

Abstract
Interleukin (IL)-12 has been shown to suppress T helper type 2 (Th2)-induced pathogenesis that is associated with allergic asthma, largely through interferon (IFN)-γ production. We have recently shown that in the absence of T-bet, the major regulator of IFN-γ expression, allergic lung inflammation is primarily associated with IL-17-associated recruitment of neutrophils into the pulmonary tract of mice. In the absence of T-bet, exogenous IL-12 was still able to suppress neutrophilic infiltration and to diminish levels of IL-17, IL-23, and IL-23R, as well as retinoic acid-related orphan receptorγt, the transcriptional regulator of the Th17 pathway. The same effects were observed in T-bet−/− IFN-γ−/− double knockout mice, showing an IFN-γ-independent effect of IL-12 in this model. IL-10 expression in the lungs of T-bet-deficient mice was significantly increased after IL-12 treatment, and inoculation of anti-IL-10R mAb completely reversed the ability of IL-12 to suppress histological inflammation, recruitment of inflammatory cell subsets into the lung, bronchiole hyperresponsiveness, and IL-17 production. We conclude that Th17-mediated allergic lung inflammation that becomes dominant in the absence of effective IFN-γ signaling can be effectively suppressed by IL-12 through an IL-10-dependent mechanism.
INTRODUCTION
Allergic asthma is characterized as a chronic inflammatory disorder of the bronchial mucosa and has been hypothesized to be the result of dysregulated CD4+ T helper type 2 (Th2) cell activity.1 Expression of Th2-type cytokines, specifically interleukin (IL)-4, IL-5, and IL-13, is increased in response to inhaled allergens and results in bronchiole hyperresponsiveness (BHR), increased concentrations of allergen-specific serum IgE, goblet cell hyperplasia, and eosinophilic infiltration into the airways.2,3 In addition to eosinophils, some studies have reported increased numbers of neutrophils in the airways of asthmatic patients, particularly in patients with severe asthma.4,5 Moreover, the proinflammatory cytokine IL-17, has been found to be expressed in the airways of patients with asthma,6,7 and the levels of expression can be correlated with disease severity.8 There is also evidence for a prominent role for IL-17 in the induction of neutrophilic airway inflammation and the progression of allergic asthma in animal models.9–11 For instance, neutralization of IL-17 during either allergen priming or allergen challenge of mice has been reported to inhibit the expansion and/or recruitment of neutrophils into the lung.9,12 In addition, it has been found that IL-17/IL-17R signaling is critically required for allergen sensitization leading to asthma.11 Taken together, these results indicate that IL-17 can have a crucial role in asthmatic pulmonary inflammation.
IL-17 is primarily produced by a distinct population of CD4+ T cells, namely Th17 cells.13 In addition to IL-17 (IL-17A), Th17 cells secrete IL-17F, IL-21, and IL-22, and have been implicated in the pathogenesis of various inflammatory conditions, including experimental autoimmune encephalitis and collagen-induced arthritis.14–16 Th17 cell differentiation is induced by transforming growth factor-β and IL-6 in mice,17–19 and IL-23 is important in the maintenance of Th17 cell lineage commitment.13,20 These cytokines induce the expression of the transcription factors, retinoic acid-related orphan receptor (ROR)γt and RORα, in naive T cells after T-cell receptor stimulation, which results in Th17 cell differentiation.21,22 Conversely, other cytokines, including IL-2, IL-4, IL-27, and interferon (IFN)-γ, have the ability to inhibit or attenuate Th17 differentiation in both humans and mice.23–26 In addition, mice lacking T-bet, the T-box transcription factor crucial for Th1 cell differentiation and IFN-γ production, produce greater numbers of IL-17+ cells,27 presumably because of reduced inhibitory IFN-γ production. Interestingly, there is now compelling evidence that in the absence of T-bet, IL-17 has a predominant role in the pathogenesis of allergic asthma. We,28 along with others,29 have shown that mice deficient in T-bet show increased Th17 pulmonary cytokine production and enhanced Th17-mediated neutrophilic inflammation in the airways after allergen sensitization and challenge. In addition, neutralization of IL-17 in T-bet-deficient mice can reverse airway inflammation after antigen challenge.28 These results suggest that T-bet is a negative regulator of Th17-mediated allergic airway inflammation.
It is well known that Th1 cells, which produce IFN-γ, can inhibit the development of Th2-mediated airway inflammation.30 In fact, IL-12 drives CD4+ T-cell differentiation toward a Th1-like phenotype31 and can reverse the polarization of Th2 cells.32 The ability of IL-12 to inhibit allergic asthma has been studied by several laboratories in murine models of allergen-induced airway inflammation. Treatment with IL-12 during the antigen-sensitization stage has been found to reduce levels of eosinophils in bronchoalveolar lavage (BAL) fluids, diminish lung pathology, and lessen BHR.33,34 In addition, IL-12 given during the antigen-challenge phase can abrogate allergic airway inflammation and BHR in sensitized mice, inhibit airway eosinophilia,35,36 and prevent increases in IL-4 and IL-5 production after airway challenge,37,38 effectively causing a shift to a Th1 cellular response, which is IFN-γ dependent. It has also been reported that IL-12 has inhibitory effects on allergic lung inflammation that are independent of IFN-γ,35,39 but the precise mechanism responsible for this activity remains unclear. Finally, although it has now been established that IL-17 can have an important role in allergic lung inflammation, the role of IL-12 in alleviating Th17/neutrophil-mediated allergic lung inflammation still remains unexplored.
In this study, we examined the ability of IL-12 to suppress Th17-cell-mediated asthma, using ovalbumin (OVA) sensitization and challenge of T-bet−/− mice as a model. Our results show that Th17 cells have an important role in antigen-induced airway inflammation in the absence of T-bet and that IL-12 suppresses this inflammation independently from IFN-γ expression. It has been previously shown that in vitro exposure of T cells and macrophages to IL-12 can result in IL-10 expression.40–42 In addition, there are reports that IL-10 negatively regulates Th17-associated cytokine production.43 Thus, we tested whether IL-12 alleviates Th17-mediated allergic lung inflammation through induction of IL-10. The results presented in this study detail a novel biological mechanism in which a Th1-inducing cytokine, IL-12, suppresses Th17 cytokine production through induction of IL-10 and independently from IFN-γ signaling. These findings expand our understanding of lung immune homeostasis and could affect attempts to develop effective therapies for allergen-induced lung inflammation.
