

Abstract
The differentially methylated imprinting control region (ICR) region upstream of the H19 gene regulates allelic Igf2 expression by means of a methylation-sensitive chromatin insulator function. We have previously shown that maternal inheritance of mutated (three of the four) target sites for the 11-zinc finger protein CTCF leads to loss of Igf2 imprinting. Here we show that a mutation in only CTCF site 4 also leads to robust activation of the maternal Igf2 allele despite a noticeably weaker interaction in vitro of site 4 DNA with CTCF compared to other ICR sites, sites 1 and 3. Moreover, maternally inherited sites 1 to 3 become de novo methylated in complex patterns in subpopulations of liver and heart cells with a mutated site 4, suggesting that the methylation privilege status of the maternal H19 ICR allele requires an interdependence between all four CTCF sites. In support of this conclusion, we show that CTCF molecules bind to each other both in vivo and in vitro, and we demonstrate strong interaction between two CTCF-DNA complexes, preassembled in vitro with sites 3 and 4. We propose that the CTCF sites may cooperate to jointly maintain both methylation-free status and insulator properties of the maternal H19 ICR allele. Considering many other CTCF targets, we propose that site-specific interactions between various DNA-bound CTCF molecules may provide general focal points in the organization of looped chromatin domains involved in gene regulation.
The neighboring Igf2 and H19 genes are expressed monoallelically from opposite chromosomes and are generally recognized as the paradigm of mammalian genomic imprinting (1, 11). The cis elements underlying their parent-of-origin-dependent expression patterns were traced to a differentially methylated region (imprinting control region [ICR]) located 2 kb upstream of the H19 transcriptional unit (5, 26-28). Subsequently, it was documented that repression of the paternal H19 allele was initiated, but not maintained by the H19 ICR, while the continuous presence of this region was required to suppress the maternal Igf2 allele (25). Arguments that this region contained a methylation-sensitive chromatin insulator to block communication between downstream enhancers and Igf2 promoters (3, 9, 12, 13) were later confirmed by experiments (10).
Despite similar functions, the mouse and human H19 ICRs display little sequence similarity with the exception of a CpG-rich element repeated four times in the mouse H19 ICR and seven times in the human H19 ICR (9). We and others have noted that the 11-zinc finger protein CTCF, which associates with all known vertebrate chromatin insulator cis elements (2, 19), interacted with these repeat elements in a methylation-sensitive manner (3, 9, 13). Importantly, CTCF interacts in vivo with only the maternal H19 ICR allele (13), which is consistent with an involvement with the in vivo insulation of the maternal Igf2 from downstream lineage-specific enhancers. To confirm an in vivo role of the CTCF target sites at the Igf2/H19 locus, we previously created a knock-in mouse with mutations in three of the four target sites. This mouse showed robust biallelic expression of Igf2 and substantial gain of methylation at the maternal H19 ICR region (22). Similar results have been reported by another group (24).
While these results underscore the dualistic properties of the CTCF target sites, i.e., they organize long-range insulators and propagate methylation-free domains, it is not known whether these effects depended on the additive or synergistic effects of all the CTCF target sites or to what degree these sites contribute individually to methylation protection and/or insulation. To gain insight into this issue, we created a knock-in mouse with a mutation only in the fourth target site. It was striking that despite the fact that CTCF interacted less efficiently with site 4 than with sites 1 and 3, there was a robust activation of the maternal Igf2 allele. The results reported here significantly add to our understanding of the complexity of the function of the maternally inherited H19 ICR and suggest that the essential features of the H19 ICR reflect cooperativity between the CTCF target sites.
MATERIALS AND METHODS
Generation of strain 121.
The targeting strategy to replace the fourth endogenous CTCF target site with a mutated version (central GTGG motif mutated to ATAT) was performed as described previously (22). Homologous recombination events of the parent embryonic stem (ES) cell clone (clone 121) were screened in F1 offspring by Southern blotting analyses as outlined in Fig. 1. The neomycin gene was deleted by mating homozygous mutant mice with a transgenic mouse strain carrying the Cre recombinase gene under the control of the β-actin promoter (17) to generate the 121* strain.
FIG. 1.
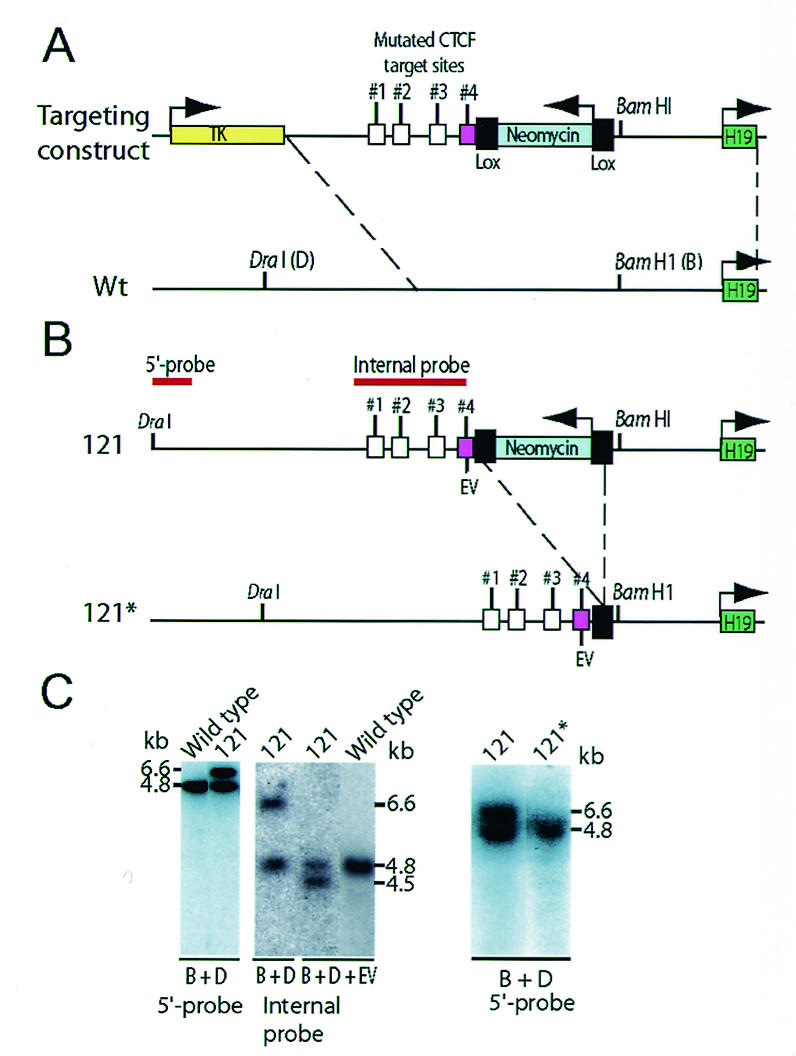
Schematic illustration of the knock-in strategy. (A) Recombination map of the targeting construct and endogenous H19 ICR. CTCF target site 4 (cerise box) was targeted in the knock-in procedure. Wt, wild-type. (B) Deletion of the neomycin gene by breeding mice of strain 121 with mice harboring a β-actin promoter-driven Cre recombinase gene to generate the 121* substrain. (C) The recombination event in clone 121 was assessed by using a probe that is positioned 5′ of the sequence covered in the targeting construct. The properly recombined insert generated a larger fragment due to the extra sequence information provided by the neomycin gene. The mutated targeting allele replaced the endogenous sequence in clone 121 as confirmed by EcoRV (EV) digestion and Southern blot analysis. The mutated allele of the 121* strain is indistinguishable from the wild-type allele when digested with BamHI (B) and DraI (D). For additional information, see the text.
