in people over 50, and the risk increases with age. A high-fat diet and family history of colorectal cancer are also risk factors (1, 2). The inactivation of adenomatous polyposis coli (apc) gene, a tumor suppressor gene, due to mutation or deletion results in the development of adenomatous polyps, which are considered precursor lesions of colonic carcinomas (3–5). Current therapies (surgery, chemotherapy and radiotherapy) while effective against localized disease often fail to control more advanced metastatic colorectal cancer. Because the incidence of colon cancer increases with advancing age, this cancer is expected to become an increasingly greater health problem as life expectancy improves. Novel treatment modalities are therefore needed to prevent and treat colorectal cancer.
Aberration of apoptosis has been implicated not only in malignant transformation but also in resistance of tumors to conventional cancer therapies (6). Thus promotion of apoptosis in colon cancer cells with novel agents could lead to tumor regression and improved prognosis. Indeed, apoptosis has been implicated in preventing the development and progression of premalignant colonic epithelial cells to colon tumors (7–9).
Synthetic triterpenoids derived from oleanolic acid (CDDO, CDDO-Im and CDDO-Me) have shown greater anti-inflammatory activity than natural oleanolic acid (10, 11). CDDOs inhibit the proliferation of diverse types of tumor cell lines, including leukemia, multiple myeloma, osteosarcoma, breast, brain, prostate, lung, and pancreatic cancer cells in culture (12–16). Although the mechanisms of the anticancer effects of CDDOs are not fully understood, cancer cell differentiation and activation of caspase-dependent and -independent apoptosis contribute to the antitumor activity of CDDOs (17, 18). In addition, CDDOs inhibit MAPK (Erk1/2) and NF-κB signaling, and modulate TGF-β/Smad and PPARγ signaling (15, 19–21). CDDOs have also shown strong chemopreventive activity in animal models of liver, breast and lung (22, 23). On the other hand, the response of colorectal cancer cells to CDDOs in vitro or in vivo has not been examined. In the present study, we investigated the response of several human colorectal cancer cell lines to CDDO, CDDO-Im and CDDO-Me in vitro. Our result demonstrated that CDDOs inhibited the growth of several colorectal cancer cell lines by inducing apoptosis. CDDO-Me induced apoptosis through the activation of procaspases-3, -8 -9 and mitochondrial depolarization. Induction of apoptosis was associated with the inhibition of prosurvival Akt, NF-κB and mammalian target of rapamycin (mTOR) signaling molecules and antiapoptotic Bcl-2, Bcl-xL, Bad and survivin.
Materials and Methods
Reagents and antibodies
Synthetic CDDO, CDDO-Im, and CDDO-Me were obtained from the National Cancer Institute, Bethesda, MD, through the Rapid Access to Intervention Development Program. Anti-caspase-3, caspase-8, and caspase-9 antibodies were purchased from BD Pharmingen (San Diego, CA, USA). Anti-NF-κB (p65), anti-Bcl-2, anti-Bcl-xL, anti-Bad and anti-survivin antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). 96 AQueous One Solution Proliferation Assay System was purchased from Promega (Madison, WI, USA).
A 100 mM solution of CDDOs was prepared in DMSO and all test concentrations were prepared by diluting the stock solution in tissue culture medium.
Cell lines
HCT 8, HCT-15, HT-29, HCT 116, Colo 201 and Colo 205 human colorectal cancer cell lines were obtained from the American Type Culture Collection (ATCC), Rockville, MD, USA. HCT116 and HT-29 cells were grown in modified McCoy 5α culture medium (ATCC Cat #30-2007), whereas HCT8, HCT-15, HCT 116, Colo 210 and Colo 205 cells were grown in RPMI-1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal calf serum (Hyclone, Logan, UT, USA), 100 U/ml penicillin G, 100 μg/ml streptomycin sulfate. All cell lines were cultured at 37°C in a humidified atmosphere consisting of 5% CO2 and 95% air, and maintained by subculturing cells twice a week.
Measurement of cell viability (MTS assay)
Cells (1×104) were seeded into each well of a 96-well plate in 100 μl of tissue culture medium. After 24 h incubation to allow cells to adhere, cells were treated with CDDOs at concentrations shown in individual experiments for 72 h. Cell viability was then determined by the colorimetric MTS assay using CellTiter 96 AQueous One Solution Proliferation Assay System from Promega (Madison, WI, USA) following instructions provided by the vendor. Percentage cytotoxicity was calculated from the loss of cell viability in cultures.