RESULTS
IL-12 suppresses IL-17 production
Previous studies have shown the ability of IL-12 to inhibit Th244 and Th1727 cytokine expression. This inhibition has been found to be mediated primarily by IFN-γ.24,31,45 However, in the absence of T-bet, Th1 cells lose their ability to produce IFN-γ, and we thus hypothesized that IL-12 would have little to no effect in suppressing Th2 or Th17 cytokine production in T-bet−/− mice. To determine whether IL-12 can suppress Th2 cytokine expression in the absence of T-bet, the levels of IL-4 and IL-5 from the lungs, as well as BAL fluids of wild-type (WT) and T-bet−/− mice were measured after OVA sensitization and challenge, with or without IL-12 treatment during the challenge phase. As expected, challenge of sensitized WT animals with OVA induced the expression of IL-4 and IL-5 in lung tissue, whereas no IFN-γ production was detected (Figure 1a). IL-12 treatment induces IFN-γ production in WT mice and suppresses the secretion of Th2-associated cytokines. Essentially identical results were obtained when BAL fluid cytokine production was measured (data not shown). However, this pattern was not observed in sensitized T-bet-deficient animals after OVA challenge. In the absence of T-bet, the levels of IL-4 and IL-5 failed to reach the levels observed in WT mice (Figure 1a). Furthermore, after IL-12 treatment, there was no induction of IFN-γ, and IL-12 treatment had only minor effects on Th2-related cytokine expression.
Figure 1.

Interleukin (IL)-12 suppresses pulmonary Th17 activity. Cytokine levels in lung homogenates obtained from unchallenged, ovalbumin (OVA)-challenged, and OVA-challenged plus IL-12-treated BALB/c T-bet+/+ (wild type, WT) and T-bet−/− mice were measured 24 h after a 5-day OVA challenge. Protein levels were normalized to lung tissue total protein. (a) IL-4, IL-5, and interferon (IFN)-γ were measured by murine cytometric bead array and (b) IL-17 levels were measured by enzyme-linked immunosorbent assay (ELISA). The data are shown as means±s.e.m.; 8 mice per group. (c) IL-17, retinoic acid-related orphan receptor (ROR)γt, and RORα transcripts were measured by quantitative real-time (RT)-PCR. Transcript expression was normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and used to calculate expression relative to untreated mice. (d, e) Expression of IL-23 protein and IL-23R transcripts, respectively, in the lungs of sensitized mice upon antigen challenge relative to non-sensitized mice after antigen challenge. Lungs were collected 24 h after a 5-day OVA challenge from both groups of mice. Levels of IL-23 were measured by ELISA, and levels of IL-23R transcripts were determined by quantitative RT-PCR. The data represent means±s.e.m.; 8 mice per group. (f) Flow cytometric detection of IFN-γ and IL-17-producing cells in lung lymphocyte preparations from OVA-challenged and OVA-challenged plus IL-12-treated T-bet+/+ (WT) and T-bet−/− mice. The data represent mean percentages of positively stained cells within the lymphocyte gate. *P < 0.05. All results are representative of at least three independent experiments with 6–8 mice per group.
Previously, we,28 along with others,29 showed that in the absence of T-bet, IL-17 is the primary mediator of antigen-induced lung inflammation. Indeed, T-bet has been shown to be a negative regulator of Th17 differentiation,46 which suggests that in the absence of T-bet, Th17 could be the default pathway. Therefore, we sought to determine whether IL-12 would have any effect on the Th17 pathway in the absence of T-bet expression. Surprisingly, although IL-12 treatment of WT mice had a subtle effect on IL-17 production or on transcript levels of IL-17A and RORγt, which was most likely due to IFN-γ production, it had a dramatic inhibitory effect in T-bet−/− mice (Figure 1b and c). In addition, IL-23 production in T-bet−/− but not WT animals was suppressed after IL-12 treatment (Figure 1d). IL-23R expression, which is necessary for the maintenance of Th17 cells,25 was increased in T-bet-deficient mice after OVA challenge, and IL-12 treatment strikingly inhibited its expression (Figure 1e). IL-17 and IFN-γ expression in lung lymphocytes was also measured by intracellular cytokine staining. After OVA challenge, WT and T-bet−/− mice contained similar numbers of IFN-γ+ cells (mean of 4.2×104 IFN-γ+ cells per lung in WT mice and 5.4×104 IFN-γ+ cells per lung in T-bet−/− mice). This suggests that there are residual lung lymphocytes in T-bet−/− mice that produce IFN-γ after allergen challenge. However, after IL-12 treatment, IFN-γ+ cell levels increased in WT but not in T-bet−/− mice. Although there was no change in numbers of IFN-γ+ cells in the absence of T-bet, the percentages of IL-17-producing lymphocytes were higher in sensitized T-bet−/− mice than in WT mice after OVA challenge, and these levels were significantly reduced after IL-12 treatment (Figure 1f). Thus, it seems that IL-12 inhibits cytokine expression as well as the generation and expansion of Th17 cells in the absence of T-bet.
IL-12 alleviates allergen-induced asthma in the absence of T-bet
To determine whether IL-12-mediated suppression of Th17 cells might influence development of asthma, T-bet−/− mice were tested for induction of allergic lung inflammation after OVA challenge and IL-12 treatment. Previously, we have shown that allergic lung inflammation and BHR observed in T-bet−/− mice after OVA sensitization and challenge is primarily mediated by the Th17 pathway.28 Similar to previous results, there was increased peribronchiolar and perivascular cellular infiltration in the lungs of both WT and T-bet−/− mice after OVA challenge in the absence of exogenous IL-12 treatment (Figure 2a and b). As shown previously, the amount of pulmonary inflammation was exacerbated in T-bet−/− mice compared with WT mice, most likely due to increases in eosinophilic infiltration in combination with increased neutrophilic infiltration that is mediated by increased IL-17 production.28 Although the amount of peribronchiolar inflammation decreased in WT mice after IL-12 treatment, dramatically decreased inflammation was also observed in OVA-sensitized and OVA-challenged T-bet−/− mice after IL-12 treatment. Although IL-12 had a more pronounced effect on eosinophilic infiltration in WT animals compared with T-bet−/− mice, it suppressed neutrophilic infiltration to a greater extent in T-bet−/− mice (Figure 2c). It was also found that IL-12 reversed BHR in both WT and T-bet−/− animals (Figure 2d). To measure BHR, animals were anesthetized, tracheostomized, and connected to a mechanical ventilator. Measurement of input impedance after challenge with increasing doses of methacholine (see the section “Materials and methods”) established a dose–response relationship between airway resistance (or Newtonian resistance (RN)) and tissue stiffness (or elastance (H)). Resistance tended to be elevated after OVA challenge of WT and T-bet−/− mice, although this was not statistically significant. However, tissue elastance was significantly elevated after OVA challenge, likely due to enhanced peripheral airway closure.47 IL-12 treatment significantly suppressed this tissue elastance to a greater extent in T-bet−/− mice compared with WT mice (Figure 2d) and caused BHR to return to baseline levels. These various measurements suggest that IL-12 influences the peripheral airways (as determined by H) to a greater extent than the central airways of the lung (as determined by RN). IL-12 treatment of naive mice in the absence of OVA challenge had no effect on lung homeostasis (data not shown). Overall, the results show that IL-12 treatment suppresses antigen-induced airway inflammation in T-bet-deficient animals at least as well as in WT animals, and suggest that IL-12 inhibits IL-17-mediated inflammatory activity independently from IFN-γ production.