Southern blot analyses.
Genomic DNA was isolated using Wizard genomic DNA isolation kit (Promega). DNA was digested with DraI and BamHI restriction enzymes, analyzed by electrophoresis on a 1% agarose gel blotted onto a Hybond N+ membrane (Amersham Pharmacia). A 1.3-kb PCR-generated fragment (bp −6750 to −5406 with respect to the transcription start site of the mouse H19 gene) outside the targeting vector sequence was used to screen for correct genomic integration of the targeting construct. The presence of the neomycin gene allowed discrimination between the mutated and wild-type alleles. To assess the presence of the mutated CTCF target site, DNA was digested with DraI, BamHI, and EcoRV and probed with an internal probe spanning the sequence −4296 to −2140 bp upstream of the H19 transcription start site, covering all four CTCF sites in the H19 ICR. The genotype of the mutant H19 ICR was confirmed by sequencing the entire 2-kb domain of tail DNA of an F1 offspring (data not shown).
ChIP analysis.
Fetal mouse liver cells (embryonic day 16 [E16]) were mechanically dispersed and cross-linked with formaldehyde. Chromatin immunopurification (ChIP) DNA was prepared from fetal mouse liver by using an affinity-purified rabbit antibody against the N-terminal portion of CTCF and analyzed for the presence of H19 ICR sequences as described previously (13).
Expression analyses.
To determine the imprinted status of Igf2, homozygous 121* mice were mated with SD7 mice a congenic strain carrying the distal end of chromosome 7 of Mus musculus spretus on a Mus musculus domesticus background (6). RNA was isolated from individual organs of 1- to 2-day-old neonatal mice using TriPure DNA or RNA isolation reagent (Roche). RNA was treated with RQ1 DNase I (Promega) to remove any contaminating genomic DNA. Reverse transcription-PCR (RT-PCR) analyses were performed using the Qiagen one-step RT-PCR kit per the manufacturer's guidelines. Primers and PCR conditions have previously been published (6). The PCR product was purified and digested with BsaAI to determine the allelic origin of Igf2 transcripts as described earlier (6, 22).
For Northern blot hybridization, total RNA was isolated using the TriPure reagent (Roche), according to the manufacturer's instructions. Approximately 10 μg of total RNA was electrophoresed on a 1% agarose-formaldehyde gel and blotted onto a Hybond N+ membrane (Amersham Biosciences). Hybridization was done with a 32P-labeled Igf2 cDNA probe (covering exons 4 to 6 and recognizing all major transcripts). Washed blots were exposed to a PhosphorImager screen (Fuji FLA 3000) for 4 h for quantitation, and later, the blots were exposed to X-ray films for 2 days. The Igf2-hybridized blots were stripped and subsequently hybridized to a glyceraldehyde-3-phosphate dehydrogenase cDNA probe (Ambion).
EMSAs.
Individual DNA fragments containing sites 1, 3, and 4, corresponding to bp −4023 to −3761, −2752 to −2427, and −2193 to −1983, with respect to the H19 transcription start site, were generated by PCR amplification using primer sets. The primer sets follow: site 1 For (5′-TCC TGC TCA CTG TCC ATT CA-3′) and site 1 Rev (5′-GGA TTC TTT GCA GAG AGT AAG-3′), site 3 For (5′-CTC AGT GGT CGA TAT GGT TT-3′) and site 3 Rev (5′-TGA GTC AAG TTC TCT GGT TC-3′), and site 4 For (5′-ACT CTC CAC GCT GTG CAG ATT T-3′) and site 4 Rev (5′-CCC ACC CTC CTG CTT CAC TTC AAA-3′). The DNA fragments containing sites 1, 3, and 4 were then subjected to restriction digestion with HpaII-DdeI, AvaII, and BglII, respectively. The gel-purified fragments were labeled with [γ-32P]ATP and subsequently purified on a Sephadex G-50 column. Binding reaction mixtures (20 μl) containing binding buffer (0.1 mM EDTA, 2 mM MgCl2, 75 mM KCl, 40 mM HEPES, 10% glycerol), CTCF, and radiolabeled DNA were analyzed by electrophoretic mobility shift assays (EMSAs) as described previously (13). For competition experiments, unlabeled competitor site 4 DNA was added at different amounts (1-, 5-, 10-, 50-, 100-, 200-, and 500-fold). CTCF-DNA interaction was quantitated by scoring free and bound DNA probe by FLA 3000 PhosphorImager analysis. The normalized ratios of bound DNA versus unbound DNA were plotted against the concentration of the unlabeled competitor DNA.