Annexin V-FITC binding
Induction of apoptosis was assessed by the binding of annexin V to phosphotidylserine, which is externalized to the outer leaflet of the plasma membrane early during induction of apoptosis. Briefly, cells treated with CDDO-Me for 20 h were resuspended in the binding buffer provided in the annexin V-FITC apoptosis detection kit II (BD Biosciences, Pharmingen, USA). Cells were reacted with 5 μl of annexin V-FITC reagent and 5 μl of propidium iodide (PI) for 30 min at room temperature in the dark. Stained cells were analyzed by fluorescent activated cell sorting (FACS) on a FACScan flow cytometer (Becton Dickinson, USA).
Isolation of nuclear proteins
Nuclear extracts were prepared as described previously (Deeb et al., 2004). Following treatment with CDDO-Me for 20 h, cells were washed three times with PBS and incubated on ice for 15 minutes in hypotonic buffer A (10 mM HEPES, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 0.5 mM PMSF, and 0.6% NP40). Cells were vortexed gently for lysis and nuclei were separated from the cytosol by centrifugation at 12,000 ×g for 1 minute. Nuclei were resuspended in buffer C (20 mM HEPES, pH 7.9, 25% glycerol, 0.4 M NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 0.5 mM PMSF) and shaken for 30 minutes at 4°C. Nuclear extracts were obtained by centrifugation at 12,000 xg and protein concentration measured by Bradford assay (Bio Rad, Richmond, CA, USA). NF-κB in nuclear extracts was detected by Western blotting as described below.
Western blotting
Total cellular proteins were isolated by detergent lysis (1% Triton-X 100 (v/v), 10 mM Tris-HCl (pH 7.5), 5 mM EDTA, 150 mM NaCl, 10% glycerol, 2 mM sodium vanadate, 5 μg/ml leupeptin, 1 μg/mL aprotinin, 1 μg/ml pepstatinin, and 10 μg/ml 4-2-aminoethyl-benzenesulfinyl fluoride). Lysates were clarified by centrifugation at 14,000 ×g for 10 min at 4°C, and protein concentrations were determined by Bradford assay. Samples (50 μg) were boiled in an equal volume of sample buffer (20% glycerol, 4% SDS, 0.2% bromophenol blue, 125 mM Tris-HCl (ph 7.5), and 640 mM 2-mercaptoethanol) and separated on 10–14% SDS-polyacrylamide gels. Proteins resolved on the gels were transferred to nitrocellulose membranes. Membranes were blocked with 5% milk in 10 mM Tris-HCl (pH 8.0), 150 mM NaCl with 0.05% Tween 20 (TPBS) and probed with protein specific antibodies to NF-κB (1:500), p-Akt (1:500), p-mTOR (1:1000), Bcl-2 (1:500), Bcl-xL (1:1000), Bad (1:1000), survivin (1:500) or β-actin (1:500) followed by HRP-conjugated secondary antibody. Immune complexes were visualized with enhanced chemiluminescence (ECL) detection system from Amersham Corp (Arlington Heights, IL, USA).
Mitochondrial depolarization assay
Loss of mitochondrial potential by CDDO-Me was determined using mitochondrial potential sensor JC-1 (Molecular Probes, Invitrogen, San Diego, CA, USA). A total of 1×106 control (untreated) and CDDO-Me treated cells in 1 ml culture medium were loaded with mitochondrial sensor JC-1 dye (10 μg/ml) for 10 minutes at 22°C and analyzed by flow cytometry. In normal cells, dye is aggregated in mitochondria, fluoresces red, and is detected in the FL2 channel. In cells with altered mitochondrial potential, the dye fails to accumulate in the mitochondria, remains as monomers in the cytoplasm, fluoresces green, and is detected in the FL1 channel.
Statistical analysis
Data are presented as means±S.D.