Figure 2.

Interleukin (IL)-12 alleviates allergen-induced asthma in the absence of T-bet. BALB/c T-bet+/+ and T-bet−/− mice (6–8 weeks old) were sensitized intraperitoneally (i.p.) on days 1 and 7 with 10 μg ovalbumin (OVA). Intranasal (i.n.) challenge with 100 μg OVA was performed for 5 consecutive days starting 1 week after the final sensitization. IL-12 (0.25 μg) was given i.n. during the first two days of the 5-day OVA challenge. Airway inflammation was assessed 24 h after the day 5 OVA challenge. (a) Representative hematoxylin and eosin (H&E)-stained lung sections from naive, OVA-challenged, and OVA-challenged plus IL-12-treated BALB/c T-bet+/+ (wild type, WT) and T-bet−/− mice (×200; 8 mice per group). (b) Scoring of H&E-stained lung sections based on the severity of peribronchial inflammation. (c) Numbers of total cellular infiltrates plus differential counts of eosinophils and neutrophils obtained from bronchoalveolar lavage (BAL). BAL cells were stained with Wright’s modified stain and enumerated according to standard morphological criteria. The data represent means±s.e.m., with at least 8 mice per group and are representative of at least three independent experiments. (d) Airway function was assessed using an invasive mechanical small animal ventilator. Newtonian resistance (RN) and tissue elastance (H) were measured in response to increasing doses of methacholine. The data are shown as means of 6 mice per group±s.e.m. and are representative of two independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
IL-12 suppresses IL-17 in the absence of IFN-γ
Although CD4+ T cells and NK cells are deficient in the production of IFN-γ in the absence of T-bet, other cells, including CD8 T cells, maintain the ability to express IFN-γ.48 To determine whether a T-bet-independent, IFN-γ-dependent pathway was responsible for the suppression of allergic lung inflammation that was observed in T-bet−/− mice after IL-12 inoculation, we produced and tested T-bet−/−/IFN-γ−/− double knockout mice. Splenocytes from these animals were cultured in the presence of lipopolysaccharide and IL-12 to confirm the absence of IFN-γ expression after stimulation. Splenocytes obtained from WT and T-bet−/−/IFN-γ+/+ mice produced IFN-γ within 60 h after in vitro stimulation, but cells from T-bet−/−/IFN-γ−/− mice failed to produce detectable levels of IFN-γ (Figure 3a). Nevertheless, OVA challenge of T-bet−/−/IFN-γ−/− animals mirrored the results obtained with T-bet−/−/IFN-γ+/+ mice, i.e., there was increased cellular infiltration into the lungs of T-bet−/−/IFN-γ−/− animals after OVA challenge and IL-12 maintained its ability to suppress this infiltration (Figure 3b). In particular, there was a significant reduction in the number of pulmonary neutrophils after IL-12 treatment, although no significant effect on recruitment of eosinophils was observed, most likely due to the loss of residual IFN-γ expression in the T-bet−/−/IFN-γ−/− animals. We (data not shown), along with others,49 have observed that IFN-γ−/− mice primed and challenged with OVA show increased pulmonary inflammation and increased BHR compared with IFN-γ+/+ mice. Therefore, we used the T-bet−/−/IFN-γ−/− animals to determine the effects of IL-12 specifically on granulocytic cellular infiltration and cytokine production. Levels of Th2-related cytokines were significantly lower in T-bet−/−/IFN-γ−/− animals after OVA challenge in the absence of IL-12 compared with WT mice, and the inhibitory effect of IL-12 on Th2 cytokine production was essentially restricted to WT animals (Figure 3c). However, IL-17 was increased in T-bet−/−/IFN-γ−/− mice after OVA challenge and its production was suppressed by IL-12 inoculation (Figure 3c). These data confirm the ability of IL-12 to inhibit Th17-mediated allergic lung inflammation in the absence of intrinsic IFN-γ expression.
Figure 3.

Interleukin (IL)-12 suppresses IL-17 in the absence of interferon (IFN)- γ. (a) Pooled splenocytes were isolated from 2–3 T-bet+/+ IFN-γ+/+ (wild type, WT), T-bet−/− IFN-γ+/+, and T-bet−/− IFN-γ−/− double knockout (DKO) mice, and 1×106 cells per ml were cultured in the presence of 125 ng ml−1 IL-12 and 100 ng ml−1 lipopolysaccharide. Levels of IFN-γ in supernatant fluids were measured by enzyme-linked immunosorbent assay (ELISA) after 24, 48, 60, and 72 h. The data represent means of 4 wells per group±s.e.m. from one experiment. (b) Numbers of total cells, eosinophils, and neutrophils in bronchoalveolar lavage (BAL) obtained from WT and DKO mice were evaluated 24 h after OVA challenge. BAL cells were stained with Wright’s modified stain and counted according to standard morphological criteria. The data represent means of 6 mice per group±s.e.m. and are representative of at least three independent experiments. (c) Cytokine levels in WT and DKO lungs after induction of allergic lung inflammation and treatment with IL-12. Cytokine levels were measured by cytometric bead array (IL-4, IL-5, and IFN-γ) and by ELISA (IL-17). The data represent means of 6 mice per group±s.e.m. and are representative of at least three independent experiments. *P < 0.05.
IL-12 induces expression of pulmonary IL-10
In addition to IFN-γ, IL-12 can induce the expression of IL-10,40 and it was recently reported that IL-10 inhibits Th17 cytokine expression.43 As IL-12 was found to suppress allergic lung inflammation in an IFN-γ-independent manner, we next determined the potential role of IL-10 in this inhibitory pathway. It was found that IL-12 inoculation induced IL-10 expression in the lungs of not only OVA-challenged WT animals but also, and to an even greater extent, in T-bet−/− mice (Figure 4). The increased production of IL-10 in the absence of T-bet may be due to the lack of IFN-γ-mediated counter-regulation. Identical patterns of IL-10 induction were seen in T-bet−/−/IFN-γ−/− mice, showing the IFN-γ independence of IL-10 induction.
Figure 4.