Methylation analyses.
For Southern blot analysis, genomic DNA was isolated from tissues of 2-day-old neonatal mice. Fifteen micrograms of DNA was digested with DraI, BamHI, and BstXI and with or without the methylation-sensitive restriction enzyme HhaI. BstXI has a unique site present in the Dom allele, but not in the SD7 allele, at −3537 bp with respect to the H19 transcription start site (W. Reik, personal communication). DNA was treated with bisulfite using a modified version (22) of an established protocol (20). DNA was purified using the Wizard DNA clean-up system (Promega) per the manufacturer's guidelines. DNA was subjected to PCR amplification using the published primers and conditions (23). Amplified fragments were cloned into the pGEMT-Easy vector (Promega) and subsequently sequenced with the BigDye 3.0 Terminator Cycle sequencing kit (Applied Biosystems).
In vivo and in vitro CTCF association analyses.
In order to elucidate the CTCF∼CTCF association in living cells, the myc-tagged full-length chicken-CTCF cDNA (16) was transfected into COS 7 cells. The nuclear lysate of transfected cells was prepared and used for coimmunoprecipitation with the anti-c-myc-tagged monoclonal antibody (9E10) (Sigma Chemical Co.). Coimmunoprecipitation was performed by a modified version of the method of Chernukhin et al. (4). Specifically, a protein A-Sepharose (Sigma Chemical Co.) slurry (20 μl) and c-myc (9E10) antibody (final concentration, 5 μg/ml) were added to the lysate. The anti-α-tubulin antibody (Sigma) and the preimmune serum were used as controls. The suspension was incubated on a rotator at 4°C overnight, the complexes were then washed three times with washing buffer (25 mM Tris-HEPES [pH 8.0], 2 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 0.25 M NaCl, 0.5% Tween 20), resuspended in 50 μl of 3× sodium dodecyl sulfate (SDS) loading buffer and boiled for 5 min. Reduced complexes were separated on an SDS-10% polyacrylamide gel and then blotted onto a polyvinylidene difluoride nylon membrane, and the Western blot was probed with the CTCF rabbit polyclonal antibody.
For EMSAs designed to detect interactions between two CTCF-DNA complexes, DNA fragments containing site 3 (corresponding to −2691 to −2399 bp; forward primer, 5′-GGTCACTGAACCCCAAAAACCAGC-3′; reverse primer, 5′-CCAGCCACTGACGATCTCGGGCTG-3′) and site 4 (corresponding to bp −2285 to −2095; forward primer, 5′-CAATGTTCATAAGGGTCATGGGTG-3′; reverse primer, 5′-CCCAACCTATGCCGCGTCTGCCGAG-3′) were PCR amplified with these primers labeled at the 5′ ends with [γ-32P]ATP and T4 kinase, gel purified, and incubated with in vitro-translated (IVT) (TNT lysate) CTCF (8) either singly or in combination by mixing two preassembled CTCF-DNA complexes in the modified EMSA buffer. The CTCF-DNA complexes were empirically adjusted to maximize CTCF binding and to reduce nonspecific background levels resulting from the fortuitous binding of proteins present in nuclear extracts and even in the TNT reticulocyte lysates to the DNA probes. The DNA probes were more than 80 to 100 bp long and were previously shown to be near a minimal DNA length required for the efficient sequence-specific CTCF-DNA interactions (18). Free and CTCF-bound probes were resolved on 5% acrylamide gels containing 1% glycerol as described previously (13). IVT CTCF was used in a single EMSA binding reaction performed by mixing it with 10 μl of the 2× modified EMSA buffer containing 100 mM HEPES (pH 7.8), 500 mM NaCl, 5 mM MgCl2, 10 mM dithiothreitol, 1 mM ZnSO4, 100 μg of poly(dI-dC) per ml, 0.02% NP-40, and 20% polyethylene glycol 10,000.
To study the interactions between two preassembled (for 30 min) complexes of CTCF and 32P-labeled DNA, one half (10 μl) of the binding reaction mixture with one hot probe was mixed together with the same portion from the binding reaction mixture with the other hot probe, and the resulting 20-μl mixtures with two DNA probes were incubated for an additional 30 min at room temperature. To obtain controls for the duration of each binding reaction mixture containing two DNA probes versus one DNA probe, the second 10-μl portion from each 30-min reaction mixture with a single hot probe was mixed with 10 μl from a 30-min binding reaction mixture containing IVT luciferase in place of the IVT CTCF and incubated for an additional 30 min before loading on the same EMSA gels. Fragments that were resolved were later eluted from the EMSA gel, treated with proteinase K, and reanalyzed on acrylamide gels as shown in Fig. 5C.
FIG. 5.

CTCF interacts with itself in vivo and in vitro, while only heterologous CTCF-DNA complexes interact in vitro. (A) DNA fragments containing CTCF target sites 3 and 4 were labeled with 32P and subjected to CTCF band shift analysis either separately or in combination. Shifted band fragments (bands 1, 2, and 3) were eluted from the gel (left gel), treated with proteinase K, and resolved on a 5% acrylamide gel (right gel). F, free; B, bound. (B) The C terminus of CTCF binds to the 11 ZF domain of CTCF. The latter was initially noticed in the preliminary pull-down assay with various portions of CTCF (data not shown). Next, the IVT radiolabeled full-length CTCF, the 11 ZF domain, and the CTCF-C domain were mixed together at a ratio of approximately 1:1:1 as shown in the Input lane and subjected to a GST pull-down assay with the three bacterially expressed GST fusion proteins, GST-ABL, GST-CTCF-C, and GST-CTCF-N, immobilized on glutathione beads. (C) CTCF interacts with itself in vivo. COS 7 cells were transfected with myc-tagged CTCF, and the myc-tagged (9E10) antibody was used for coimmunoprecipitation. Immunoprecipitates (Immunoprec.) and control lysates were resolved on SDS-10% polyacrylamide gels and probed with the anti-CTCF antibody. The positions of the endogenous CTCF (16) and myc-tagged exogenous CTCF (15) are indicated to the left of the blot. No signal was observed in the controls obtained with the preimmune serum and anti-α-tubulin antibody.