Results
CDDOs inhibit the growth of colorectal cancer cells
To test the effect of CDDOs on proliferation of colorectal cancer cells, 1×104 HT-29, HCT-15, HCT 8, HCT 116, Colo 201 or Colo 205 cells were plated in each well of a 96-well microtiter plate for 24 h and then treated with CDDO, CDDO-Im, or CDDO-Me (0.625 to 10 μM) in triplicate for 72 h. The viability of cultures was determined by MTS assay. As shown in Figure 1A, CDDO, the parent synthetic analog of oleanolic acid was not very effective in inhibiting the growth of most of the cell lines at concentration of 5 μM and less. Some growth inhibition of HCT-15 (41%) and HCT-116 (35%) cells was observed at 10 μM CDDO. On the other hand, compared to CDDO, CDDO-Im and CDDO-Me exhibited stronger growth inhibitory activity at lower concentrations against all cell lines. CDDO-Im significantly reduced the growth of HT-29 (50–76%), HCT-15 (44–74%), Colo 201 (40–57%) and Colo 205 (35–79%) at 2.5–10 μM. The growth inhibitory effect of CDDO-Me was similar to that of CDDO-Im. It reduced the growth of HT-29 (47–82%), HCT-15 (58–85%), HCT 116 (14–79%), Colo 201 (33–77%) and Colo 205 (30–74%) cells at concentrations of 1.25 to 10 μM. Taken together; these data indicate that of the three synthetic oleanane triterpenoids, CDDO was least active in inhibiting the growth of colorectal cancer cells. On the other hand, CDDO-Im and CDDO-Me are almost equally effective in inhibiting the growth of most of the cancer cell lines over a concentration range of 1.25 μM to 10 μM.
Figure 1.

Effect of CDDO, CDDO-Im and CDDO-Me on the viability of colorectal cancer cells. A: 1×104 colon cancer cells (HT-29, HCT-15, HCT 8, HCT 116, Colo 201 or Colo 205) were seeded in each well of a microtiter plate in 0.1 ml of culture medium. Cells were allowed to adhere for 24 h before treating with CDDO or CDDO-Im or CDDO-Me at concentrations of 0 to 10 μM in triplicates for 72 h. Cell viability was measured by MTS assay using CellTiter AQueous assay system from Promega. Similar results were obtained in 4 independent experiments. B: Morphological changes in cell cultures (HT-29 and HCT-15 cells) treated or not with CDDO-Me for 72 h as visualized by light microscopy. Similar results were obtained in two independent experiments.
Microscopic examination of cancer cell cultures (HT-29 and HCT-15) showed partial rounding of cells at 1.25 μM and complete detachment and significant cell death (trypan blue dye exclusion) at higher concentrations of CDDO-Me (2.5 and 10 μM) after treatment for 72 h (Figure 1B).
CDDO-Me induces apoptosis in HT-29 and HCT-15 cells
Whether CDDO-Me kills colorectal cancer cells by inducing apoptosis was investigated next. We first measured the binding of annexin V-FITC and cleavage of PARP-1 in HT-29 and HCT-15 cells treated with CDDO-Me for 20 h. As shown in Figure 2A, a small percentage of untreated HT-29 and HCT-15 cells bound annexin V-FITC (<5%). In the case of HT-29 cells, following treatment with CDDO-Me at 1.25 and 2.5 μM the percentage of annexin V-FITC-binding cells increased to 46–51% and then decreased to 22% at 5 μM CDDO-Me. The percentage of annexin V-FITC-binding HCT-15 was 47, 20 and 5 at 1.25, 2.5 and 5 μM CDDO-Me, respectively.
Figure 2.

Treatment with CDDO-Me induces apoptosis in colorectal cancer cells. A: Binding of annexin V-FITC. HT-29 and HCT-15 cells were treated with CDDO-Me at concentrations of 1.25 to 5 μM for 20 h. Cells were then reacted with 5 μl of annexin V-FITC reagent for 30 min at room temperature. The percentage of annexin V-FITC positive tumor cells was determined by flow cytometry. Results are presented as percentage of annexin V-FITC-binding cells. B: Cleavage of PARP-1. Cells were treated with CDDO-Me as described above and PARP-1 was analyzed by immunoblotting. Each experiment was repeated at least twice.
In addition, the native PARP-1 (110 kDa) protein was almost completely cleaved at 2.5 and 5 μM CDDO-Me with the appearance of 89 kDa cleavage product in both cell lines (Figure 2B). These data demonstrate that CDDO-Me kills colorectal cancer cells by inducing apoptosis.