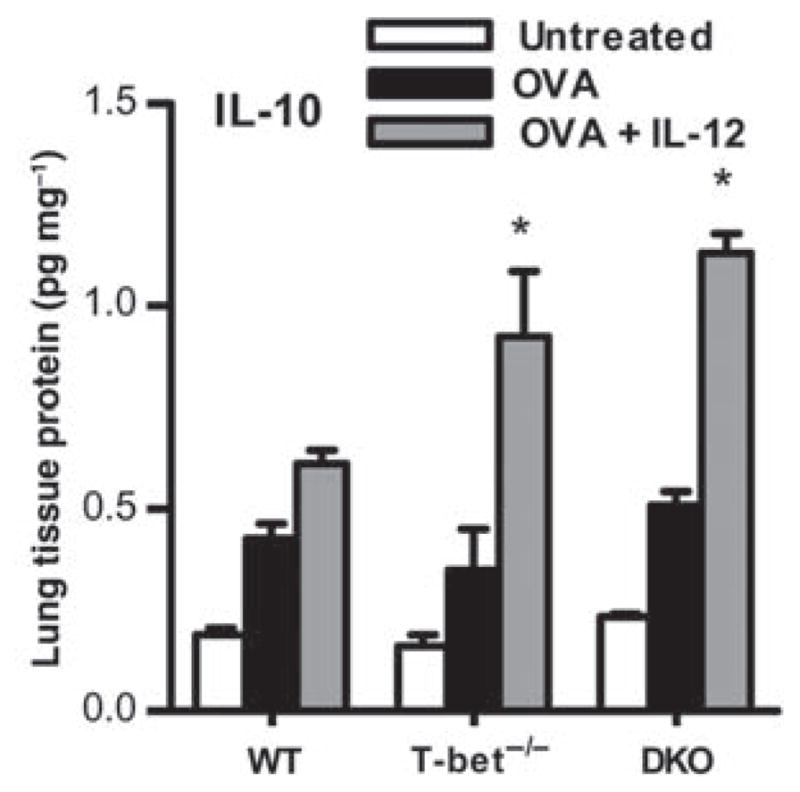
Interleukin (IL)-12 induces expression of IL-10. BALB/c wild-type (WT), T-bet−/−, and double knockout (DKO) mice were sensitized with ovalbumin (OVA) in alum, and 2 weeks later were challenged intranasally (i.n.) for 5 consecutive days with OVA. Twenty-four hours after the last OVA inoculation, homogenates were prepared from lung tissue, protein levels were normalized to lung total protein, and IL-10 was measured by enzyme-linked immunosorbent assay (ELISA). The data represent means of 6 mice per group±s.e.m. and are representative of at least three independent experiments *P <0.05.
IL-10 is responsible for IL-12-induced suppression of IL-17-mediated inflammation
To directly test the role of IL-10 in IL-12-mediated suppression of T-bet−/− allergic lung inflammation, anti-mIL-10R1 mAb was coadministered during OVA + IL-12 challenge. This IL-10R mAb has been previously found to be effective in neutralizing the in vivo effects of IL-10.50 It was found that the anti-inflammatory effect of IL-12 in T-bet−/− mice could be completely reversed by coadministration of anti-IL-10R1 mAb and the effects were observed primarily in the lung peribronchial regions (Figure 5a). IL-12-mediated inhibition of inflammatory cell infiltration that involved both eosinophils and neutrophils, was also reversed by IL-10R mAb treatment (Figure 5c). OVA-sensitized and OVA-challenged T-bet−/− mice that received anti-IL-10R mAb alone demonstrated peribronchial inflammation and eosinophilic infiltration similar to that observed in mice that were challenged with OVA but not treated with anti-IL-10R mAb (Figure 5a–c). Thus, in the absence of exogenous IL-12 treatment, endogenous IL-10 likely has a minor role in regulating allergic lung inflammation. In addition, inhibition of BHR after IL-12 treatment was lost in the presence of IL-10R mAb (Figure 5d). To determine whether IL-12/IL-10 had an effect on Th17 pathway differentiation, levels of IL-6 were determined (Figure 5e). Indeed, IL-10 that was induced by IL-12 treatment causes the suppression of IL-6 levels in the lung. Finally, treatment with anti-IL-10R mAb resulted in little to no effect of IL-12 on IL-17 levels in T-bet−/− mice (Figure 5f). In all cases, IL-12 maintained its activity in the presence of an isotype-matched, irrelevant antibody, demonstrating the specificity of the observed effects. These results indicate that IL-12 alleviates Th17-mediated pulmonary inflammation through induction of inhibitory IL-10 expression.
Figure 5.

Interleukin (IL)-10 is responsible for IL-12-induced suppression of IL-17-mediated inflammation. Ovalbumin (OVA)-sensitized and OVA-challenged BALB/c T-bet−/− mice were left untreated or treated with IL-12, IL-12 in the presence of neutralizing anti-IL-10R mAb (α-IL-10R), IL-12 in the presence of isotype control mAb (IgG), or neutralizing anti-IL-10R mAb alone. (a) Representative hematoxylin and eosin (H&E)-stained lung sections from naive and OVA-challenged BALB/c T-bet−/− mice (×200; 4 mice per group). Neutralizing anti-IL-10R mAb (α-IL-10R mAb) and IgG isotype control mAb were administered on days 1 through 3 during OVA intranasal (i.n.) challenge. (b) H&E-stained lung sections were scored for levels of peribronchial inflammation in each of the treatment groups. (c) Numbers of eosinophils and neutrophils in bronchoalveolar lavage (BAL) fluids were evaluated 24 h after OVA challenge. BAL cells were stained with Wright’s modified stain and counted according to standard morphological criteria. The data represent means±s.e.m.; 6 mice per group. (d) Methacholine dose–response curves 24 h after 5 days of i.n. OVA challenge are shown. The data represent means±s.e.m.; 6 mice per group. Levels of (e) IL-6, (f) IL-17, and (g) IL-10 in T-bet−/− lungs after induction of allergic lung inflammation and treatment with neutralizing anti-IL-10R mAb. Cytokine levels were measured by ELISA. The data represent means of at least 6 mice per group±s.e.m. and are representative of at least two independent experiments. *P < 0.05.
DISCUSSION
Our results show that IL-12 treatment in a murine model of OVA-induced, Th17-mediated allergic lung inflammation can prevent airway inflammation, inflammatory cell infiltration, and BHR in an IFN-γ-independent but IL-10-dependent manner. Th2 lymphocytes are believed to be key players in allergic asthma,51 and IL-12, which inhibits Th2 activity while promoting Th1 development through the T-bet transcription factor,45,52,53 has been targeted as a potential therapeutic. We,28 along with others,29 have previously shown in the OVA-induced asthma model that in the absence of functional T-bet, Th2 cells have a relatively minor role, and allergic lung inflammation is primarily mediated by Th17 cells. Other investigators have used a house dust mite asthma model to similarly demonstrate induction of IL-17A as well as increased BHR in A/J mice responding to allergen challenge; BHR is inhibited in this model by anti-IL-17A mAb (M Wills-Karp, personal communication). Our current findings show that IL-12 is highly effective in inhibiting such Th17-mediated pulmonary disease, through induction of IL-10 expression.