For the glutathione S-transferase (GST) bead pull-down assay shown in Fig. 5B, a pGEX-CTCF-C construct was made by in-frame ligation of a ∼0.8-kb NdeI-XhoI fragment of the human CTCF cDNA (8) from a pCITE4b-based plasmid into the EcoRI and XhoI ends of the pGEX5X-1 vector (Pharmacia) after the NdeI and EcoRI ends had been filled in with the Klenow fragment. The resulting bacterial expression vector encoded the GST-CTCF-C fusion protein that contained the C-terminal part of human CTCF (CTCF-C) beginning from the middle of the 11th Zn finger and extending to the stop codon. To express the GST-CTCF-C protein in bacteria and to purify it when it was left immobilized on the beads, we used the GST purification kit (Pharmacia) and other methods described in detail earlier by Klenova et al. (15). The purity and concentration of the isolated GST-CTCF-C protein on glutathione beads were both verified by SDS-polyacrylamide gel electrophoresis, and the concentration was adjusted to 1 mg per ml of beads during the final washings and final dilution of the beads in phosphate-buffered saline. The [35S]methionine-labeled proteins, including the full-length polypeptide, CTCF-N (the N-terminal region starting from the first Met and extending downstream to the first Zn finger), the 11 ZF domain (domain with all 11 zinc fingers), and CTCF-C (the C-terminal domain of CTCF), have been produced from the pCITE4b- or pET16b-based constructs described earlier (8, 15) and used as the DNA templates in the Promega coupled transcription-translation TNT system for 35S-labeled proteins. A construct for GST-Abl SH3 fusion protein used as an unrelated control for GST-CTCF-C was obtained from the laboratory of P. Soriano (Fred Hutchison Cancer Research Center, Seattle, Wash.).
Binding buffer A (BB-A) for the pull-down assays contained 20 mM HEPES (pH 7.0), 100 mM KCl, 2 mM MgCl2, 1 mM EDTA, 10% glycerol, 1 mM dithiothreitol, 0.1% NP-40, and 1% calf serum. For the binding reaction, 5 to 10 mCi of each 35S-labeled protein was mixed in 300 μl of BB-A with the GST beads taken at volumes giving equal amounts of each immobilized protein as measured by 35S counts of the corresponding SDS-polyacrylamide gel band. Reaction mixtures were sealed, incubated in a cold room for 2 h in a rotating tube platform, and loaded into the upper chambers of the catcher tubes (CytoSignal), which allowed us to wash the beads (with BB-A containing higher salt concentrations [100 to 300 mM KCl] and no MgCl2) by repeatedly collecting the buffer pushed from the upper chamber to the lower chamber by a low-speed centrifugation without losing any material. Radiolabeled proteins from the beads that had been washed six times were eluted by 50 μl of the SDS-polyacrylamide gel loading buffer, run together with the rainbow markers on the 12.5% polyacrylamide Bio-Rad minigels, and visualized on X-ray films by fluorographic enhancement of the 35S signal.
RESULTS
Generation of mice with a single mutated CTCF target site at the H19 ICR.
Mouse ES cells were electroporated with the targeting vector, and a homologous recombination event was identified by Southern blotting, as outlined in Fig. 1A. The ES cell clone (clone 121) with an identified single-site mutation at the fourth CTCF target site (Fig. 1A) (confirmed by sequencing the entire ICR of F1 offspring [results not shown]) was injected into blastocysts, and chimeras were obtained. The neomycin selection cassette flanked with loxP sites was deleted by mating with β-actin Cre deleter mice (Fig. 1A). Mice were bred through both parental germ lines and crossed with mice of the SD7 strain for polymorphism, and tissues were collected from 1-day-old neonatal mice for the assays. ChIP assays revealed that CTCF interacted with both regions A (sites 1 and 2) and B (sites 3 and 4) within the maternally inherited, mutated H19 ICR in mouse fetal liver (data not shown).
A single CTCF target site mutation leads to loss of Igf2 imprinting.
RT-PCR analysis was performed to assess the imprinting status of Igf2. A BsaAI polymorphic site between M. m. domesticus and SD7 mice allowed identification of the allelic origin (6). Figure 2A shows that biallelic expression of Igf2 could be documented in a wide range of tissues after maternal transmission of the mutation, while the imprinted status of Igf2 was maintained after paternal transmission of the mutant H19 ICR allele. In all instances, we could not document any changes in the imprinted state of the H19 gene (data not shown).
FIG. 2.