CDDO-Me activates procaspases in colorectal cancer cells
As further evidence that CDDO-Me induces apoptosis we examined the effect of CDDO-Me on the activation of procaspases-3, -8 and -9. Western blot analysis of cell lysates of HT-29 and HCT-15 cells treated with CDDO-Me showed almost complete processing of procaspases-3, -8 and -9 in both cell lines at 5.0 and 2.5 μM CDDO-Me (Figure 3). The reduction in the levels of these procaspases was also measurable at 1.25 but not at 0.625 μM CDDO-Me.
Figure 3.

Treatment with CDDO-Me cleaves procaspases-3, -8 and -9. HT-29 and HCT-15 cells were treated with CDDO-Me at concentrations of 0 to 5 μM. After incubation for 20 h, cellular lysates prepared from untreated (control) and treated cells were fractionated on 10% SDS-PAGE gel (50 μg/lane). Proteins were transferred from the gel to nitrocellulose membrane and first reacted with antibody to caspase-3, caspase-8 and caspase-9 or β-actin (loading control) followed by HRP-conjugated second antibody. Signals were visualized with enhanced chemiluminescence. Numbers above signal bands denote percentage suppression compared to control (0 CDDO-Me).
CDDO-Me induces mitochondrial depolarization
The cleavage of procaspase-9 suggested the involvement of mitochondrion in induction of apoptosis in colorectal cancer cells by CDDO-Me. To further confirm it we evaluated the mitochondrial depolarization in cells treated with CDDO-Me from the fluorescent shift of cells loaded with mitochondrial probe JC-1. There was significant change in the mitochondrial potential after treatment of both cell lines with CDDO-Me for 20 h. The percentage of HT-29 cells with green fluorescence increased from 1% at 0 CDDO-Me to 37% at 5 μM CDDO-Me in a dose-dependent manner (Figure 4). On the other hand, only a weak mitochondrial depolarizing effect of CDDO-Me was seen in HCT-15 cells (e.g. 17% cells at 5 μM CDDO-Me, Figure 4). Together, the cleavage of PARP-1, activation of procaspases and mitochondrial depolarization demonstrated induction of apoptosis by CDDO-Me in colorectal cells both through the non-mitochondrial and mitochondrial pathways.
Figure 4.

CDDO-Me induces mitochondrial depolarization in colon cancer cells. HT-29 and HCT-15 cells were treated with CDDO-Me at 0 to 5 μM for 20 h, then 1×106 cells were resuspended in 1 ml culture medium and loaded with mitochondrial potential sensor JC-1 (10 μg/ml) for 10 minutes at 22°C. Cells were analyzed by flow cytometry for fluorescence emission. Data are shown as flow cytographs of cells fluorescing red (FL2 channel) or green (FL1 channel). Histograms showing the percentage of cells with loss of mitochondrial potential difference. Similar results were obtained in two separate experiments.
CDDO-Me inhibits prosurvival Akt, mTOR and NF-κB signaling proteins in colorectal cancer cells
Akt and NF-κB are major antiapoptotic pathways that confer survival advantage and resistance to various forms of anticancer therapies. In addition, increase in mTOR activity promotes tumor growth and inhibition of mTOR inhibits proliferation and survival of tumor cells. We investigated the effect of CDDO-Me on these antiapoptotic signaling proteins in colorectal cancer cells. HT-29 and HCT-15 cells were treated with CDDO-Me at 0 to 5 μM for 20 h and p-Akt (ser473), p-mTOR (ser2448), and nuclear NF-κB (p65) expression was analyzed by immunoblotting. CDDO-Me significantly to completely inhibited p-Akt, p-mTOR and NF-κB in both cell lines at concentrations of 5 and 2.5 μM (60% to 90% reduction, Figure 5A). The levels of these proteins were only slightly reduced at lower concentrations.
Figure 5.