IL-12 has been shown to be an effective therapeutic in many diseases, such as cancer, because of it ability to activate multiple inflammatory mediators.54 We found that IL-12 treatment of WT mice after allergen sensitization and challenge induced increased levels of IFN-γ and, in turn, suppressed pulmonary Th2 cytokine production. These results parallel those reported in numerous allergen-induced murine animal models.33,35,37–39 However, in the absence of T-bet, there were little to no increases in lung IFN-γ levels after IL-12 treatment, yet significant inhibition of allergic lung inflammation was still observed. To rule out the possibility that IL-12 treatment induced low-level expression of IFN-γ by T-bet−/− cells other than CD4+ T cells and that this IFN-γ was responsible for the observed inhibition of allergic lung inflammation in T-bet−/− mice, mice deficient in both T-bet and IFN-γ were generated and used to show that levels of IL-17 secretion, as well as Th17-associated transcript levels, were significantly decreased by IL-12 treatment in the complete absence of IFN-γ. Our results further showed that IL-12 specifically and profoundly alleviates tissue stiffness/elastance in T-bet−/− mice, suggesting a reduction of inflammation, primarily in the peripheral airways, which led to reduced BHR. IL-12 lost the ability to suppress eosinophilic airway infiltration in the absence of IFN-γ and T-bet, but maintained the ability to inhibit neutrophilic infiltration. Similarly, although IL-12 was unable to suppress Th2 cytokine production in mice lacking both T-bet and IFN-γ, it maintained the capacity to suppress IL-17 production. Overall, our results suggest that transcription factor T-bet negatively regulates Th17 differentiation and that IL-12 can suppress Th17 functional activity independently from IFN-γ. Interestingly, recent results have shown the ability of IL-12 to repolarize Th17 cells toward a Th1 phenotype;55 however, this does not account for the findings reported in this study as the suppression of allergic lung inflammation by IL-12 occurred in the absence of IFN-γ.
It was observed that pulmonary levels of the anti-inflammatory cytokine, IL-10, were increased in OVA-sensitized and OVA-challenged animals after IL-12 treatment. Previous studies have shown that IL-12 induces IL-10 production in an IFN-γ-56,57 and a T-bet-independent58 manner. Previous results have also shown the ability of IL-10 to suppress allergic lung inflammation.59,60 Thus, to investigate the potential role of IL-10 in IL-12-mediated suppression of Th17 allergic lung inflammation, neutralizing anti-IL-10R mAb was administered to T-bet−/− mice during OVA challenge and IL-12 treatment. It was found that anti-IL-10R mAb treatment completely reversed the beneficial effects of IL-12. Both allergic lung inflammation and the levels of lung IL-17A failed to be suppressed by IL-12 treatment in the presence of the anti-IL-10R mAb. Other investigators have now found that bleomycin-induced lung inflammation, a system similarly mediated primarily by the Th17 pathway, is exacerbated in IL-10 knockout mice (TA Wynn, personal communication), highlighting the essential anti-inflammatory role of IL-10 in the lung. As CD4+CD25+ regulatory T cells have the ability to produce IL-10 and have been implicated in preventing T-cell responses to allergens, 61 the expression of Foxp3, a marker for these T-regulatory cells,62 was assessed on CD4+ T cells isolated from the lung, and it was found that IL-12 treatment had no effect on numbers of Foxp3+ cells (data not shown). Nevertheless, it should be recognized that IL-12 treatment may influence expression of other proinflammatory or anti-inflammatory cytokines or chemokines such as keratinocyte chemoattractant or transforming growth factor-β, which could also contribute to the suppression of inflammation. Overall, our results show that IL-12 treatment ameliorates allergic inflammation in the absence of T-bet through an IL-10-dependent mechanism.
These findings show that IL-12 has therapeutic anti-inflammatory effects in the absence of T-bet expression, a condition in which the activation of the IFN-γ gene is severely impaired.63–65 Interestingly, recent evidence suggests that the deficient expression of T-bet is associated with allergic asthma in humans.66–68 It is possible that activation of both Th17 and Th2 cells is involved in the pathogenesis of disease, especially in T-bet-deficient patients. Therefore, the effects of IL-12 presented in this study may be applicable to understanding the pulmonary immune response to allergens and for the development of potential therapeutics.
In summary, we have shown that IL-12 can suppress IL-17-mediated inflammation through enhanced pulmonary IL-10 expression. Although IL-12 serves as an adjuvant for the preferential development of Th1 cytokine responses, the results suggest that molecules that mimic the effects of IL-12 and/or IL-10 might, in addition to inhibiting Th2 responses, alleviate Th17 responses and thus be particularly active in decreasing allergic lung inflammation in certain asthmatic individuals who have deficiencies in T-bet expression.
METHODS
Mice
BALB/c WT mice were obtained from Charles River Laboratory (Wilmington, MA) through a contract with the National Cancer Institute. BALB/c T-bet−/− and BALB/c IFN-γ−/− mice were obtained from the Jackson Laboratory (Bar Harbor, ME). Mice deficient in both T-bet and IFN-γ were generated by backcrossing T-bet−/− mice with IFN-γ−/− mice at least eight times on to a BALB/c background. DNA from tail samples was used for PCR genotyping according to Jax Mice (Jax, Bar Harbor, ME) genotyping protocols. All experimental animals were 6–8 weeks of age and housed in microisolator cages under pathogen-free conditions at the Albany Medical College. All animal experiments were approved by the Institutional Animal Care and Use Committee.
Allergic airway inflammation
Allergic airway inflammation was induced as described previously.28,69 Mice were sensitized by intraperitoneal injection of 10 μg of OVA (Sigma-Aldrich, St Louis, MO) in 4 mg of alum (Reheis, Berkeley Heights, NJ) on days 7 and 14. One week after the second sensitization and after light anesthesia had been achieved, the sensitized mice were challenged intranasally with 100 μg OVA in saline for 5 consecutive days. For the first 2 days of OVA challenge, 0.25 μg of recombinant, murine IL-12p70 (provided by Wyeth Vaccines, Pearl River, NY) or saline was also administered intranasally. Twenty-four hours after the final OVA challenge, airway responsiveness was assessed, and the lung tissue and BAL fluids were collected.
Airway responsiveness measurement
Bronchial responsiveness to aerosolized methacholine was measured 24 h after the final OVA challenge as described previously,28,70 using anesthetized and tracheostomized mice connected to a computer-controlled small-animal mechanical ventilator (flexiVent; SCIREQ Montreal, QC, Canada). Measurements of Newtonian resistance and tissue stiffness/elastance were recorded in response to increasing doses of methacholine.
Histology
After perfusion through the right ventricle, lungs were inflated through the trachea with 1 ml of 5% buffered formalin and then fixed in the same solution by immersion. Blocks of lung tissue were cut from around the main bronchus and embedded in paraffin blocks, and 5-μm tissue sections were fixed to microscope slides and stained with hematoxylin and eosin (Fisher Scientific, Pittsburgh, PA). Slides were blinded and six to eight peribronchial regions per mouse were scored for inflammatory cellular infiltrate severity around airways at ×200 magnification as described previously.28,71
Inflammatory cells isolated from BAL fluids were counted using a hemocytometer, and cytospin slides were prepared and stained with Wright–Giemsa stain (HEMA 3; Biochemical Sciences, Swedesboro, NJ). Differential cell counting for at least 200 cells was performed under oil-immersion light microscopy.