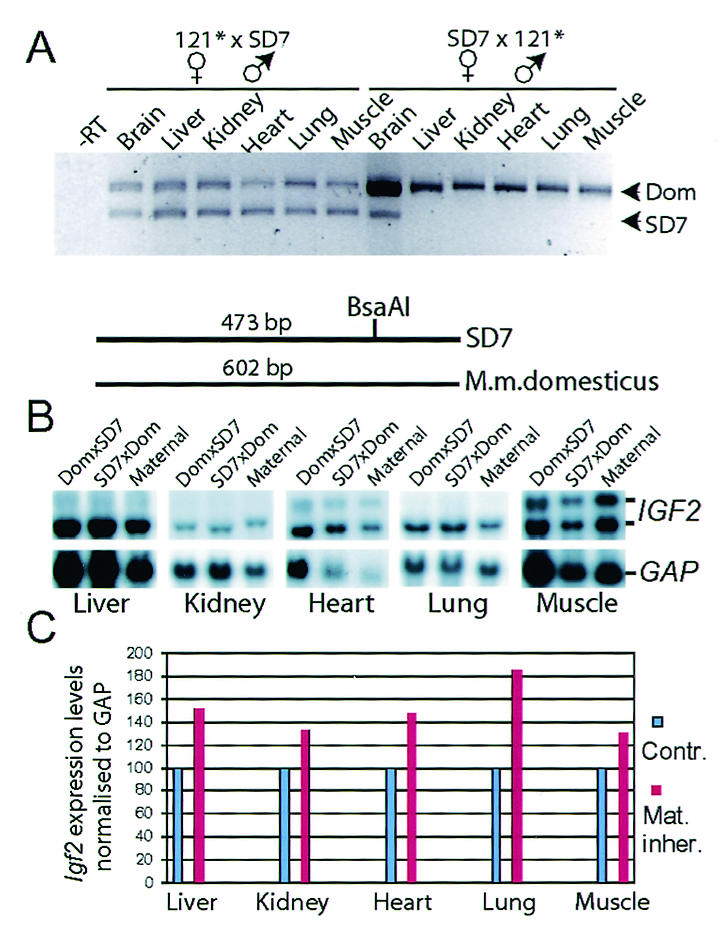
Expression analyses of Igf2. (A) RT-PCR analyses of Igf2 expression pattern in various organs dissected from neonatal offspring after reciprocal crosses between 121 and SD7 mice. A polymorphic BsaAI site in exon 4 of Igf2, present only in SD7 mice, was used to discriminate between the alleles. The animal crosses are presented in the order of female to male. The −RT lane contained RNA and primers but no reverse transcriptase for a control. (B) Northern blot analysis of Igf2 expression in neonatal mice harboring the mutated allele on the maternal chromosome. The blot was stripped and reprobed with the glyceraldehyde-3-phosphate dehydrogenase (GAP) probe to confirm RNA loading. (C) Graph showing Igf2 expression after the levels were normalized to GAP expression. The level of wild-type Igf2 expression was set at 100%. Contr., contributed; Mat. inher., maternally inherited.
To ascertain whether biallelic expression of Igf2 generated a higher level of Igf2 expression, we performed Northern blot hybridization analysis on total cellular RNA extracted from neonatal tissues. Figure 2B shows that the Igf2 mRNA levels, normalized against glyceraldehyde-3-phosphate dehydrogenase mRNA levels, were significantly higher when the mutant H19 ICR allele was inherited maternally and that Igf2 mRNA was found in a wide range of tissues. This effect on Igf2 expression was accompanied with a decrease in H19 expression (not shown).
Differential binding efficiencies between the CTCF target sites.
Given the importance of site 4 in in vivo chromatin insulation, we wanted to examine the possibility that this site displayed a particularly high affinity towards CTCF. To this end, we labeled fragments with sites 1, 3, and 4 with the CTCF target site positioned in the middle and examined the ability of a cold fragment encompassing site 4 to compete out CTCF binding in band shift assays. Surprisingly, this approach revealed that site 4 displayed a markedly weaker binding efficiency to CTCF than sites 1 and 3 (Fig. 3). We conclude that the loss of Igf2 imprinting with a mutated site 4 inherited maternally cannot be explained by binding properties alone at individual CTCF target sites.
FIG. 3.
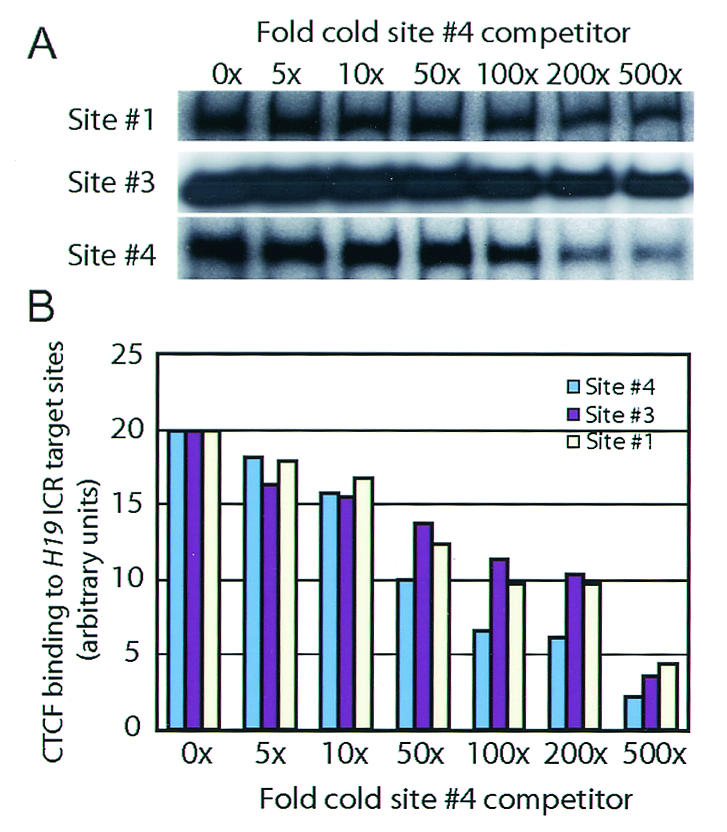
Binding efficiencies of the H19 ICR CTCF target sites. (A) CTCF target sites 1, 2, and 4 were labeled with 32P and competed with cold site 4 in band shift analyses. (B) The graph depicts the mean values for two independent experiments describing the degree of competition on individual sites in relation to fold increases in the amount of the cold competitor.
The single-site mutation leads to complex epigenetic changes at the H19 ICR.
Because the single-site mutation of site 4 resulted in loss of Igf2 imprinting and our demonstration of a direct physical interaction between CTCF-DNA complexes, it was essential to examine whether the methylation states of sites 1 to 3 were affected in the 121* mice. The methylation patterns were first determined by Southern blot analysis of neonatal tissue DNA specimens, which were isolated from tissues of reciprocal parental crosses between mutant and SD7 mice. Genomic DNA was isolated from single liver, lung, and brain tissue samples, while five or six tissue samples from the same cross were pooled for DNA isolation of heart, kidney, and muscle tissues. The DNA specimens were (in triplicate) restricted with DraI, BamHI, BstX1, and the methylation-sensitive enzyme HhaI, which has a single site in region A harboring CTCF sites 1 and 2 and five sites in region B, including one at site 3 and another between sites 3 and 4 (Fig. 4A). Figure 4A shows a representative analysis revealing that paternal transmission of the mutant H19 ICR allele did not affect its normal allelic methylation pattern. Conversely, there was a diverse pattern of gain of DNA methylation of the maternally inherited mutant H19 ICR allele: while no methylation gain was observed in the brain tissue, a moderate de novo methylation was observed for liver and kidney tissue, and a more extensive methylation pattern was observed for heart, lung, and muscle tissue, at a region harboring CTCF target sites 3 and 4 (Fig. 4A).