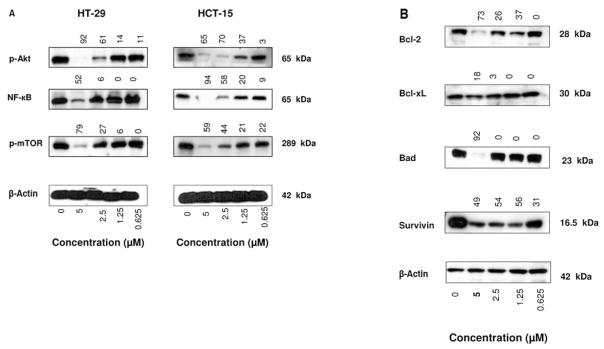
CDDO-Me inhibits prosurvival p-Akt, NF-κB and p-mTOR and antiapoptotic Bcl-2, Bcl-xL, Bad and survivin. HT-29 and HCT-15 cells were treated with CDDO-Me at 0 to 5 μM for 20 h. After treatment, cell lysates were prepared and analyzed by Western blotting using anti-p-Akt antibody, anti-NF-κB (p65) antibody, anti-p-mTOR antibodies (A) or anti-Bcl-2, Bcl-xL, Bad or survivin (B) or anti-β-actin antibody (loading control). Numbers above the signal bands denote percent suppression compared to control (0 CDDO-Me).
CDDO-Me inhibits anti-apoptotic Bcl-2, Bcl-xL, Bad and survivin in HT-29 cells
To determine whether CDDO-Me modulates the levels of anti-apoptotic proteins Bcl-2, Bcl-xL, Bad and survivin in colorectal cancer cells, HT-29 cells were treated with CDDO-Me for 20 h and levels of these proteins were analyzed by immunoblotting. As shown in Figure 5B, treatment with CDDO-Me significantly reduced the levels of Bcl-2 (60%–80%), Bcl-xL (20%–54%), Bad (90%) and survivin (50% to 80%) at 1.25 to 5 μM concentrations, suggesting that reduction in the levels of these antiapoptotic proteins by CDDO-Me is in part responsible for induction of apoptosis.
Discussion
Triterpenes or triterpenoids are members of a larger family of structurally related compounds known as cyclosqualenoids that are widely distributed in the plant kingdom (24). Oleanolic acid and ursolic acid are naturally occurring triterpenoids that have been used in traditional Asian medicine for centuries as antibacterial, antifungal, anti-cancer, and anti-inflammatory agents (25–27). However, recent studies have shown that naturally occurring triterpenoids possess only weak anti-inflammatory and antitumorigenic activities. In contrast, synthetic triterpenoids derived from oleanolic acid and ursolic acid exhibit potent anti-inflammatory, antitumorigenic, antiproliferative, and apoptosis inducing activities. Indeed, CDDO, CDDO-Im and CDDO-Me have shown strong antitumorigenic activity against a wide range of cancer cells in vitro (12–21) and in vivo (22, 23). The anticancer activity of synthetic triterpenoids for colorectal cancer has not been investigated. In the present study, we provide evidence that some of the synthetic CDDOs have potent growth inhibitory and apoptosis-inducing effects on human colorectal cancer cells. Our results demonstrate that while the parent compound, CDDO, has minimal growth inhibitory activity against most of the colon cancer cell lines, its methyl ester (CDDO-Me) and imidazole (CDDO-Im) derivatives exhibit potent growth inhibitory activity against colon cancer cells. At concentrations ranging from 1.25 μM to 10 μM, CDDO-Im and CDDO-Me were equally effective in inhibiting the growth of colon cancer cells and both were more effective than CDDO. These findings are consistent with previous reports in which CDDO-Im and CDDO-Me were shown to be more effective than CDDO for antitumor activity in other cancer models (11, 15, 19).
Two major pathways of apoptotic cell death program, namely receptor-mediated (extrinsic) and chemical-induced mitochondrial (intrinsic) apoptosis have been identified. In both cases, caspases, a family of cysteine proteases, play an important role in apoptotic cell death (28, 29). Binding of the death ligands (e.g. TNF-α, FasL, TRAIL) with their cognate receptors activates initiator caspase-8 which then cleaves and activates effector caspases-3, -6, and -7 leading to apoptosis (28). In chemical-induced (chemotherapeutic agents) apoptosis, undefined signals induce the release of cytochrome c from mitochondria, which in conjunction with Apaf-1 causes activation of initiator caspase-9. Activated caspase-9, in turn, activates effector caspases-3, -6, and -7 (29). CDDO-Me caused the cleavage of initiator procaspase-8 and the effector procaspase-3 in HT-29 and HTC-15, suggesting that the death receptor-signaling pathway of apoptosis might be involved in CDDO-Me-induced apoptosis. CDDO-Me also induced the cleavage of procaspase-9 and mitochondrial depolarization, indicating that the intrinsic pathway of apoptosis is also activated in tumor cells treated with CDDO-Me. The induction of both pathways of apoptosis by CDDO-Me in colon cancer cells is consistent with the previous reports showing activation of both pathways of apoptosis by CDDOs in other tumor types (16, 18).