Cytokine levels
Identical portions of the lung tissues (entire right lobe from each mouse) were snap frozen in liquid nitrogen and stored at −80°C for cytokine analysis. After thawing, the tissues were homogenized and supernatant fluids were harvested. Protein concentrations of IFN-γ, IL-4, and IL-5 in the lung homogenates and in BAL fluids were assessed by cytometric bead array analysis using a BD FACSArray (BD Biosciences, Franklin Lakes, NJ). The concentrations of IL-6, IL-10, IL-17, and IL-23 were assessed by ELISA (enzyme-linked immunosorbent assay), for IL-6 and IL-10 by BD OptEIA (BD Biosciences), for IL-17 by the DuoSet ELISA (R&D Systems, Minneapolis, MN), and for IL-23 by the Elisa Ready-Set-Go! Kit (eBioScience, SanDiego, CA). Cytokine protein levels in homogenized lung tissues were normalized to lung tissue total protein for each group of mice.
RT-PCR
Total cellular RNA was extracted from the lungs with TriZol reagent (Invitrogen, Carlsbad, CA), cDNA was prepared using oligo(dT) primer and SuperScript RT (Invitrogen), and real time (RT)-PCR was performed for IL-17A, RORγt, RORα, and IL-23R using Taq DNA polymerase. The primer pairs used were described previously.21,72 RT-PCR for GAPDH (glyceraldehyde 3-phosphate dehydrogenase) was performed to control for sample-to-sample variation in RNA isolation and integrity, RNA input, and reverse transcription. The ΔΔCt method was used to normalize transcript levels to GAPDH and to calculate the expression relative to untreated mice. Threshold cycle values were validated with different concentrations of RNA to ensure that the levels of fold induction remained constant. All PCR amplifications were performed at least three times with independent RNA preparations using a Bio-Rad iCycler Optical System and an iQ SYBR green RT-PCR kit (Bio-Rad Laboratories, Hercules, CA).
Flow cytometry
The lungs were digested with 2.5 mg ml−1 collagenase D, 0.25 mg ml −1 DNaseI (Roche, Mannheim, Germany), and 1 mM MgCl2 for 1 h at 37°C, and lymphocytes were isolated by passage through nylon mesh and density gradient centrifugation on Lympholyte M (Cedarlane Laboratories, Hornby, Ontario, Canada). The cells were washed, fixed with 2% paraformaldehyde, and permeabilized for intracellular staining with phycoerythrin-conjugated anti-mouse IL-17 mAb (BD Pharmingen, Franklin Lakes, NJ) and Alexa flour-conjugated anti-mouse IFN-γ mAb (BD Biosciences), at 4°C for 30 min. The stained cells were stored in the dark at 4 °C and analyzed within 24 h on a FACSCanto flow cytometer using the FACSDiva (BD Biosciences) software. The lymphocyte population was gated on the basis of forward and side scatter, and 10,000 gated events per sample were collected. The data are reported as the percentages of cells within the lymphocyte gate.
Cell culture
Cell activation assays were performed using splenocytes prepared by mechanical disruption, followed by NH4Cl lysis of red blood cells. After washing in RPMI 1640 medium, the cells were suspended in complete medium (RPMI 1640 containing penicillin, streptomycin, and 10% fetal calf serum). Cells were plated in triplicate at 1×106 cells per ml in 24-well plates and incubated for 72h at 37°C in 5% CO2 in the presence of 125 ng ml−1 IL-12 and 100ngml−1 lipopolysaccharide. Supernatant fluids from each culture were harvested at 24, 48, 60, and 72 h.
Anti-IL-10R mAb treatment
To neutralize pulmonary IL-10, rat anti-murine IL-10R1 mAb (Bio X Cell, West Lebanon, NH) or control rat IgG (Jackson Immunoresearch Laboratories, West Grove, PA) was inoculated intranasally. (20 μg per mouse) on days 1 and 2 during OVA challenge, and intraperitoneally (200 μg per mouse) on days 1, 3, and 5 of OVA challenge.
Statistical analysis
Single pairs of experimental groups were compared by Student’s t-test, and comparisons of more than two groups were performed by ANOVA (analysis of variance) with Tukey’s multiple comparison posttest. Probability values for significance were set at 0.05. Values for all measurements are expressed as the mean±s.e.m.
Acknowledgments
We thank the Immunology Core of the Center for Immunology and Microbial Disease for histology and flow cytometry services. This work was supported by NIH grant no. RO1 AI41715 (to DWM)
Footnotes
DISCLOSURE
The author declared no conflict of interest.