FIG. 4.

CpG methylation analyses. (A) Southern blot analysis of genomic DNA isolated from various neonatal organs of reciprocal crosses between 121 and SD7 mice. The various tissues were digested with HhaI (+) or not digested with HhaI (−). The positions of molecular size markers (in kilobases) are indicated to the right of the blots. The parental alleles were discriminated by exploiting a polymorphic BstXI site, which is shown in the map below the blots. (B) Bisulfite sequencing analyses of the maternally transmitted mutant H19 ICR allele covering all four CTCF target sites. Region A contained sites 1 and 2, while region B contained sites 3 and 4. The wild-type and mutant alleles were discriminated by known single-base polymorphism in region A and by the introduced point mutation in region B. Filled circles represent methylated CpGs, and open circles represent unmethylated CpGs. (C) Graphical representation of the percentage of individual CpG methylation in H19 ICR.
Given the modest resolution of the Southern blot hybridization analyses, we performed bisulfite sequencing analyses at individual CpGs at or around all four CTCF target sites of the mutant allele upon maternal inheritance. Figure 4B and C show that target site 4 has gained de novo methylation, albeit to different degrees, in both liver and heart tissue. We were surprised to note, however, that site 3 also gained methylation on the maternal allele when the neighboring site 4 was mutated in a subpopulation of liver cells. Moreover, in some instances, wild-type site 3 was heavily methylated, while mutated site 4 was unmethylated on the maternal chromosome (Fig. 4B and C). We interpret these data to mean that there is a cooperativity between sites 3 and 4 and that the deletion of one site sets the stage for de novo methylation of either site in a stochastic manner. In line with this deduction, we observe that while the Southern blot hybridization analyses revealed considerable methylation at site 3 in heart tissue, based on pooled tissue specimens, the bisulfite methylation analysis of an individual organ revealed a variant methylation pattern.
In contrast to the more or less dramatic differences in methylation states at sites 3 and 4 of region B, the CTCF target sites (sites 1 and 2) of region A were moderately affected by mutation of site 4 (Fig. 4B and C). Although we note that the methylation pattern of sites 1 and 2 appears to be reciprocal between liver and heart tissue (Fig. 4B and C), the data are not extensive enough to allow a firm conclusion on these lines.
CTCF interactions in vitro and in cultured cells.
To explore the possibility of physical interactions between CTCF-DNA complexes, we tested fragments containing sites 3 and 4 independently and in combination in CTCF band shift assays. Figure 5A shows that neither of the sites by themselves revealed any interaction between CTCF-DNA complexes, since the CTCF-DNA interaction was represented by a single band in each instance. Importantly, when CTCF-DNA complexes representative for each of these sites were mixed, an additional and prominent low-mobility band (band 3 in Fig. 5A) could be observed. After elution and purification, it was evident that both sites 3 and 4 were included in this band. We conclude that CTCF-DNA complexes within the H19 ICR interact in a heterologous manner, thereby supporting our proposal that CTCF target sites interact within the H19 ICR to organize chromatin insulator and methylation protection features.
The mechanism underlying the heterologous, but not homologous, association of the CTCF-DNA complexes might reflect conformational changes in CTCF that varies between sites 3 and 4. This supposition draws support from our previous observation that zinc finger utilization differs between sites 3 and 4 (13). Hence, the CTCF-DNA complexes at site 3 and 4 might display a different repertoire of zinc fingers not engaged in DNA binding but available for protein-protein interactions. Support for this interpretation is provided in Fig. 5B, which shows an analysis of the ability of various GST baits to interact with CTCF and its derivatives. Of these three baits, only the beads with immobilized CTCF-C domain showed efficient specific binding to the 11 ZF domain alone or in the context of the full-length CTCF, retaining nearly all of the input of 35S-labeled full-length CTCF and 11 ZF domain proteins during the extensive washing procedure, which completely removed each of the 35S-labeled proteins from the beads with GST-CTCF-N or GST-ABL protein. Since the C-terminal CTCF domain (in GST-CTCF-C) binds to the individual 11 ZF domain and to the full-length CTCF, it is likely that the C terminus in the context of the full-length CTCF is not shielding intramolecularly the same regions of the 11 ZF domain (or any sets of individual zinc fingers) that are required for binding to the C terminus intermolecularly.
To test whether CTCF can interact with itself in living cells, the pHHc-myc plasmid (15) was transfected into COS 7 cells. The cell lysates produced were then used for the coimmunoprecipitation experiment with anti-myc-tagged antibody (9E10). As shown in Fig. 5C, two bands appear after immunoprecipitation with the anti-myc-tagged CTCF antibodies. The ∼175-kDa upper band represents the myc-tagged CTCF (15), and the second lower band (∼130 kDa [16]) represents the endogenous CTCF from the COS 7 cell lysate. No signal was observed in the immunoprecipitates obtained with the preimmune serum and anti-α-tubulin antibodies (Fig. 5C). These analyses document that two CTCF molecules can interact in living cells.