Akt/PBK is a major antiapoptotic pathway which is frequently hyperactivated in most cancers (30). Activated p-Akt promotes cell growth and survival by inactivating downstream substrates such as Bad, procaspase-9, and Forkhead transcription factors (31, 32). Antiapoptotic NF-κB and progrowth mTOR signaling pathways are downstream targets of activated Akt/PBK and control the expression of genes and cellular processes involved cell proliferation, oncogenesis, angiogenesis, and apoptosis (33, 34). CDDO-Me inhibited the expression of p-Akt, NF-κB and p-mTOR in HT-29 and HCT-15, indicating that the inhibition of these prosurvival signaling proteins is essential for induction of apoptosis by CDDO-Me. NF-κB-regulated Bcl-2, Bcl-xL, Bad and survivin are major antiapoptotic proteins that render cancer cells resistant to chemotherapeutic agents and ionizing radiation. The inhibition of these antiapoptotic proteins by CDDO-Me indicated that they are also targets of CDDO-Me in colorectal cancer cells. Overall, the growth inhibition and induction of apoptosis in colon cancer cells by CDDO-Me suggest that CDDO-Me is a novel agent for therapeutic development to treat colorectal cancer.
Acknowledgments
This work was supported by NIH grant 1R01 130948 to S.C.G.
References
- 1.Giovannucci E, Willett WC. Dietary factors and risk of colon cancer. Ann Med. 1994;26:443–452. doi: 10.3109/07853899409148367. [DOI] [PubMed] [Google Scholar]
- 2.Rao CV, Hirose Y, Indranie C, Reddy BS. Modulation of experimental colon tumorigenesis by types and amounts of dietary fatty acids. Cancer Res. 2001;61:1927–1933. [PubMed] [Google Scholar]
- 3.Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247:322–324. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- 4.Kinzler KW, Vogelstein B. Cancer-susceptibility genes: gatekeepers and caretakers. Nature. 1997;386:761–763. doi: 10.1038/386761a0. [DOI] [PubMed] [Google Scholar]
- 5.Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, Gould KA, Dove WF. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science. 1992;256:668–670. doi: 10.1126/science.1350108. [DOI] [PubMed] [Google Scholar]
- 6.Kerr JF, Winterford CM, Harmon BV. Apoptosis. Its significance in cancer and cancer therapy. Cancer. 1994;73:2013–2026. doi: 10.1002/1097-0142(19940415)73:8<2013::aid-cncr2820730802>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 7.Hall PA, Coates PJ, Ansari B, Hopwood D. Regulation of cell number in the mammalian gastrointestinal tract: the importance of apoptosis. J Cell Sci. 1994;107:3569–3577. doi: 10.1242/jcs.107.12.3569. [DOI] [PubMed] [Google Scholar]
- 8.Bedi A, Pasricha PJ, Akhtar AJ, Barber JP, Bedi GC, Giardiello FM, Zehnbauer BA, Hamilton SR, Jones RJ. Inhibition of apoptosis during development of colorectal cancer. Cancer Res. 1995;55:1811–1826. [PubMed] [Google Scholar]
- 9.Swamy MV, Patlolla JM, Steele VE, Kopelovich L, Reddy BS, Rao CV. Chemoprevention of familial adenomatous polyposis by low doses of atorvastatin and celecoxib given individually and in combination to APCM in mice. Cancer Res. 2006;66:7370–7377. doi: 10.1158/0008-5472.CAN-05-4619. [DOI] [PubMed] [Google Scholar]