References
- 1.Wills-Karp M. Immunologic basis of antigen-induced airway hyperresponsiveness. Annu Rev Immunol. 1999;17:255–281. doi: 10.1146/annurev.immunol.17.1.255. [DOI] [PubMed] [Google Scholar]
- 2.Nakajima H, Takatsu K. Role of cytokines in allergic airway inflammation. Int Arch Allergy Immunol. 2007;142:265–273. doi: 10.1159/000097357. [DOI] [PubMed] [Google Scholar]
- 3.Herrick CA, Bottomly K. To respond or not to respond: T cells in allergic asthma. Nat Rev Immunol. 2003;3:405–412. doi: 10.1038/nri1084. [DOI] [PubMed] [Google Scholar]
- 4.Jatakanon A, et al. Neutrophilic inflammation in severe persistent asthma. Am J Respir Crit Care Med. 1999;160:1532–1539. doi: 10.1164/ajrccm.160.5.9806170. [DOI] [PubMed] [Google Scholar]
- 5.Louis R, et al. The relationship between airways inflammation and asthma severity. Am J Respir Crit Care Med. 2000;161:9–16. doi: 10.1164/ajrccm.161.1.9802048. [DOI] [PubMed] [Google Scholar]
- 6.Laan M, Palmberg L, Larsson K, Linden A. Free, soluble interleukin-17 protein during severe inflammation in human airways. Eur Respir J. 2002;19:534–537. doi: 10.1183/09031936.02.00280902. [DOI] [PubMed] [Google Scholar]
- 7.Molet S, et al. IL-17 is increased in asthmatic airways and induces human bronchial fibroblasts to produce cytokines. J Allergy Clin Immunol. 2001;108:430–438. doi: 10.1067/mai.2001.117929. [DOI] [PubMed] [Google Scholar]
- 8.Chakir J, et al. Airway remodeling-associated mediators in moderate to severe asthma: effect of steroids on TGF-beta, IL-11, IL-17, and type I and type III collagen expression. J Allergy Clin Immunol. 2003;111:1293–1298. doi: 10.1067/mai.2003.1557. [DOI] [PubMed] [Google Scholar]
- 9.Hellings PW, et al. Interleukin-17 orchestrates the granulocyte influx into airways after allergen inhalation in a mouse model of allergic asthma. Am J Respir Cell Mol Biol. 2003;28:42–50. doi: 10.1165/rcmb.4832. [DOI] [PubMed] [Google Scholar]
- 10.Nakae S, et al. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity. 2002;17:375–387. doi: 10.1016/s1074-7613(02)00391-6. [DOI] [PubMed] [Google Scholar]
- 11.Schnyder-Candrian S, et al. Interleukin-17 is a negative regulator of established allergic asthma. J Exp Med. 2006;203:2715–2725. doi: 10.1084/jem.20061401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He R, Oyoshi MK, Jin H, Geha RS. Epicutaneous antigen exposure induces a Th17 response that drives airway inflammation after inhalation challenge. Proc Natl Acad Sci USA. 2007;104:15817–15822. doi: 10.1073/pnas.0706942104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. 2006;24:677–688. doi: 10.1016/j.immuni.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 14.Cua DJ, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 15.Murphy CA, et al. Divergent pro- and anti-inflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003;198:1951–1957. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng Y, et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648–651. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
- 17.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 18.Bettelli E, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 19.Mangan PR, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 20.Langrish CL, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ivanov II, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 22.Yang XO, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. 2008;28:29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Acosta-Rodriguez EV, et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol. 2007;8:639–646. doi: 10.1038/ni1467. [DOI] [PubMed] [Google Scholar]
- 24.Annunziato F, et al. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007;204:1849–1861. doi: 10.1084/jem.20070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harrington LE, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 26.Stumhofer JS, et al. Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nat Immunol. 2006;7:937–945. doi: 10.1038/ni1376. [DOI] [PubMed] [Google Scholar]
- 27.Park H, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Durrant DM, Gaffen SL, Riesenfeld EP, Irvin CG, Metzger DW. Development of allergen-induced airway inflammation in the absence of T-bet regulation is dependent on IL-17. J Immunol. 2009;183:5293–5300. doi: 10.4049/jimmunol.0803109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fujiwara M, et al. T-bet inhibits both TH2 cell-mediated eosinophil recruitment and TH17 cell-mediated neutrophil recruitment into the airways. J Allergy Clin Immunol. 2007;119:662–670. doi: 10.1016/j.jaci.2006.12.643. [DOI] [PubMed] [Google Scholar]
- 30.Coyle AJ, et al. Mice lacking the IFN-gamma receptor have impaired ability to resolve a lung eosinophilic inflammatory response associated with a prolonged capacity of T cells to exhibit a Th2 cytokine profile. J Immunol. 1996;156:2680–2685. [PubMed] [Google Scholar]
- 31.McDyer JF, Wu CY, Seder RA. The regulation of IL-12: its role in infectious, autoimmune, and allergic diseases. J Allergy Clin Immunol. 1998;102:11–15. doi: 10.1016/s0091-6749(98)70047-8. [DOI] [PubMed] [Google Scholar]
- 32.Smits HH, et al. IL-12-induced reversal of human Th2 cells is accompanied by full restoration of IL-12 responsiveness and loss of GATA-3 expression. Eur J Immunol. 2001;31:1055–1065. doi: 10.1002/1521-4141(200104)31:4<1055::aid-immu1055>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 33.Kips JC, et al. Interleukin-12 inhibits antigen-induced airway hyperresponsiveness in mice. Am J Respir Crit Care Med. 1996;153:535–539. doi: 10.1164/ajrccm.153.2.8564093. [DOI] [PubMed] [Google Scholar]
- 34.Sur S, et al. Immunomodulatory effects of IL-12 on allergic lung inflammation depend on timing of doses. J Immunol. 1996;157:4173–4180. [PubMed] [Google Scholar]
- 35.Gavett SH, et al. Interleukin 12 inhibits antigen-induced airway hyperresponsiveness, inflammation, and Th2 cytokine expression in mice. J Exp Med. 1995;182:1527–1536. doi: 10.1084/jem.182.5.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iwamoto I, Kumano K, Kasai M, Kurasawa K, Nakao A. Interleukin-12 prevents antigen-induced eosinophil recruitment into mouse airways. Am J Respir Crit Care Med. 1996;154:1257–1260. doi: 10.1164/ajrccm.154.5.8912732. [DOI] [PubMed] [Google Scholar]
- 37.Schwarze J, et al. Local treatment with IL-12 is an effective inhibitor of airway hyperresponsiveness and lung eosinophilia after airway challenge in sensitized mice. J Allergy Clin Immunol. 1998;102:86–93. doi: 10.1016/s0091-6749(98)70058-2. [DOI] [PubMed] [Google Scholar]
- 38.Matsuse H, et al. Intranasal IL-12 produces discreet pulmonary and systemic effects on allergic inflammation and airway reactivity. Int Immunopharmacol. 2003;3:457–468. doi: 10.1016/S1567-5769(02)00250-3. [DOI] [PubMed] [Google Scholar]
- 39.Bruselle GG, et al. Role of IFN-gamma in the inhibition of the allergic airway inflammation caused by IL-12. Am J Respir Cell Mol Biol. 1997;17:767–771. doi: 10.1165/ajrcmb.17.6.2820. [DOI] [PubMed] [Google Scholar]