DISCUSSION
We report here that the mutation of a single CTCF target site (site 4) in the mouse H19 ICR leads to robust activation of the maternal Igf2 allele. This result was unexpected, since we show here that site 4 displays a weaker interaction to CTCF than sites 1 and 3 and since site 3 displays efficient chromatin insulation in episomal assays (13). On the other hand, the presence of multiple CTCF target sites in the H19 ICRs (four in mouse and seven in humans) implies cooperative contributions of each site to the ICR chromatin insulator and its methylation-free status. This perception was borne out by our demonstration here that site 3 became hypermethylated in a subpopulation of liver cells with a mutated site 4 on the maternal allele. Moreover, we demonstrate that the individual CTCF-DNA complexes for sites 3 and 4 interact with each other in vitro only in a heterologous manner potentially involving a pairwise interaction between the C-terminal end of one CTCF molecule with the zinc finger domain of another and that CTCF interacts with itself in vivo. This implies that the dimerization potential is controlled by DNA binding. It is possible that specific binding sites may allow zinc fingers to be engaged in protein-protein interactions, supporting the interaction of CTCF-bound site 3 with CTCF-bound site 4. Interaction of DNA-bound CTCF molecules may be a general mechanism to organize looped chromatin domains, which for example may cause a functional scaffold for active loops, insulated loops, or a more-complex structure like the active chromatin hub (21). Therefore, for the H19 ICR, we propose that CTCF target sites physically interact in a cooperative manner to manifest the methylation-free domain of the maternal H19 ICR allele and that this property is essential to insulate the maternal Igf2 from downstream enhancers. We also conclude that, contrary to popular beliefs, the gain in CpG methylation on the maternal allele does not seem to be due to linear spreading of methylation from a distant point but was restricted to CpGs at or near the CTCF target sites. Although it is uncertain whether the gain of methylation of the maternally transmitted allele in the soma is relevant to the mechanisms of the imprint acquisition in the male germ line, the observations here are compatible with a focal recruitment of the de novo methylation machinery to the H19 ICR.
Our proposal of a cooperativity of CTCF target sites in chromatin insulation is at odds with a previous report that the imprinted state of Igf2 was maintained despite deletion of sites 3 and 4 (7). To explain this discrepancy, there are several, not mutually exclusive possibilities that come to our minds. For example, genetic deletions might perturb local chromatin conformation to uncover cis-acting elements normally made unavailable by nucleosomes. Such cis elements could potentiate or antagonize the desired effects and yet go unnoticed. Indeed, the entire wild-type H19 ICR allele is covered by positioned nucleosomes, and upsetting their phasing by insertional mutagenesis abolished the insulator function and CTCF binding to its wild-type target site (14). Alternatively, the polarity of the H19 ICR insulator function (12) might be controlled by cis elements other than CTCF target sites. If these are deleted or mutated without intent, such as in large deletion knockout strategies, the polarity of the insulator might switch with ensuing loss of Igf2 imprinting.
Our results indicate strongly that the CTCF target sites within the H19 ICR cooperate to manifest both methylation protection and insulator functions. Since we have also shown that the in vitro binding strength of CTCF to several hundred novel CTCF target sites from mouse fetal liver was directly proportional to their insulating properties (R. Mukhopadhyay et al., unpublished data), we conclude that strong insulation properties might be restricted to regions in the genome displaying in essence a constitutive binding of CTCF. This might be achieved by either multiple CTCF target sites with weaker affinity or by single cis elements, which interact with CTCF in an essentially irreversible manner. However, these considerations fail to explain the strong insulation of sites 1 and 3 in transient-transfection approaches (9, 13) but absence of insulation in the endogenous position. Therefore, we propose that the CTCF target site functions at the H19 ICR are context dependent and involve an aspect of higher-order chromatin conformation that is not recapitulated at ectopic sites.
Acknowledgments
We are grateful to Wolf Reik for generously providing both SD7 mice and sequence information of the M. musculus spretus H19 ICR, to Gail Martin for providing the Cre-transgenic mouse line, and to Ragnar Mattsson at the Lund University transgenic core facility for help with blastocyst injections.
This work was supported in part by the Swedish Science Research Council (VR), the Swedish Cancer Research Foundation (CF), the Swedish Pediatric Cancer Foundation, and the Wallenberg and Lundberg Foundations, as well as Stiftelsen Wenner-Grenska Samfundet, and the intramural research funding from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (to V.L.) and the Association for International Cancer Research (IC, EK).
REFERENCES
- 1.Bartolomei, M. S., and S. M. Tilghman. 1997. Genomic imprinting in mammals. Annu. Rev. Genet. 31:493-525. [DOI] [PubMed] [Google Scholar]
- 2.Bell, A., A. West, and G. Felsenfeld. 1999. The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell 98:387-396. [DOI] [PubMed] [Google Scholar]
- 3.Bell, A. C., and G. Felsenfeld. 2000. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 405:482-485. [DOI] [PubMed] [Google Scholar]
- 4.Chernukhin, I., S. Shamsuddin, A. Robinson, A. Carne, A. Paul, A. El-Kady, V. Lobanenkov, and E. Klenova. 2000. Physical and functional interaction between two pluripotent proteins, the Y-box DNA/RNA-binding factor, YB-1, and the multivalent zinc finger factor, CTCF. J. Biol. Chem. 275:29915-29921. [DOI] [PubMed] [Google Scholar]
- 5.Davis, T., G. Yang, J. McCarrey, and M. Bartolomei. 2000. The H19 methylation imprint is erased and re-established differentially on the parental alleles during male germ cell development. Hum. Mol. Genet. 9:2885-2894. [DOI] [PubMed] [Google Scholar]