- 10.Honda T, Rounds BV, Gribble GW, Suh N, Wang Y, Sporn MB. Design and synthesis of 2-cyano-3, 12-dioxoolean-1, 9-dien-28-oic acid, a novel and highly active inhibitor of nitric oxide production in mouse macrophages. Biorg Med Chem Lett. 1998;8:2711–2714. doi: 10.1016/s0960-894x(98)00479-x. [DOI] [PubMed] [Google Scholar]
- 11.Honda T, Rounds BV, Bore L, Favaloro FG, Gibble GW, Suh N, Wang Y, Sporn MB. Novel synthetic oleanane triterpenoids: a series of highly active inhibitors of nitric oxide production in mouse macrophages. Bioorg Chem Lett. 1999;9:3429–3434. doi: 10.1016/s0960-894x(99)00623-x. [DOI] [PubMed] [Google Scholar]
- 12.Gao X, Deeb D, Jiang H, Liu Y, Dulchavsky S, Gautam S. Synthetic triterpenoids inhibit growth and induce apoptosis in human glioblastoma and neuroblastoma cells through inhibition of prosurvival Akt, NF-κB and Notch1 signaling. J Neurooncol. 2007;84:147–157. doi: 10.1007/s11060-007-9364-9. [DOI] [PubMed] [Google Scholar]
- 13.Deeb D, Gao X, Dulchavsky SA, Gautam SC. CDDO-Me induces apoptosis and inhibits Akt, mTOR and NF-κB in prostate cancer cells. Anticancer Res. 2007;27:3035–3044. [PubMed] [Google Scholar]
- 14.Konopleva M, Tsao T, Ruvolo P, Stiouf I, Estrov Z, Leysath CE, Zhao S, Harris D, Chang S, Jackson CE, Munsell M, Suh N, Gribble G, Honda T, May WS, Sporn MB, Andreeff M. Novel triterpenoid CDDO-Me is a potent inducer of apoptosis and differentiation in acute myelogenous leukemia. Blood. 2002;99:326–335. doi: 10.1182/blood.v99.1.326. [DOI] [PubMed] [Google Scholar]
- 15.Shishodia S, Sethi G, Konopleva M, Andreeff M, Aggarwal BB. A synthetic triterpenoid, CDDO-Me, inhibits IkappaBalpha kinase and enhances apoptosis induced by TNF and chemotherapeutic agents through down-regulation of expression of nuclear factor kappaB-regulated gene products in human leukemic cells. Clin Cancer Res. 2006;12:1828–1838. doi: 10.1158/1078-0432.CCR-05-2044. [DOI] [PubMed] [Google Scholar]
- 16.Ikeda T, Sporn M, Honda T, Gribble GW, Kufe D. The novel triterpenoid CDDO and its derivatives induce apoptosis by disruption of intracellular redox balance. Cancer Res. 2003;63:5551–5558. [PubMed] [Google Scholar]
- 17.Konopleva M, Tsao T, Estrov Z, Lee RM, Wang RY, Jackson CE, McQueen T, Monaco G, Munsell M, Belmont J, Kantarjian H, Sporn MB, Andreeff M. The synthetic triterpenoid 2-cyano-3, 12-dioxooleana-1, 9-dien-28-oic acid induces caspase-dependent and -independent apoptosis in acute myelogenous leukemia. Cancer Res. 2004;64:7927–7935. doi: 10.1158/0008-5472.CAN-03-2402. [DOI] [PubMed] [Google Scholar]
- 18.Ito Y, Pandey P, Sporn MB, Datta R, Kharbanda S, Kufe D. The novel triterpenoid CDDO induces apoptosis and differentiation of human osteosarcoma cells by a caspase-8 dependent mechanism. Mol Pharmacol. 2001;59:1094–1099. doi: 10.1124/mol.59.5.1094. [DOI] [PubMed] [Google Scholar]
- 19.Konopleva M, Contractor R, Kurinna SM, Chen W, Andreeff M, Ruvolo PP. The novel triterpenoid CDDO-Me suppresses MAPK pathways and promotes p38 activation in acute myeloid leukemia cells. Leukemia. 2005;19:1350–1354. doi: 10.1038/sj.leu.2403828. [DOI] [PubMed] [Google Scholar]
- 20.Suh N, Roberts AB, Birkey Reffey S, Miyazono K, Itoh S, ten Dijke P, Heiss EH, Place AE, Risingsong R, Williams CR, Honda T, Gribble GW, Sporn MB. Synthetic triterpenoids enhance transforming growth factor beta/Smad signaling. Cancer Res. 2003;63:1371–1376. [PubMed] [Google Scholar]