- 40.Daftarian PM, Kumar A, Kryworuchko M, Diaz-Mitoma F. IL-10 production is enhanced in human T cells by IL-12 and IL-6 and in monocytes by tumor necrosis factor-alpha. J Immunol. 1996;157:12–20. [PubMed] [Google Scholar]
- 41.Meyaard L, Hovenkamp E, Otto SA, Miedema F. IL-12-induced IL-10 production by human T cells as a negative feedback for IL-12-induced immune responses. J Immunol. 1996;156:2776–2782. [PubMed] [Google Scholar]
- 42.Jeannin P, Delneste Y, Seveso M, Life P, Bonnefoy JY. IL-12 synergizes with IL-2 and other stimuli in inducing IL-10 production by human T cells. J Immunol. 1996;156:3159–3165. [PubMed] [Google Scholar]
- 43.Gu Y, et al. Interleukin 10 suppresses Th17 cytokines secreted by macrophages and T cells. Eur J Immunol. 2008;38:1807–1813. doi: 10.1002/eji.200838331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Djuretic IM, et al. Transcription factors T-bet and Runx3 cooperate to activate Ifng and silence Il4 in T helper type 1 cells. Nat Immunol. 2007;8:145–153. doi: 10.1038/ni1424. [DOI] [PubMed] [Google Scholar]
- 45.Paul WE, Seder RA. Lymphocyte responses and cytokines. Cell. 1994;76:241–251. doi: 10.1016/0092-8674(94)90332-8. [DOI] [PubMed] [Google Scholar]
- 46.Mathur AN, et al. T-bet is a critical determinant in the instability of the IL-17-secreting T-helper phenotype. Blood. 2006;108:1595–1601. doi: 10.1182/blood-2006-04-015016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wagers S, Lundblad LK, Ekman M, Irvin CG, Bates JH. The allergic mouse model of asthma: normal smooth muscle in an abnormal lung? J Appl Physiol. 2004;96:2019–2027. doi: 10.1152/japplphysiol.00924.2003. [DOI] [PubMed] [Google Scholar]
- 48.Pearce EL, et al. Control of effector CD8+ T cell function by the transcription factor eomesodermin. Science. 2003;302:1041–1043. doi: 10.1126/science.1090148. [DOI] [PubMed] [Google Scholar]
- 49.Koch M, et al. Role of local pulmonary IFN-gamma expression in murine allergic airway inflammation. Am J Respir Cell Mol Biol. 2006;35:211–219. doi: 10.1165/rcmb.2005-0293OC. [DOI] [PubMed] [Google Scholar]
- 50.Sundstedt A, O’Neill EJ, Nicolson KS, Wraith DC. Role for IL-10 in suppression mediated by peptide-induced regulatory T cells in vivo. J Immunol. 2003;170:1240–1248. doi: 10.4049/jimmunol.170.3.1240. [DOI] [PubMed] [Google Scholar]
- 51.Robinson DS, et al. Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. N Engl J Med. 1992;326:298–304. doi: 10.1056/NEJM199201303260504. [DOI] [PubMed] [Google Scholar]
- 52.Manetti R, et al. Interleukin 12 induces stable priming for interferon gamma (IFN-gamma) production during differentiation of human T helper (Th) cells and transient IFN-gamma production in established Th2 cell clones. J Exp Med. 1994;179:1273–1283. doi: 10.1084/jem.179.4.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schulz EG, Mariani L, Radbruch A, Hofer T. Sequential polarization and imprinting of type 1 T helper lymphocytes by interferon-gamma and interleukin-12. Immunity. 2009;30:673–683. doi: 10.1016/j.immuni.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 54.Portielje JE, et al. Subcutaneous injection of interleukin 12 induces systemic inflammatory responses in humans: implications for the use of IL-12 as vaccine adjuvant. Cancer Immunol Immunother. 2005;54:37–43. doi: 10.1007/s00262-004-0574-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee YK, et al. Late developmental plasticity in the T helper 17 lineage. Immunity. 2009;30:92–107. doi: 10.1016/j.immuni.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Windhagen A, Anderson DE, Carrizosa A, Williams RE, Hafler DA. IL-12 induces human T cells secreting IL-10 with IFN-gamma. J Immunol. 1996;157:1127–1131. [PubMed] [Google Scholar]
- 57.Jankovic D, et al. In the absence of IL-12, CD4(+) T cell responses to intracellular pathogens fail to default to a Th2 pattern and are host protective in an IL-10(−/−) setting. Immunity. 2002;16:429–439. doi: 10.1016/s1074-7613(02)00278-9. [DOI] [PubMed] [Google Scholar]
- 58.Saraiva M, et al. Interleukin-10 production by Th1 cells requires interleukin-12-induced STAT4 transcription factor and ERK MAP kinase activation by high antigen dose. Immunity. 2009;31:209–219. doi: 10.1016/j.immuni.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Henry E, et al. Dendritic cells genetically engineered to express IL-10 induce long-lasting antigen-specific tolerance in experimental asthma. J Immunol. 2008;181:7230–7242. doi: 10.4049/jimmunol.181.10.7230. [DOI] [PubMed] [Google Scholar]
- 60.Campbell JD, et al. Peptide immunotherapy in allergic asthma generates IL-10-dependent immunological tolerance associated with linked epitope suppression. J Exp Med. 2009;206:1535–1547. doi: 10.1084/jem.20082901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ling EM, et al. Relation of CD4+CD25+ regulatory T-cell suppression of allergen-driven T-cell activation to atopic status and expression of allergic disease. Lancet. 2004;363:608–615. doi: 10.1016/S0140-6736(04)15592-X. [DOI] [PubMed] [Google Scholar]
- 62.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 63.Mullen AC, et al. Role of T-bet in commitment of TH1 cells before IL-12-dependent selection. Science. 2001;292:1907–1910. doi: 10.1126/science.1059835. [DOI] [PubMed] [Google Scholar]
- 64.Szabo SJ, et al. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 65.Szabo SJ, Sullivan BM, Peng SL, Glimcher LH. Molecular mechanisms regulating Th1 immune responses. Annu Rev Immunol. 2003;21:713–758. doi: 10.1146/annurev.immunol.21.120601.140942. [DOI] [PubMed] [Google Scholar]
- 66.Finotto S, et al. Development of spontaneous airway changes consistent with human asthma in mice lacking T-bet. Science. 2002;295:336–338. doi: 10.1126/science.1065544. [DOI] [PubMed] [Google Scholar]
- 67.Raby BA, et al. T-bet polymorphisms are associated with asthma and airway hyperresponsiveness. Am J Respir Crit Care Med. 2006;173:64–70. doi: 10.1164/rccm.200503-505OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Munthe-Kaas MC, et al. T cell-specific T-box transcription factor haplotype is associated with allergic asthma in children. J Allergy Clin Immunol. 2008;121:51–56. doi: 10.1016/j.jaci.2007.07.068. [DOI] [PubMed] [Google Scholar]
- 69.Alouani S, Juillard P, Chvatchko Y. Murine model of allergic lung inflammation. Methods Mol Biol. 2000;138:285–293. doi: 10.1385/1-59259-058-6:285. [DOI] [PubMed] [Google Scholar]
- 70.Takeda K, Haczku A, Lee JJ, Irvin CG, Gelfand EW. Strain dependence of airway hyperresponsiveness reflects differences in eosinophil localization in the lung. Am J Physiol Lung Cell Mol Physiol. 2001;281:L394–L402. doi: 10.1152/ajplung.2001.281.2.L394. [DOI] [PubMed] [Google Scholar]
- 71.Doherty TA, Soroosh P, Broide DH, Croft M. CD4+ cells are required for chronic eosinophilic lung inflammation but not airway remodeling. Am J Physiol Lung Cell Mol Physiol. 2009;296:L229–L235. doi: 10.1152/ajplung.90543.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Morishima N, Mizoguchi I, Takeda K, Mizuguchi J, Yoshimoto T. TGF-beta is necessary for induction of IL-23R and Th17 differentiation by IL-6 and IL-23. Biochem Biophys Res Commun. 2009;386:105–110. doi: 10.1016/j.bbrc.2009.05.140. [DOI] [PubMed] [Google Scholar]