- 6.Dean, W., L. Bowden, A. Aitchison, J. Klose, J. Meneses, W. Reik, and R. Feil. 1998. Altered imprinted gene methylation and expression in completely ES cell-derived mouse fetuses: association with aberrant phenotypes. Development 125:2273-2282. [DOI] [PubMed] [Google Scholar]
- 7.Drewell, R., J. Brenton, J. Ainscough, S. Barton, K. Hilton, K. Arney, L. Dandolo, and M. Surani. 2000. Deletion of a silencer element disrupts H19 imprinting independently of a DNA methylation epigenetic switch. Development 127:3419-3428. [DOI] [PubMed] [Google Scholar]
- 8.Filippova, G., S. Fagerlie, E. Klenova, C. Myers, Y. Dehner, G. Goodwin, P. Neiman, S. Collins, and V. Lobanenkov. 1996. An exceptionally conserved transcriptional repressor, CTCF, employs different combinations of zinc fingers to bind diverged promoter sequences of avian and mammalian c-myc oncogenes. Mol. Cell. Biol. 16:2802-2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hark, A. T., C. J. Schoenherr, D. J. Katz, R. S. Ingram, J. M. Levorse, and S. M. Tilghman. 2000. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 405:486-489. [DOI] [PubMed] [Google Scholar]
- 10.Holmgren, C., K. Kanduri, G. Dell, A. Ward, R. Mukhopadhyay, M. Kanduri, V. Lobanenkov, and R. Ohlsson. 2001. CpG methylation regulates the Igf2/H19 insulator. Curr. Biol. 11:1128-1130. [DOI] [PubMed] [Google Scholar]
- 11.Horsthemke, B., M. A. Surani, T. C. James, and R. Ohlsson. 1999. The mechanisms of genomic imprinting, p. 91-118. In R. Ohlsson (ed.), Genomic imprinting: an interdisciplinary approach, vol. 25. Springer-Verlag, Berlin, Germany. [DOI] [PubMed] [Google Scholar]
- 12.Kanduri, C., C. Holmgren, G. Franklin, M. Pilartz, E. Ullerås, M. Kanduri, L. Liu, V. Ginjala, E. Ulleras, R. Mattsson, and R. Ohlsson. 2000. The 5′-flank of the murine H19 gene in an unusual chromatin conformation unidirectionally blocks enhancer-promoter communication. Curr. Biol. 10:449-457. [DOI] [PubMed] [Google Scholar]
- 13.Kanduri, C., V. Pant, D. Loukinov, E. Pugacheva, C.-F. Qi, A. Wolffe, R. Ohlsson, and V. V. Lobanenkov. 2000. Functional interaction of CTCF with the insulator upstream of the H19 gene is parent of origin-specific and methylation-sensitive. Curr. Biol. 10:853-856. [DOI] [PubMed] [Google Scholar]
- 14.Kanduri, M., C. Kanduri, P. Mariano, A. Vostrov, W. Quitsche, V. Lobanenkov, and R. Ohlsson. 2002. Multiple nucleosome positioning sites regulate the CTCF-mediated insulator function of the H19 imprinting control region. Mol. Cell. Biol. 22:3339-3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klenova, E., I. Chernukhin, A. El-Kady, R. Lee, E. Pugacheva, D. Loukinov, G. Goodwin, D. Delgado, G. Filippova, J. Leon, H. R. Morse, P. Neiman, and V. Lobanenkov. 2001. Functional phosphorylation sites in the C-terminal region of the multivalent multifunctional transcriptional factor CTCF. Mol. Cell. Biol. 21:2221-2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klenova, E. M., R. H. Nicolas, A. F. Carne, R. E. Lee, V. V. Lobanenkov, and G. H. Goodwin. 1997. Molecular weight abnormalities of the CTCF transcription factor: CTCF migrates aberrantly in SDS-PAGE and the size of the expressed protein is affected by the UTRs and sequences within the coding region of the CTCF gene. Nucleic Acids Res. 25:466-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lewandoski, M., and G. Martin. 1997. Cre-mediated chromosome loss in mice. Nat. Genet. 17:223-225. [DOI] [PubMed] [Google Scholar]
- 18.Lobanenkov, V., R. Nicolas, V. Adler, H. Paterson, E. Klenova, A. Polotskaja, and G. Goodwin. 1990. A novel sequence-specific DNA binding protein, which interacts with three regularly spaced direct repeats of the CCCTC-motif in the 5′-flanking sequence of the chicken c-myc gene. Oncogene 5:1743-1753. [PubMed] [Google Scholar]
- 19.Ohlsson, R., R. Renkawitz, and V. Lobanenkov. 2001. CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet. 17:520-527. [DOI] [PubMed] [Google Scholar]
- 20.Olek, A., J. Oswald, and J. Walter. 1996. A modified and improved method for bisulphite based cytosine methylation analysis. Nucleic Acids Res. 24:5064-5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Palstra, R. J., B. Tolhuis, E. Splinter, R. Nijmeijer, F. Grosveld, and W. de Laat. 2003. The beta-globin nuclear compartment in development and erythroid differentiation. Nat. Genet. 35:190-194. [DOI] [PubMed] [Google Scholar]
- 22.Pant, V., P. Mariano, C. Kanduri, A. Mattsson, V. Lobanenkov, R. Heuchel, and R. Ohlsson. 2003. The nucleotides responsible for the direct physical contact between the chromatin insulator protein CTCF and the H19 imprinting control region manifest parent of origin-specific long-distance insulation and methylation-free domains. Genes Dev. 17:586-590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reed, M. R., C. F. Huang, A. D. Riggs, and J. R. Mann. 2001. A complex duplication created by gene targeting at the imprinted H19 locus results in two classes of methylation and correlated Igf2 expression phenotypes. Genomics 74:186-196. [DOI] [PubMed] [Google Scholar]
- 24.Schoenherr, C., J. Levorse, and S. Tilghman. 2003. CTCF maintains differential methylation at the Igf2/H19 locus. Nat. Genet. 33:66-69. [DOI] [PubMed] [Google Scholar]
- 25.Srivastava, M., S. Hsieh, A. Grinberg, L. Williams-Simons, S. P. Huang, and K. Pfeifer. 2000. H19 and Igf2 monoallelic expression is regulated in two distinct ways by a shared cis acting regulatory region upstream of H19. Genes Dev. 14:1186-1195. [PMC free article] [PubMed] [Google Scholar]
- 26.Thorvaldsen, J. L., K. L. Duran, and M. S. Bartolomei. 1998. Deletion of the H19 differentially methylated domain results in loss of imprinted expression of H19 and Igf2. Genes Dev. 12:3693-3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tremblay, K., K. Duran, and M. Bartolomei. 1997. A 5′ 2-kilobase-pair region of the imprinted mouse H19 gene exhibits exclusive paternal methylation throughout development. Mol. Cell. Biol. 17:4322-4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tremblay, K. D., J. R. Saam, R. S. Ingram, S. M. Tilghman, and M. S. Bartolomei. 1995. A paternal-specific methylation imprint marks the alleles of the mouse H19 gene. Nat. Genet. 9:407-413. [DOI] [PubMed] [Google Scholar]