- 21.Chintharlapalli S, Papineni S, Konopleva M, Andreef M, Samudio I, Safe S. 2-Cyano-3, 12-dioxoolean-1, 9-dien-28-oic acid and related compounds inhibit growth of colon cancer cells through peroxisome proliferator-activated receptor gamma-dependent and -independent pathways. Mol Pharmacol. 2005;68:119–128. doi: 10.1124/mol.105.011437. [DOI] [PubMed] [Google Scholar]
- 22.Xiaoyang L, Konopleva M, Zeng Z, Ruvolo V, Stephens LC, Schober W, McQueen T, Dietrich M, Madden TL, Andreeff M. The novel triterpenoid C-28 methyl ester of 2-cyano-3, 12-dioxoolen-1, 9-dien-28-oic acid inhibits metastatic murine breast tumor growth through inactivation of STAT3 signaling. Cancer Res. 2007;67:4210–4218. doi: 10.1158/0008-5472.CAN-06-3629. [DOI] [PubMed] [Google Scholar]
- 23.Liby K, Royce DB, Williams CR, Risingsong R, Yore MM, Honda T, Gribble GW, Dmitrovsky E, Sporn TA, Sporan MB. The synthetic triterpenoid CDDO-methyl ester and CDDO-amide prevent lung cancer induced by vinyl carbomate in A/J mice. Cancer Res. 2007;67:2414–2419. doi: 10.1158/0008-5472.CAN-06-4534. [DOI] [PubMed] [Google Scholar]
- 24.Sporn MB, Suh N. Chemoprevention of cancer. Carcinogenesis. 2007;21:525–530. doi: 10.1093/carcin/21.3.525. [DOI] [PubMed] [Google Scholar]
- 25.Huang MT, Ho CT, Wang ZY, Ferraro T, Lou YR, Stauber K, Ma W, Georgiadis C, Laskin JD, Conney AH. Inhibition of skin tumorigenesis by rosemary and its constituents carnosol and ursolic acid. Cancer Res. 1994;54:701–708. [PubMed] [Google Scholar]
- 26.Nishino H, Nishino A, Takayasu J, Hasegawa T, Iwashima A, Hirabayashi K, Iwata S, Shibata S. Inhibition of the tumor-promoting action of 12-O-tetradecanoylphorbol-13-accetate by some oleanane-type triterpenoid compounds. Cancer Res. 1988;48:5210–5215. [PubMed] [Google Scholar]
- 27.Ryu SY, Oak MH, Yoon SK, Cho DI, Yoo GS, Kim TS, Kim KM. Anti-allergic and anti-inflammatory triterpenes from the herb of Prunella vulgaris. Planta Med. 2000;66:358–360. doi: 10.1055/s-2000-8531. [DOI] [PubMed] [Google Scholar]
- 28.Ashkenazi A, Dixit VM. Death receptors: Signaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 29.Sun X-M, MacFarlane M, Zhuang J, Wolf BB, Green DR, Cohen GM. Distinct caspase cascades are initiated in receptor-mediated and chemical-induced apoptosis. J Biol Chem. 1999;274:5053–5060. doi: 10.1074/jbc.274.8.5053. [DOI] [PubMed] [Google Scholar]
- 30.Marte BM, Downward J. PKB/Akt: connecting phosphoino-sitide3-kinase to cell survival and beyond. Trends Biochem Sci. 1997;22:355–358. doi: 10.1016/s0968-0004(97)01097-9. [DOI] [PubMed] [Google Scholar]
- 31.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of Bad couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 32.Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Standbridge E, Frisch S, Reed JC. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 33.Mayo MW, Baldwin AS. The transcription factor NF-κB: control of oncogenesis and cancer therapy resistance. Biochim Biophys Acta. 2000;1470:M55–M62. doi: 10.1016/s0304-419x(00)00002-0. [DOI] [PubMed] [Google Scholar]
- 34.Hidalgo M, Rowinsky EK. The rapamycin-sensitive signal transduction pathway as a target for cancer therapy. Oncogene. 2000;19:6680–6686. doi: 10.1038/sj.onc.1204091. [DOI] [PubMed] [Google Scholar]